
Small Molecule Immunomodulators as Next-Generation Therapeutics for Glioblastoma



Abstract
Simple Summary
Abstract
1. Introduction
2. Immune Checkpoints
2.1. PD-1/PD-L1
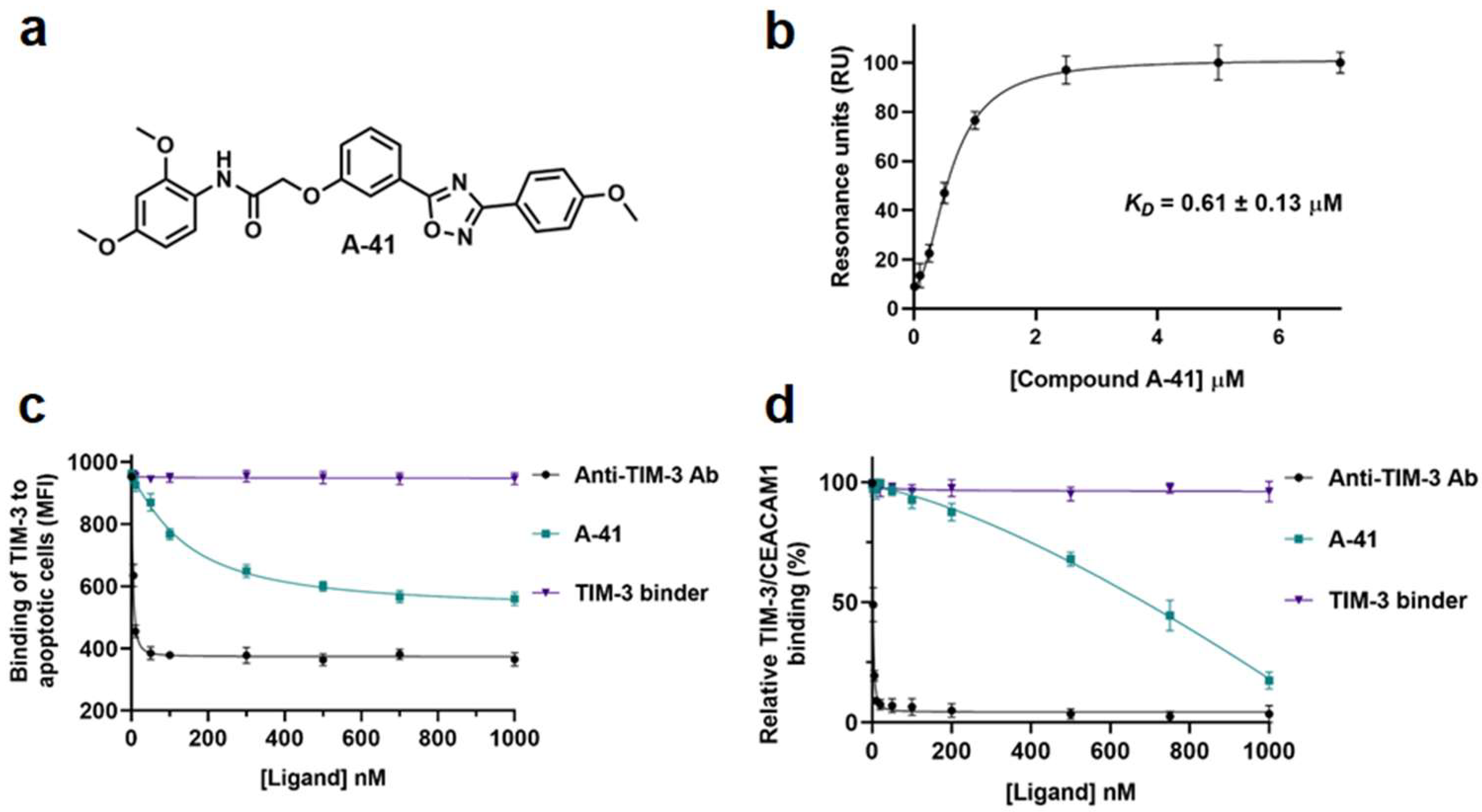
2.2. TIM-3
2.3. LAG-3
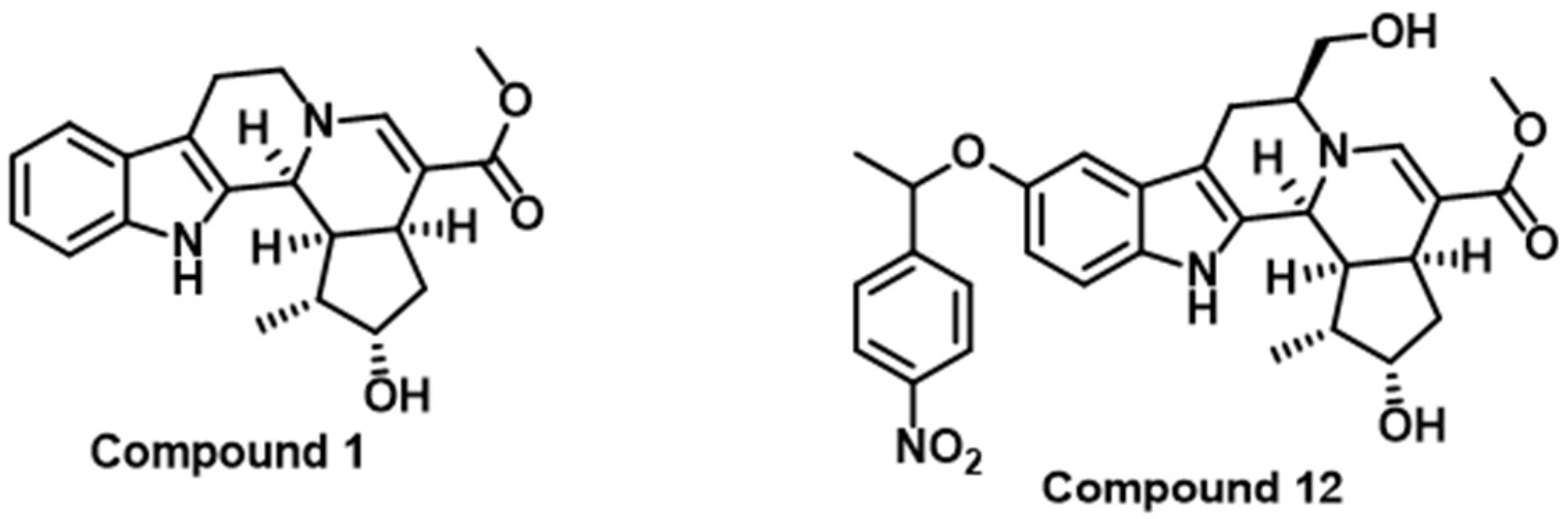
2.4. CTLA-4
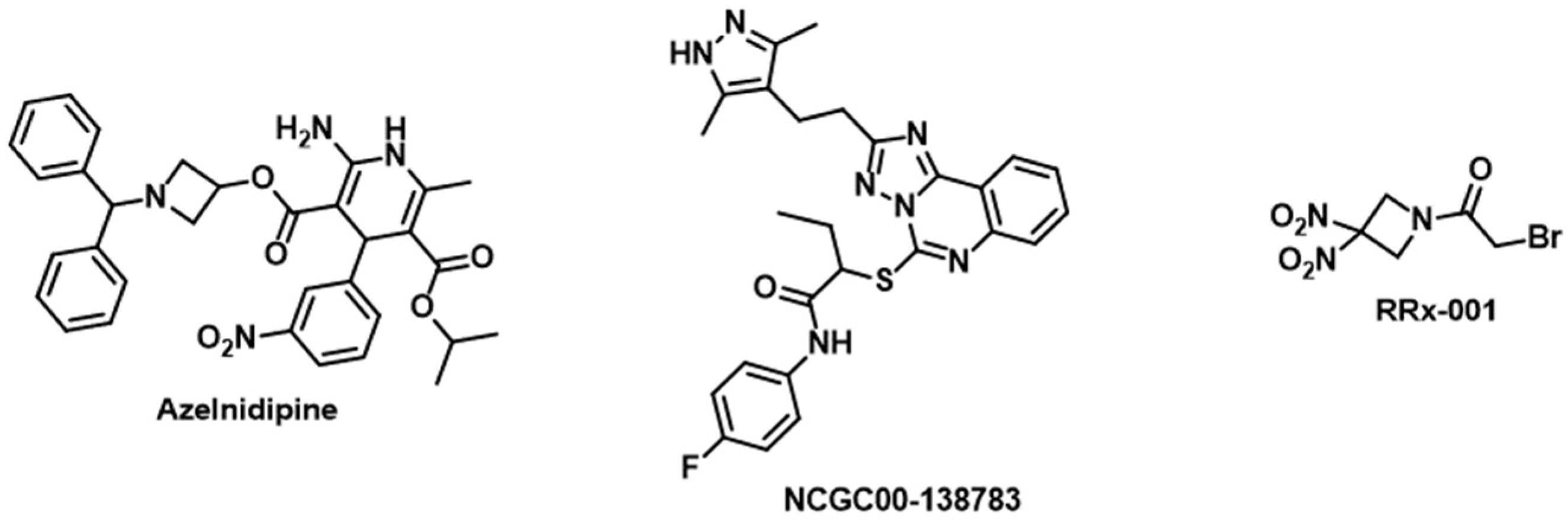
2.5. TIGIT
2.6. VISTA
2.7. ICOS
3. TAM-Related Targets
3.1. CCL2/CCR2 Axis
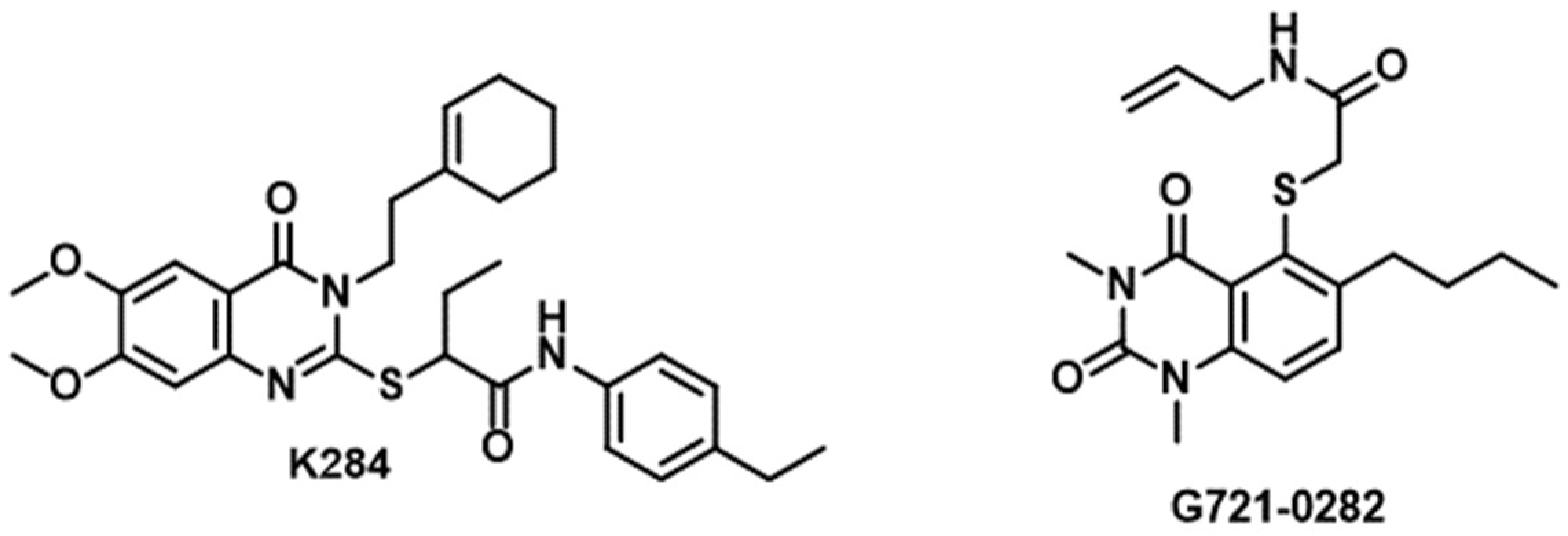
3.2. CHI3L1/Gal-3
3.3. SLIT2/ROBO
3.4. CD47
3.5. CSF-1/CSF-1R
3.6. IL-6/IL-6R
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2020, 10, 574012. [Google Scholar] [CrossRef] [PubMed]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 21, 329, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.; Odia, Y.; Khosla, A.A.; Ahluwalia, M.S. Key Clinical Principles in the Management of Glioblastoma. JCO Oncol. Pract. 2023, 19, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Marenco-Hillembrand, L.; Wijesekera, O.; Suarez-Meade, P.; Mampre, D.; Jackson, C.; Peterson, J.; Trifiletti, D.; Hammack, J.; Ortiz, K.; Lesser, E.; et al. Trends in glioblastoma: Outcomes over time and type of intervention: A systematic evidence based analysis. J. Neurooncol. 2020, 147, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.; Cree, I.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System tumours: A practical update on what neurosurgeons need to know—A minireview. Acta Neurochir. 2022, 164, 2453–2464. [Google Scholar] [CrossRef]
- Bale, T.A.; Rosenblum, M.K. The 2021 WHO Classification of Tumors of the Central Nervous System: An update on pediatric low-grade gliomas and glioneuronal tumors. Brain Pathol. 2022, 32, e13060. [Google Scholar] [CrossRef]
- van der Meulen, M.; Ramos, R.C.; Mason, W.P.; Deimling, A.; Maas, S.L.N. Opinion & Special Article: Glioma Classification. Neurology 2022, 99, 903–908. [Google Scholar]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef] [PubMed]
- van Solinge, T.S.; Nieland, L.; Chiocca, E.A.; Broekman, M.L.D. Advances in local therapy for glioblastoma—Taking the fight to the tumour. Nat. Rev. Neurol. 2022, 18, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Tang, Q.; Ren, L.; Liu, J.; Li, W.; Fu, W.; Wang, J.; Du, G. A narrative review of research progress on drug therapies for glioblastoma multiforme. Ann. Transl. Med. 2021, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Waqar, M.; Trifiletti, D.M.; McBain, C.; O’Connor, J.; Coope, D.J.; Akkari, L.; Quinones-Hinojosa, A.; Borst, G.R. Early Therapeutic Interventions for Newly Diagnosed Glioblastoma: Rationale and Review of the Literature. Curr. Oncol. Rep. 2022, 24, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Tonn, J.C.; Brada, M.; Pentheroudakis, G. High-grade malignant glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21, 190–193. [Google Scholar] [CrossRef]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef]
- Gramatzki, D.; Roth, P.; Rushing, E.J.; Weller, J.; Andratschke, N.; Hofer, S.; Korol, D.; Regli, L.; Pangalu, A.; Pless, M.; et al. Bevacizumab may improve quality of life, but not overall survival in glioblastoma: An epidemiological study. Ann. Oncol. 2018, 29, 1431–1436. [Google Scholar] [CrossRef]
- Nava, F.; Tramacere, I.; Fittipaldo, A.; Bruzzone, M.G.; DiMeco, F.; Fariselli, L.; Finocchiaro, G.; Pollo, B.; Salmaggi, A.; Silvani, A.; et al. Survival effect of first- and second-line treatments for patients with primary glioblastoma: A cohort study from a prospective registry, 1997–2010. Neuro Oncol. 2014, 16, 719–727. [Google Scholar] [CrossRef]
- Löber-Handwerker, R.; Döring, K.; Bock, C.; Rohde, V.; Malinova, V. Defining the impact of adjuvant treatment on the prognosis of patients with inoperable glioblastoma undergoing biopsy only: Does the survival benefit outweigh the treatment effort? Neurosurg. Rev. 2022, 45, 2339–2347. [Google Scholar] [CrossRef]
- Kudulaiti, N.; Zhou, Z.; Luo, C.; Zhang, J.; Zhu, F.; Wu, J. A nomogram for individualized prediction of overall survival in patients with newly diagnosed glioblastoma: A real-world retrospective cohort study. BMC Surg. 2021, 21, 238. [Google Scholar] [CrossRef]
- Baid, U.; Rane, S.U.; Talbar, S.; Gupta, S.; Thakur, M.H.; Moiyadi, A.; Mahajan, A. Overall Survival Prediction in Glioblastoma with Radiomic Features Using Machine Learning. Front. Comput. Neurosci. 2020, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Renrick, A.N.; Dunbar, Z.T.; Shanker, A. Update on the current revolution in cancer immunotherapy. Immunotherapy 2019, 11, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Paucek, R.D.; Baltimore, D.; Li, G. The Cellular Immunotherapy Revolution: Arming the Immune System for Precision Therapy. Trends Immunol. 2019, 40, 292–309. [Google Scholar] [CrossRef]
- Kelly, P.N. The Cancer Immunotherapy Revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Bausart, M.; Préat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef]
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenges. Front. Immunol. 2021, 12, 676301. [Google Scholar] [CrossRef]
- Habashy, K.J.; Mansour, R.; Moussalem, C.; Sawaya, R.; Massaad, M.J. Challenges in glioblastoma immunotherapy: Mechanisms of resistance and therapeutic approaches to overcome them. Br. J. Cancer 2022, 127, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J. Phase II trials in the era of glioblastoma immunotherapy: New mechanisms of action, familiar challenges in trial design and tumor response assessment. Neuro Oncol. 2023, 25, 1098–1099. [Google Scholar] [CrossRef] [PubMed]
- Sener, U.; Ruff, M.W.; Campian, J.L. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7046. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Buerki, R.A.; Chheda, Z.S.; Okada, H. Immunotherapy of Primary Brain Tumors: Facts and Hopes. Clin. Cancer Res. 2018, 24, 5198–5205. [Google Scholar] [CrossRef]
- Wang, X.; Lu, J.; Guo, G.; Yu, J. Immunotherapy for recurrent glioblastoma: Practical insights and challenging prospects. Cell Death Dis. 2021, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.J.; Chen, J.S.; Jain, S.; Morshed, R.A.; Haddad, A.F.; Gill, S.; Beniwal, A.; Aghi, M.K. Immunotherapy Resistance in Glioblastoma. Front. Genet. 2021, 12, 750675. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Kreatsoulas, D.; Bolyard, C.; Wu, B.X.; Cam, H.; Giglio, P.; Li, Z. Translational landscape of glioblastoma immunotherapy for physicians: Guiding clinical practice with basic scientific evidence. J. Hematol. Oncol. 2022, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ishikawa, E.; Sugii, N.; Matsuda, M. Therapeutic Strategies for Overcoming Immunotherapy Resistance Mediated by Immunosuppressive Factors of the Glioblastoma Microenvironment. Cancers 2020, 12, 1960. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.; Nie, K.; Pong, W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.-P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Green, J.J. Overcoming delivery barriers in immunotherapy for glioblastoma. Drug Deliv. Transl. Res. 2021, 11, 2302–2316. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.-Y.; Fang, D.; Latha, D.; et al. Opening of the Blood–Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clin. Cancer Res. 2021, 27, 4325–4337. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Haumann, R.; Videira, J.C.; Kaspers, G.J.L.; van Vuurden, D.G.; Hulleman, E. Overview of Current Drug Delivery Methods Across the Blood–Brain Barrier for the Treatment of Primary Brain Tumors. CNS Drugs 2020, 34, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Nah, S.Y.; Lee, K.; Choi, N.; Kim, H.N. Triculture Model of In Vitro BBB and its Application to Study BBB-Associated Chemosensitivity and Drug Delivery in Glioblastoma. Adv. Funct. Mater. 2022, 32, 2106860. [Google Scholar] [CrossRef]
- Straehla, J.P.; Hajal, C.; Safford, H.C.; Offeddu, G.S.; Boehnke, N.; Dacoba, T.; Wyckoff, J.; Kamm, R.; Hammond, P. A predictive microfluidic model of human glioblastoma to assess trafficking of blood–brain barrier-penetrant nanoparticles. Proc. Natl. Acad. Sci. USA 2022, 119, e2118697119. [Google Scholar] [CrossRef] [PubMed]
- Blethen, K.E.; Arsiwala, T.A.; Fladeland, R.A.; Sprowls, S.A.; Panchal, D.; Adkins, C.E.; Kielkowski, B.N.; Earp, L.E.; Glass, M.J.; Pritt, T.A.; et al. Modulation of the blood-tumor barrier to enhance drug delivery and efficacy for brain metastases. Neuro-Oncol. Adv. 2021, 3, v133–v143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qu, H.; Xue, X. Blood–brain barrier penetrating liposomes with synergistic chemotherapy for glioblastoma treatment. Biomater. Sci. 2022, 10, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Kurawattimath, V.; Wilson, B.; Geetha, K.M. Nanoparticle-based drug delivery across the blood-brain barrier for treating malignant brain glioma. OpenNano 2023, 10, 100128. [Google Scholar] [CrossRef]
- Ou, Z.; Li, X.; You, Y.; Liu, D.; Wang, J. Interpreting the Therapeutic Efficiency of Multifunctional Hybrid Nanostructure against Glioblastoma. ACS Omega 2023, 8, 12259–12267. [Google Scholar] [CrossRef]
- Luo, H.; Shusta, E.V. Blood-Brain Barrier Modulation to Improve Glioma Drug Delivery. Pharmaceutics 2020, 12, 1085. [Google Scholar] [CrossRef]
- Mathew, E.N.; Berry, B.C.; Yang, H.W.; Carroll, R.S.; Johnson, M.D. Delivering Therapeutics to Glioblastoma: Overcoming Biological Constraints. Int. J. Mol. Sci. 2022, 23, 1711. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Feng, Y.; Gao, S.; Mu, Q.; Liu, C. Nanotherapeutics Overcoming the Blood-Brain Barrier for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 786700. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.A.; Noorani, B.; Alqahtani, F.; Bhalerao, A.; Raut, S.; Sivandzade, F.; Cucullo, L. Understanding the brain uptake and permeability of small molecules through the BBB: A technical overview. J. Cereb. Blood Flow Metab. 2021, 41, 1797–1820. [Google Scholar] [CrossRef] [PubMed]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Kouhi, A.; Pachipulusu, V.; Kapenstein, T.; Hu, P.; Epstein, A.L.; Khawli, L.A. Brain Disposition of Antibody-Based Therapeutics: Dogma, Approaches and Perspectives. Int. J. Mol. Sci. 2021, 22, 6442. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hao, Z.; Mao, F.; Guo, D. Small Molecule Inhibitors in Adult High-Grade Glioma: From the Past to the Future. Front. Oncol. 2022, 12, 911876. [Google Scholar] [CrossRef]
- Hamer, P.C.D.W. Small molecule kinase inhibitors in glioblastoma: A systematic review of clinical studies. Neuro Oncol. 2010, 12, 304–316. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804. [Google Scholar] [CrossRef]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed]
- Mitusova, K.; Peltek, O.O.; Karpov, T.E.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S. Overcoming the blood–brain barrier for the therapy of malignant brain tumor: Current status and prospects of drug delivery approaches. J. Nanobiotechnol. 2022, 20, 412. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Ahsan, S.M.; Kumar, J.M.; Kondapi, A.K.; Rao, N.M. Overcoming blood brain barrier with a dual purpose Temozolomide loaded Lactoferrin nanoparticles for combating glioma (SERP-17-12433). Sci. Rep. 2017, 7, 6602. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chan, H.L.; Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 2019, 26, 3009–3025. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Gupta, M.; Sahasranaman, S. Immune Checkpoint inhibitors: An introduction to the next-generation cancer immunotherapy. J. Clin. Pharmacol. 2016, 56, 157–169. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, R.; Yang, A.G.; Zheng, G. Diversity of immune checkpoints in cancer immunotherapy. Front. Immunol. 2023, 14, 1121285. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Pesce, S.; Trabanelli, S.; Di Vito, C.; Greppi, M.; Obino, V.; Guolo, F.; Minetto, P.; Bozzo, M.; Calvi, M.; Zaghi, E.; et al. Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes. Cancers 2020, 12, 3504. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.Y.; Choi, J.; Jackson, C.; Lim, M. Combination immunotherapy strategies for glioblastoma. J. Neurooncol. 2021, 151, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Bausart, M.; Vanvarenberg, K.; Ucakar, B.; Lopes, A.; Vandermeulen, G.; Malfanti, A.; Préat, V. Combination of DNA Vaccine and Immune Checkpoint Blockades Improves the Immune Response in an Orthotopic Unresectable Glioblastoma Model. Pharmaceutics 2022, 14, 1025. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, V.A.; Dmello, C.; McGrail, D.J.; Brat, D.J.; Lee-Chang, C.; Heimberger, A.B.; Chand, D.; Stupp, R.; Sonabend, A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Investig. 2023, 133, e163447. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Scheffel, T.B.; Grave, N.; Vargas, P.; Diz, F.M.; Rockenbach, L.; Morrone, F.B. Immunosuppression in Gliomas via PD-1/PD-L1 Axis and Adenosine Pathway. Front. Oncol. 2021, 10, 617385. [Google Scholar] [CrossRef]
- Yang, T.; Kong, Z.; Ma, W. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: Clinical studies, challenges and potential. Hum. Vaccin. Immunother. 2021, 17, 546–553. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Sasikumar, P.G.; Ramachandra, M. Small Molecule Agents Targeting PD-1 Checkpoint Pathway for Cancer Immunotherapy: Mechanisms of Action and Other Considerations for Their Advanced Development. Front. Immunol. 2022, 13, 752065. [Google Scholar] [CrossRef] [PubMed]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Błaszkiewicz, U.; Pustuła, M.; Butera, R.; Dömling, A.; Holak, T.A. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef]
- Koblish, H.K.; Wu, L.; Wang, L.S.; Liu, P.C.C.; Wynn, R.; Rios-Doria, J.; Spitz, S.; Liu, H.; Volgina, A.; Zolotarjova, N.; et al. Characterization of INCB086550: A Potent and Novel Small-Molecule PD-L1 Inhibitor. Cancer Discov. 2022, 12, 1482–1499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, Y.; Yu, C.; Du, H.; Liu, J.; Li, H.; Huang, S.; Zhu, Q.; Xu, Y.; Zou, Y. Discovery of Novel Small-Molecule Inhibitors of PD-1/PD-L1 Interaction via Structural Simplification Strategy. Molecules 2021, 26, 3347. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, M.; Musielak, B.; Kocik, J.; Skalniak, L.; Sala, D.; Czub, M.; Magiera-Mularz, K.; Rodriguez, I.; Myrcha, M.; Stec, M.; et al. Di-bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J. Med. Chem. 2020, 63, 11271–11285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, S.; Plewka, J.; Wu, C.; Zhu, M.; Yu, Q.; Musielak, B.; Wang, X.; Awadasseid, A.; Magiera-Mularz, K.; et al. Design, Synthesis, and Antitumor Activity Evaluation of 2-Arylmethoxy-4-(2,2′-dihalogen-substituted biphenyl-3-ylmethoxy) Benzylamine Derivatives as Potent PD-1/PD-L1 Inhibitors. J. Med. Chem. 2023, 66, 10579–10603. [Google Scholar] [CrossRef] [PubMed]
- Ważyńska, M.A.; Butera, R.; Requesens, M.; Plat, A.; Zarganes-Tzitzikas, T.; Neochoritis, C.G.; Plewka, J.; Skalniak, L.; Kocik-Krol, J.; Musielak, B.; et al. Design, Synthesis, and Biological Evaluation of 2-Hydroxy-4-phenylthiophene-3-carbonitrile as PD-L1 Antagonist and Its Comparison to Available Small Molecular PD-L1 Inhibitors. J. Med. Chem. 2023, 66, 9577–9591. [Google Scholar] [CrossRef] [PubMed]
- Butera, R.; Ważyńska, M.; Magiera-Mularz, K.; Plewka, J.; Musielak, B.; Surmiak, E.; Sala, D.; Kitel, R.; de Bruyn, M.; Nijman, H.W.; et al. Design, Synthesis, and Biological Evaluation of Imidazopyridines as PD-1/PD-L1 Antagonists. ACS Med. Chem. Lett. 2021, 12, 768–773. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Komrokji, R.S.; Brunner, A.M. TIM-3 pathway dysregulation and targeting in cancer. Expert Rev. Anticancer Ther. 2021, 21, 523–534. [Google Scholar] [CrossRef]
- Liu, Z.; Han, H.; He, X.; Li, S.; Wu, C.; Yu, C.; Wang, S. Expression of the galectin-9-Tim-3 pathway in glioma tissues is associated with the clinical manifestations of glioma. Oncol. Lett. 2016, 11, 1829–1834. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.A.; Talagayev, V.; Pach, S.; Wolber, G.; Gabr, M.T. Discovery of Small-Molecule TIM-3 Inhibitors for Acute Myeloid Leukemia Using Pharmacophore-Based Virtual Screening. J. Med. Chem. 2023, 66, 11464–11475. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Workman, C.J.; Vignali, D.A.A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 2023, 24, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.A.; Rehman, A.U.; Gabr, M.T. Discovery of First-in-Class Small Molecule Inhibitors of Lymphocyte Activation Gene 3 (LAG-3). ACS Med. Chem. Lett. 2023, 14, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.A.; Zhang, L.; Gabr, M.T. Development of a high-throughput TR-FRET screening assay for LAG-3/FGL1 interaction. SLAS Discov. 2023, 28, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020, 20, 7. [Google Scholar] [CrossRef]
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: The Ipi-Glio trial protocol. BMC Cancer 2020, 20, 198. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ichinohe, K.; Sugawara, A.; Kida, S.; Murase, S.; Zhang, J.; Yamada, O.; Hattori, T.; Oshima, Y.; Kikuchi, H. Development of Indole Alkaloid-Type Dual Immune Checkpoint Inhibitors Against CTLA-4 and PD-L1 Based on Diversity-Enhanced Extracts. Front. Chem. 2021, 9, 766107. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Lucca, L.E.; Lerner, B.A.; Park, C.; DeBartolo, D.; Harnett, B.; Kumar, V.P.; Ponath, G.; Raddassi, K.; Huttner, A.; Hafler, D.A.; et al. Differential expression of the T-cell inhibitor TIGIT in glioblastoma and MS. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e712. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Du, J.; Wang, H.; Chen, C.; Jiao, L.; Cheng, X.; Zhou, X.; Chen, S.; Gou, S.; Zhao, W.; et al. Repositioning liothyronine for cancer immunotherapy by blocking the interaction of immune checkpoint TIGIT/PVR. Cell Commun. Signal 2020, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiao, L.; Qian, Y.; Dong, Q.; Sun, Y.; Zheng, W.V.; Zhao, W.; Zhai, W.; Qiu, L.; Wu, Y.; et al. Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules 2021, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Tagliamento, M.; Agostinetto, E.; Borea, R.; Brandão, M.; Poggio, F.; Addeo, A.; Lambertini, M. VISTA: A Promising Target for Cancer Immunotherapy? Immunotargets Ther. 2021, 10, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Ghouzlani, A.; Lakhdar, A.; Rafii, S.; Karkouri, M.; Badou, A. The immune checkpoint VISTA exhibits high expression levels in human gliomas and associates with a poor prognosis. Sci. Rep. 2021, 11, 21504. [Google Scholar] [CrossRef]
- Gabr, M.T.; Gambhir, S.S. Discovery and Optimization of Small-Molecule Ligands for V-Domain Ig Suppressor of T-Cell Activation (VISTA). J. Am. Chem. Soc. 2020, 142, 16194–16198. [Google Scholar] [CrossRef]
- Amatore, F.; Gorvel, L.; Olive, D. Role of Inducible Co-Stimulator (ICOS) in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 141–150. [Google Scholar] [CrossRef]
- Wang, J.; Shi, F.; Shan, A. Transcriptome profile and clinical characterization of ICOS expression in gliomas. Front. Oncol. 2022, 12, 946967. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, S.A.; Świderek, K.; Gabr, M.T. First-in-class small molecule inhibitors of ICOS/ICOSL interaction as a novel class of immunomodulators. RSC Med. Chem. 2023, 14, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Zweemer, A.J.; Nederpelt, I.; Vrieling, H.; Hafith, S.; Doornbos, M.L.; de Vries, H.; Abt, J.; Gross, R.; Stamos, D.; Saunders, J.; et al. Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Mol. Pharmacol. 2013, 84, 551–561. [Google Scholar] [CrossRef]
- Wu, X.; Singh, R.; Hsu, D.K.; Zhou, Y.; Yu, S.; Han, D.; Shi, Z.; Huynh, M.; Campbell, J.J.; Hwang, S.T. A Small Molecule CCR2 Antagonist Depletes Tumor Macrophages and Synergizes with Anti-PD-1 in a Murine Model of Cutaneous T-Cell Lymphoma (CTCL). J. Investig. Dermatol. 2020, 140, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.F.; Zhu, T.; Mao, C.X.; Liu, Z.X.; Wang, Z.B.; Mao, X.Y.; Li, L.; Yin, J.Y.; Zhou, H.H.; Liu, Z.Q. PPIC, EMP3 and CHI3L1 Are Novel Prognostic Markers for High Grade Glioma. Int. J. Mol. Sci. 2016, 17, 1808. [Google Scholar] [CrossRef]
- Steponaitis, G.; Skiriutė, D.; Kazlauskas, A.; Golubickaitė, I.; Stakaitis, R.; Tamašauskas, A.; Vaitkienė, P. High CHI3L1 expression is associated with glioma patient survival. Diagn. Pathol. 2016, 11, 42. [Google Scholar] [CrossRef]
- Ku, B.M.; Lee, Y.K.; Ryu, J.; Jeong, J.Y.; Choi, J.; Eun, K.M.; Shin, H.Y.; Kim, D.G.; Hwang, E.M.; Yoo, J.C.; et al. CHI3L1 (YKL-40) is expressed in human gliomas and regulates the invasion, growth and survival of glioma cells. Int. J. Cancer 2011, 128, 1316–1326. [Google Scholar] [CrossRef]
- Shao, R.; Taylor, S.L.; Oh, D.S.; Schwartz, L.M. Vascular heterogeneity and targeting: The role of YKL-40 in glioblastoma vascularization. Oncotarget 2015, 6, 40507–40518. [Google Scholar] [CrossRef]
- Chen, W.J.; Zhang, X.; Han, H.; Lv, J.N.; Kang, E.M.; Zhang, Y.L.; Liu, W.P.; He, X.S.; Wang, J.; Wang, G.H.; et al. The different role of YKL-40 in glioblastoma is a function of MGMT promoter methylation status. Cell Death Dis. 2020, 11, 668. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Jiang, Y.; Li, Z.; Wu, L.; Santiago, U.; Zou, H.; Cai, C.; Sharma, V.; Guan, Y.; McCarl, L.H.; et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J. Clin. Investig. 2021, 131, e147552. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Yu, J.E.; Kim, K.C.; Lee, D.C.; Son, D.; Lee, H.; Jung, J.; Kim, N.; Ham, Y.; Yun, J.; et al. A small molecule targeting CHI3L1 inhibits lung metastasis by blocking IL-13Rα2-mediated JNK-AP-1 signals. Mol. Oncol. 2022, 16, 508–526. [Google Scholar] [CrossRef] [PubMed]
- Ham, H.J.; Lee, Y.S.; Lee, H.P.; Ham, Y.W.; Yun, J.; Han, S.B.; Hong, J.T. G721-0282 Exerts Anxiolytic-Like Effects on Chronic Unpredictable Mild Stress in Mice Through Inhibition of Chitinase-3-Like 1-Mediated Neuroinflammation. Front. Cell Neurosci. 2022, 16, 793835. [Google Scholar] [CrossRef] [PubMed]
- Brose, K.; Bland, K.S.; Wang, K.H.; Arnott, D.; Henzel, W.; Goodman, C.S.; Tessier-Lavigne, M.; Kidd, T. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 1999, 96, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Barak, R.; Yom-Tov, G.; Guez-Haddad, J.; Gasri-Plotnitsky, L.; Maimon, R.; Cohen-Berkman, M.; McCarthy, A.A.; Perlson, E.; Henis-Korenblit, S.; Isupov, M.N.; et al. Structural Principles in Robo Activation and Auto-inhibition. Cell 2019, 177, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Q.; Zhou, D.L.; Lei, Y.; Zheng, L.; Chen, S.X.; Gou, H.J.; Gu, Q.L.; He, X.D.; Lan, T.; Qi, C.L.; et al. Slit2/Robo1 signaling promotes intestinal tumorigenesis through Src-mediated activation of the Wnt/β-catenin pathway. Oncotarget 2015, 6, 3123–3135. [Google Scholar] [CrossRef]
- Zhou, W.J.; Geng, Z.H.; Spence, J.R.; Geng, J.G. Induction of intestinal stem cells by R-spondin 1 and Slit2 augments chemoradioprotection. Nature 2013, 501, 107–111. [Google Scholar] [CrossRef]
- Geraldo, L.H.; Xu, Y.; Jacob, L.; Pibouin-Fragner, L.; Rao, R.; Maissa, N.; Verreault, M.; Lemaire, N.; Knosp, C.; Lesaffre, C.; et al. SLIT2/ROBO signaling in tumor-associated microglia and macrophages drives glioblastoma immunosuppression and vascular dysmorphia. J. Clin. Investig. 2021, 131, e141083. [Google Scholar] [CrossRef]
- Yang, H.; Xun, Y.; You, H. The landscape overview of CD47-based immunotherapy for hematological malignancies. Biomark. Res. 2023, 11, 15. [Google Scholar] [CrossRef]
- Ma, D.; Liu, S.; Lal, B.; Wei, S.; Wang, S.; Zhan, D.; Zhang, H.; Lee, R.S.; Gao, P.; Lopez-Bertoni, H.; et al. Extracellular Matrix Protein Tenascin C Increases Phagocytosis Mediated by CD47 Loss of Function in Glioblastoma. Cancer Res. 2019, 79, 2697–2708. [Google Scholar] [CrossRef] [PubMed]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [PubMed]
- Burgess, T.L.; Amason, J.D.; Rubin, J.S.; Duveau, D.Y.; Lamy, L.; Roberts, D.D.; Farrell, C.L.; Inglese, J.; Thomas, C.J.; Miller, T.W. A homogeneous SIRPα-CD47 cell-based, ligand-binding assay: Utility for small molecule drug development in immuno-oncology. PLoS ONE 2020, 15, e0226661. [Google Scholar] [CrossRef] [PubMed]
- Cabrales, P. RRx-001 Acts as a Dual Small Molecule Checkpoint Inhibitor by Downregulating CD47 on Cancer Cells and SIRP-α on Monocytes/Macrophages. Transl. Oncol. 2019, 12, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- De, I.; Steffen, M.D.; Clark, P.A.; Patros, C.J.; Sokn, E.; Bishop, S.M.; Litscher, S.; Maklakova, V.I.; Kuo, J.S.; Rodriguez, F.J.; et al. CSF1 Overexpression Promotes High-Grade Glioma Formation without Impacting the Polarization Status of Glioma-Associated Microglia and Macrophages. Cancer Res. 2016, 76, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Almahariq, M.F.; Quinn, T.J.; Kesarwani, P.; Kant, S.; Miller, C.R.; Chinnaiyan, P. Inhibition of Colony-Stimulating Factor-1 Receptor Enhances the Efficacy of Radiotherapy and Reduces Immune Suppression in Glioblastoma. In Vivo 2021, 35, 119–129. [Google Scholar] [CrossRef]
- Denny, W.A.; Flanagan, J.U. Small-molecule CSF1R kinase inhibitors; review of patents 2015-present. Expert Opin. Ther. Pat. 2021, 31, 107–117. [Google Scholar] [CrossRef]
- Uciechowski, P.; Dempke, W.C.M. Interleukin-6: A Masterplayer in the Cytokine Network. Oncology 2020, 98, 131–137. [Google Scholar] [CrossRef]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.P.T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef]
- Liu, Q.; Li, G.; Li, R.; Shen, J.; He, Q.; Deng, L.; Zhang, C.; Zhang, J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J. Neurooncol. 2010, 100, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; He, Z.; Duan, H.; Zhang, D.; Li, J.; Yang, H.; Dorsey, J.F.; Zou, W.; Nabavizadeh, S.A.; Bagley, S.J.; et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat. Commun. 2021, 12, 3424. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Kim, D.; Park, J.; Kim, Y.K.; Park Choo, H.Y.; Woo, H.A. Novel Small Molecule Inhibitors Targeting the IL-6/STAT3 Pathway or IL-1β. Molecules 2022, 27, 2696. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kashiwakura, I.; Yamaguchi, M.; Yoshino, H.; Tanaka, T.; Ikeda, K.; Ye, Z.; Komatsu, H.; Matsuzaki, T.; Hosoda, M. Discovery of a Novel Small-molecule Interleukin-6 Inhibitor Through Virtual Screening Using Artificial Intelligence. Med. Chem. 2022, 18, 694–700. [Google Scholar] [CrossRef]
- Nada, H.; Sivaraman, A.; Lu, Q.; Min, K.; Kim, S.; Goo, J.I.; Choi, Y.; Lee, K. Perspective for Discovery of Small Molecule IL-6 Inhibitors through Study of Structure-Activity Relationships and Molecular Docking. J. Med. Chem. 2023, 66, 4417–4433. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Mechanism |
|---|---|---|
| BMS202 | PD-L1 | Directly binds PD-L1 and induces PD-L1 dimerization [91] |
| INCB086550 | PD-L1 | Induces PD-L1 dimerization and internalization, resulting in blocking PD-L1/PD-1 [93] |
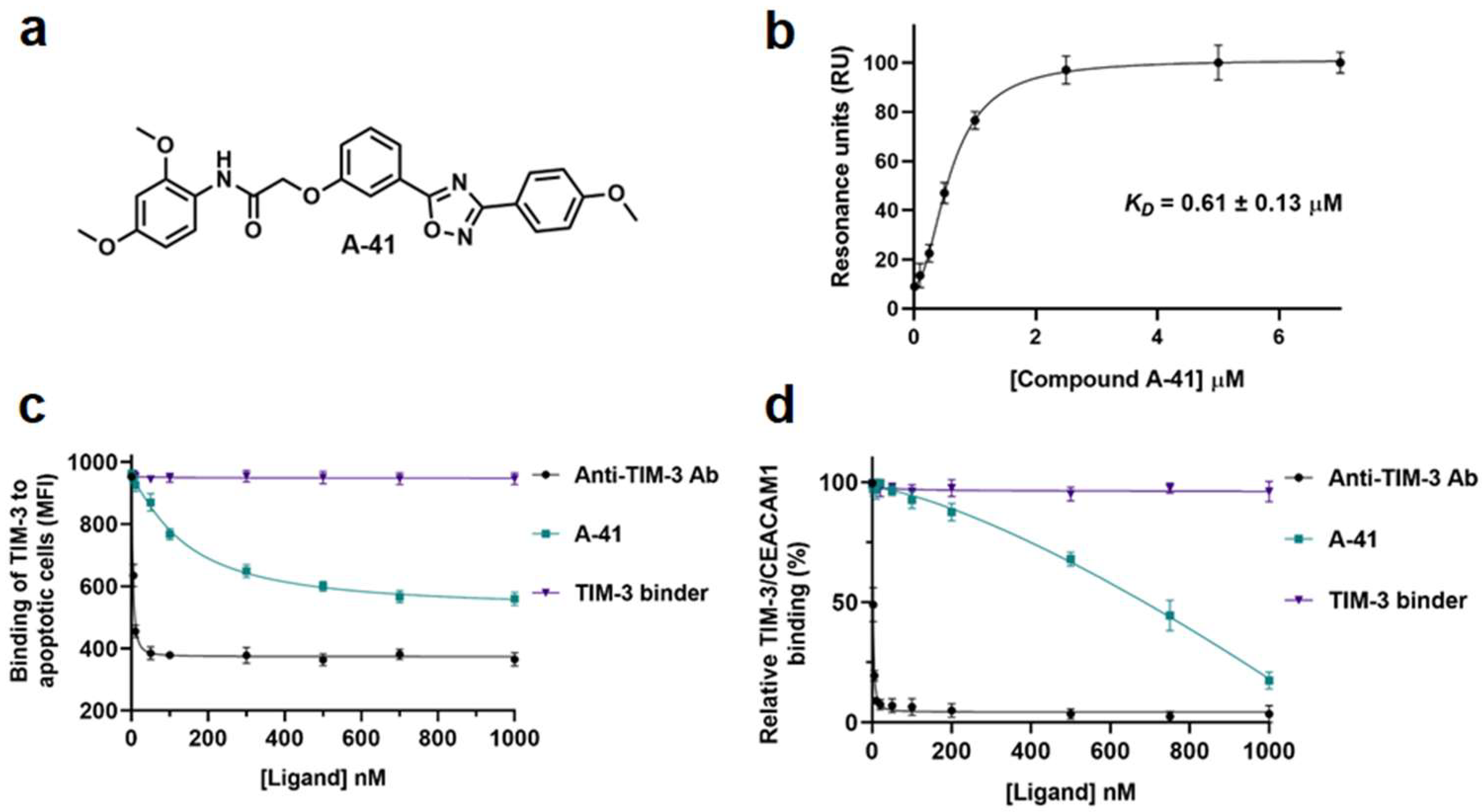
| A-41 | TIM-3 | Directly binds TIM-3 and blocks TIM-3/ligand interaction [102] |
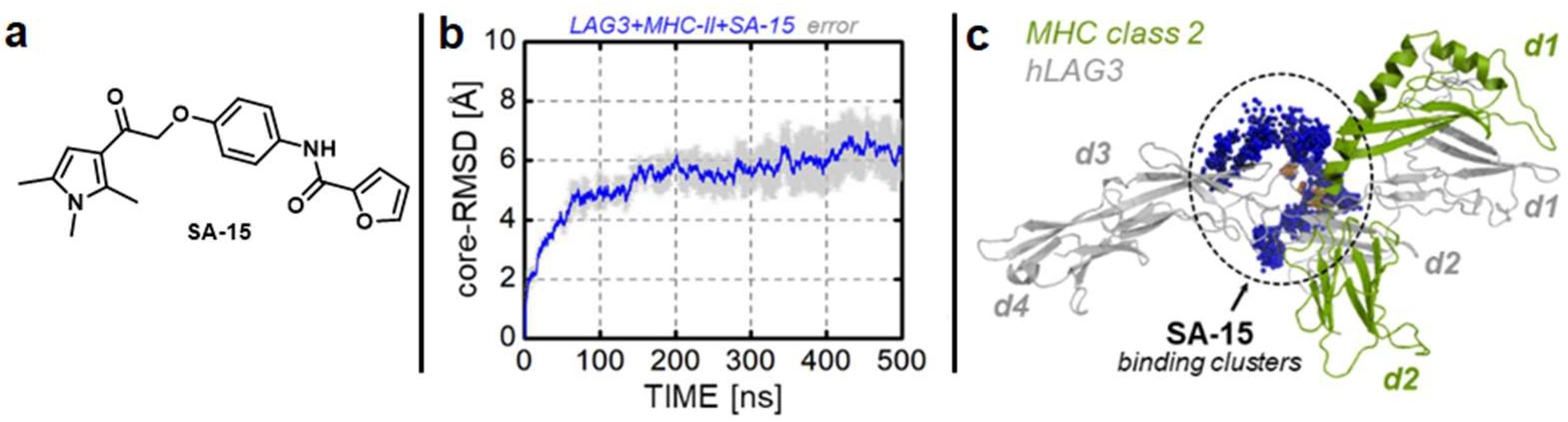
| SA-15 | LAG-3 | Directly binds LAG-3 and blocks key LAG-3 interactions [105] |
| Compound 1 | CTLA-4 | Inhibits CTLA-4 gene expression [110] |
| Compound 12 | CTLA-4 and PD-L1 | Suppresses CTLA-4 and PD-L1 gene expression as well as protein expression on cell surface [110] |
| III | VISTA | Binds VISTA and blocks key VISTA interactions [119] |
| AG-120 | ICOS | Binds near the ICOS/ICOSL interface and inhibits the interaction [120] |
| K284 | CHI3L1 | Binds CHI3L1 and prevents the binding of CHI3L1 to its receptor [133] |
| G721-0282 | CHI3L1 | Decreases the chronic unpredictable mild stress-elevated levels of CHI3L1 [134] |
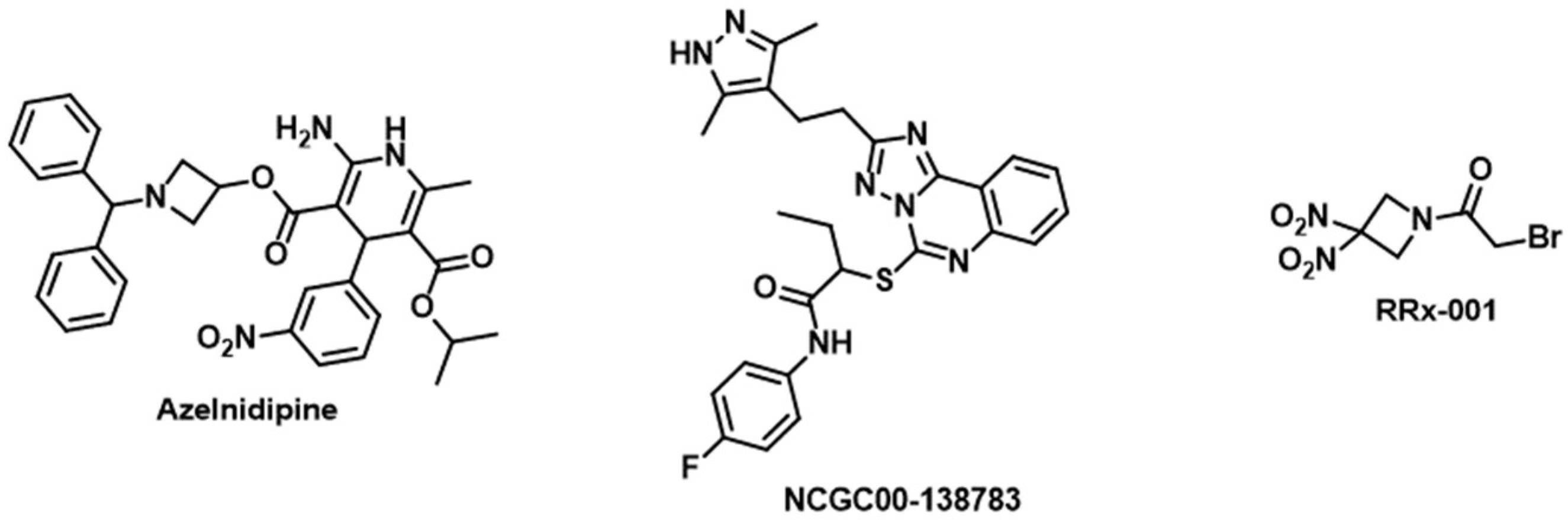
| Azelnidipine | CD47 and TIGIT | Dual inhibitor of CD47/SIRPα and TIGIT/PVR interactions [116] |
| NCGC00-138783 | CD47 | Blocks CD47/SIRPα interaction in cell-based assay [143] |
| RRx-001 | CD47 | Decreases the expression levels of CD47 and SIRPα on tumor cells and monocytes/macrophages, respectively [144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdel-Rahman, S.A.; Gabr, M. Small Molecule Immunomodulators as Next-Generation Therapeutics for Glioblastoma. Cancers 2024, 16, 435. https://doi.org/10.3390/cancers16020435
Abdel-Rahman SA, Gabr M. Small Molecule Immunomodulators as Next-Generation Therapeutics for Glioblastoma. Cancers. 2024; 16(2):435. https://doi.org/10.3390/cancers16020435
Chicago/Turabian StyleAbdel-Rahman, Somaya A., and Moustafa Gabr. 2024. "Small Molecule Immunomodulators as Next-Generation Therapeutics for Glioblastoma" Cancers 16, no. 2: 435. https://doi.org/10.3390/cancers16020435
APA StyleAbdel-Rahman, S. A., & Gabr, M. (2024). Small Molecule Immunomodulators as Next-Generation Therapeutics for Glioblastoma. Cancers, 16(2), 435. https://doi.org/10.3390/cancers16020435