
Dose Optimization of Targeted Therapies for Oncologic Indications


Abstract
Simple Summary
Abstract
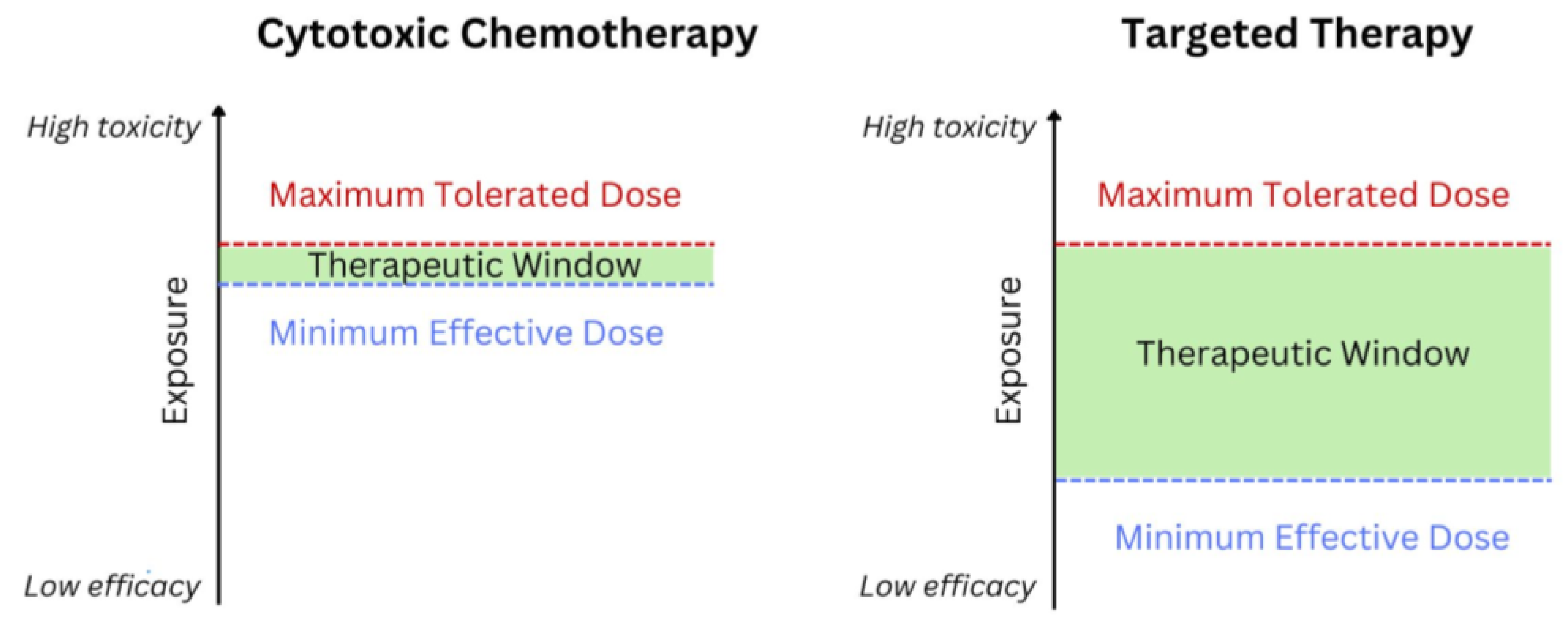
1. Introduction
2. Investigator and Sponsor-Initiated Dose Optimization in the Post-Marketing Setting
2.1. Case Study 1
2.2. Case Study 2
2.3. Case Study 3
3. FDA Strategies for Managing Dose Optimization in the Post-Marketing Setting
3.1. Case Study 4
3.2. Case Study 5
4. Shifting Dose Optimization into the Pre-Marketing Setting
Case Study 6
5. Recommendations from the FDA Guidance on Dose Optimization of Oncology Drugs
6. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Project Optimus. Available online: https://www.fda.gov/about-fda/oncology-center-excellence/project-optimus (accessed on 27 April 2024).
- Food and Drug Administration. Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases. Guidance for Industry. 2023. Available online: https://www.fda.gov/media/164555/download (accessed on 27 April 2024).
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH-E4 Guideline for Industry: Dose-Response Information to Support Drug Registration. 1994. Available online: https://www.fda.gov/media/71279/download (accessed on 27 April 2024).
- Mittapalli, R.K.; Guo, C.; Drescher, S.K.; Yin, D. Oncology dose optimization paradigms: Knowledge gained and extrapolated from approved oncology therapeutics. Cancer Chemother. Pharmacol. 2022, 90, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Loeser, A.; Kim, J.S.; Peppercorn, J.; Burkard, M.E.; Niemierko, A.; Juric, D.; Kalinsky, K.; Rugo, H.; Glenn, L.; Hodgdon, C.; et al. The Right Dose: Results of a Patient Advocate-Led Survey of Individuals with Metastatic Breast Cancer Regarding Treatment-Related Side Effects and Views About Dosage Assessment to Optimize Quality of Life. JCO Oncol. Pract. 2024, OP2300539. Available online: https://ascopubs.org/doi/10.1200/OP.23.00539 (accessed on 27 April 2024). [CrossRef]
- Loeser, A.L.; Gao, L.; Bardia, A.; Burkard, M.E.; Kalinsky, K.M.; Peppercorn, J.; Rugo, H.S.; Carlson, M.; Cowden, J.; Glenn, L.; et al. Patient-centered dosing: Oncologists’ perspectives about treatment-related side effects and individualized dosing for patients with metastatic breast cancer (MBC). Breast Cancer Res. Treat. 2022, 196, 549–563. [Google Scholar] [CrossRef]
- Food and Drug Administration. Approval Letter for Ibrutinib. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2014/205552Orig2s000ltr.pdf (accessed on 27 April 2024).
- Food and Drug Administration. Prescribing Information for Ibrutinib. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205552Orig2lbl.pdf (accessed on 27 April 2024).
- Food and Drug Administration. Medical Review Package for Ibrutinib. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205552Orig2s000MedR.pdf (accessed on 27 April 2024).
- Hou, J.Z.; Ryan, K.; Du, S.; Fang, B.; Marks, S.; Page, R.; Peng, E.; Szymanski, K.; Winters, S.; Le, H. Real-world ibrutinib dose reductions, holds and discontinuations in chronic lymphocytic leukemia. Future Oncol. 2021, 17, 4959–4969. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Nabhan, C.; Thompson, M.C.; Lamanna, N.; Brander, D.M.; Hill, B.; Howlett, C.; Skarbnik, A.; Cheson, B.D.; Zent, C.; et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: A real-world analysis. Haematologica 2018, 103, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Bose, P.; Cruz, N.D.; Jiang, Y.; Wu, Q.; Thompson, P.A.; Feng, S.; Kroll, M.H.; Qiao, W.; Huang, X.; et al. A pilot study of lower doses of ibrutinib in patients with chronic lymphocytic leukemia. Blood 2018, 132, 2249–2259. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, R.B.; Schenkel, C.; Levit, L.A.; Hu, B.; Lei, X.J.; Harvey, R.D.; Morrison, V.A.; Pollastro, T.; Waterhouse, D.; Weekes, C.; et al. Oncologists’ Perspectives on Individualizing Dose Selection for Patients with Metastatic Cancer. JCO Oncol. Pract. 2022, 18, e1807–e1817. [Google Scholar] [CrossRef]
- Food and Drug Administration. Approval Letter for Pazopanib. 2009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2009/022465s000ltr.pdf (accessed on 27 April 2024).
- Food and Drug Administration. Prescribing Information for Pazopanib. 2009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022465lbl.pdf (accessed on 27 April 2024).
- Hurwitz, H.I.; Dowlati, A.; Saini, S.; Savage, S.; Suttle, A.B.; Gibson, D.M.; Hodge, J.P.; Merkle, E.M.; Pandite, L. Phase I trial of pazopanib in patients with advanced cancer. Clin. Cancer Res. 2009, 15, 4220–4227. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Medical Review Package for Pazopanib. 2009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022465s000_MedR.pdf (accessed on 27 April 2024).
- Lubberman, F.J.E.; Gelderblom, H.; Hamberg, P.; Vervenne, W.L.; Mulder, S.F.; Jansman, F.G.A.; Colbers, A.; van der Graaf, W.T.A.; Burger, D.M.; Luelmo, S.; et al. The Effect of Using Pazopanib with Food vs. Fasted on Pharmacokinetics, Patient Safety, and Preference (DIET Study). Clin. Pharmacol. Ther. 2019, 106, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Approval Letter for Regorafenib. 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/203085Orig1s000ltr.pdf (accessed on 27 April 2024).
- Food and Drug Administration. Prescribing Information for Regorafenib. 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203085lbl.pdf (accessed on 27 April 2024).
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Büchert, M.; Fasol, U.; Unger, C.; Krätzschmar, J.; Heinig, R.; Boix, O.; et al. A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin. Cancer Res. 2012, 8, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Medical Review Package for Regorafenib. 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203085Orig1s000MedR.pdf (accessed on 27 April 2024).
- Bekaii-Saab, T.S.; Ou, F.S.; Ahn, D.H.; Boland, P.M.; Ciombor, K.K.; Heying, E.N.; Dockter, T.J.; Jacobs, N.L.; Pasche, B.C.; Cleary, J.M.; et al. Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): A randomised, multicentre, open-label, phase 2 study. Lancet Oncol. 2019, 20, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. NCCN Guidelines Version 4.2023 Colon Cancer. Available online: https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf (accessed on 27 April 2024).
- Bekaii-Saab, T.; Khan, N.; Ostojic, H.; Jiao, X.; Chen, G.; Lin, W.; Bruno, A. Real-world dosing of regorafenib and outcomes among patients with metastatic colorectal cancer: A retrospective analysis using US claims data. Ann. Oncol. 2022, 33 (Suppl. 4), S285–S286. [Google Scholar] [CrossRef]
- Food and Drug Administration. Approval Letter for Niraparib. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/208447Orig1s000Ltr.pdf (accessed on 27 April 2024).
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Prescribing Information for Niraparib. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208447lbl.pdf (accessed on 27 April 2024).
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Food and Drug Administration. Multi-Discipline Review Package for Niraparib. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208447Orig1s000MultidisciplineR.pdf (accessed on 27 April 2024).
- Berek, J.S.; Matulonis, U.A.; Peen, U.; Ghatage, P.; Mahner, S.; Redondo, A.; Lesoin, A.; Colombo, N.; Vergote, I.; Rosengarten, O.; et al. Safety and dose modification for patients receiving niraparib. Ann. Oncol. 2018, 29, 1784–1792. [Google Scholar] [CrossRef]
- Food and Drug Administration. Prescribing Information for Niraparib. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208447s015s017lbledt.pdf (accessed on 27 April 2024).
- Heiss, B.; Pan, L.; Akalu, A.; Vallejo, J.; Cheng, J.; Balcazar, P.; Rahman, N.A.; Shord, S.S.; Shah, M.; Pazdur, R.; et al. Dosage optimization in drug development: An FDA Project Optimus analysis of postmarketing requirements issued to repair the cracks. J. Clin. Oncol. 2023, 41 (Suppl. 16), 1598. [Google Scholar] [CrossRef]
- Collins, G.; McKelvey, B.; Andrews, H.S.; Allen, J.D.; Stewart, M.D. An Analysis of Dosing-Related Postmarketing Requirements for Novel Oncology Drugs Approved by the U.S. Food and Drug Administration, 2012–2022. Clin. Cancer Res. 2023, 30, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Approval Letter for Futibatinib. 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/214801Orig1s000Correctedltr.pdf (accessed on 27 April 2024).
- Bahleda, R.; Meric-Bernstam, F.; Goyal, L.; Tran, B.; He, Y.; Yamamiya, I.; Benhadji, K.A.; Matos, I.; Arkenau, H.T. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1-4 inhibitor in patients with advanced solid tumors. Ann. Oncol. 2020, 31, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; Kelley, R.K.; Cassier, P.A.; et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N. Engl. J. Med. 2023, 388, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Prescribing Information for Futibatinib. 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214801Orig1s000lbledt.pdf (accessed on 27 April 2024).
- Food and Drug Administration. Multi-Discipline Review Package for Futibatinib. 2022. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/214801Orig1s000MultidisciplineR.pdf (accessed on 27 April 2024).
- Food and Drug Administration. Approval Letter for Melphalan Flufenamide. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/214383Orig1s000ltr.pdf (accessed on 27 April 2024).
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and Dexamethasone in Heavily Pretreated Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Bringhen, S.; Voorhees, P.; Plesner, T.; Mellqvist, U.H.; Reeves, B.; Paba-Prada, C.; Zubair, H.; Byrne, C.; Chauhan, D.; et al. Melflufen plus dexamethasone in relapsed and refractory multiple myeloma (O-12-M1): A multicentre, international, open-label, phase 1-2 study. Lancet Haematol. 2020, 7, e395–e407. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Prescribing Information for Melphalan Flufenamide. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214383s000lbl.pdf (accessed on 27 April 2024).
- Food and Drug Administration and Oncopeptides. Combined FDA and Applicant Oncologic Drugs Advisory Committee Briefing Document. 22 September 2023. Available online: https://www.fda.gov/media/161678/download (accessed on 27 April 2024).
- Schjesvold, F.H.; Dimopoulos, M.A.; Delimpasi, S.; Robak, P.; Coriu, D.; Legiec, W.; Pour, L.; Špička, I.; Masszi, T.; Doronin, V.; et al. Melflufen or pomalidomide plus dexamethasone for patients with multiple myeloma refractory to lenalidomide (OCEAN): A randomised, head-to-head, open-label, phase 3 study. Lancet Haematol. 2022, 9, e98–e110. [Google Scholar] [CrossRef] [PubMed]
- Oncopeptides. Press Release: Oncopeptides Withdraws Pepaxto® in US, Scale down Organization and Focus on R&D. 22 October 2021. Available online: https://oncopeptides.com/en/media/press-releases/oncopeptides-withdraws-pepaxto-in-us-scale-down-organization-and-focus-on-rd (accessed on 27 April 2024).
- Oncopeptides. Press Release: Oncopeptides Rescinds Voluntary Withdrawal of Pepaxto in the US—No Intent to Market in the US at This Time. 21 January 2022. Available online: https://oncopeptides.com/en/media/press-releases/oncopeptides-rescinds-voluntary-withdrawal-of-pepaxto-in-the-us-no-intent-to-market-in-the-us-at-this-time/ (accessed on 27 April 2024).
- Food and Drug Administration Center for Drug Evaluation and Research. Final Summary Minutes of the Oncologic Drugs Advisory Committee Meeting September 22–23 2022. Available online: https://www.fda.gov/media/164048/download (accessed on 27 April 2024).
- Food and Drug Administration. Notice of Proposed Withdrawal of Approval; PEPAXTO (Melphalan Flufenamide) for Injection; NDA 214383. 7 July 2023. Available online: https://downloads.regulations.gov/FDA-2023-N-3167-0002/attachment_1.pdf (accessed on 27 April 2024).
- Food and Drug Administration. FDA Issues Final Decision to Withdraw Approval of Pepaxto (Melphalan Flufenamide). 23 February 2024. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-issues-final-decision-withdraw-approval-pepaxto-melphalan-flufenamide (accessed on 27 April 2024).
- BIO. Clinical Development Success Rates and Contributing Factors 2011–2020. 2021. Available online: https://go.bio.org/rs/490-EHZ-999/images/ClinicalDevelopmentSuccessRates2011_2020.pdf (accessed on 27 April 2024).
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Tracey, A. Pazdur at FDA-ASCO workshop: Failure to optimize the dose is akin to building a house on quicksand. Cancer Lett. 2022, 48. Available online: https://cancerletter.com/regulatory-news/20220506_8/ (accessed on 27 April 2024).
- Food and Drug Administration. Expedited Programs for Serious Conditions—Drugs and Biologics. Guidance for Industry. 2014. Available online: https://www.fda.gov/media/86377/download (accessed on 27 April 2024).
- Shah, M.; Rahman, A.; Theoret, M.R.; Pazdur, R. The Drug-Dosing Conundrum in Oncology—When Less Is More. N. Engl. J. Med. 2021, 385, 1445–1447. [Google Scholar] [CrossRef]
- Fourie Zirkelbach, J.; Shah, M.; Vallejo, J.; Cheng, J.; Ayyoub, A.; Liu, J.; Hudson, R.; Sridhara, R.; Ison, G.; Amiri-Kordestani, L.; et al. Improving Dose-Optimization Processes Used in Oncology Drug Development to Minimize Toxicity and Maximize Benefit to Patients. J. Clin. Oncol. 2022, 40, 3489–3500. [Google Scholar] [CrossRef]
- Simon, R.; Freidlin, B.; Rubinstein, L.; Arbuck, S.G.; Collins, J.; Christian, M.C. Accelerated titration designs for phase I clinical trials in oncology. J. Natl. Cancer Inst. 1997, 89, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.B.; Yap, C.; Carvajal, R.; Lee, S.M. Would the Recommended Dose Have Been Different Using Novel Dose-Finding Designs? Comparing Dose-Finding Designs in Published Trials. JCO Precis. Oncol. 2021, 5, PO.21.00136. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Collette, L.; Paoletti, X.; Rizzo, E.; Massard, C.; Olmos, D.; Fowst, C.; Levy, B.; Mancini, P.; Lacombe, D.; et al. Towards new methods for the determination of dose limiting toxicities and the assessment of the recommended dose for further studies of molecularly targeted agents--dose-Limiting Toxicity and Toxicity Assessment Recommendation Group for Early Trials of Targeted therapies, an European Organisation for Research and Treatment of Cancer-led study. Eur. J. Cancer 2014, 50, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, R.K.; Yin, D.; Beaupre, D.; Palaparthy, R. Starting dose selection and dose escalation for oncology small molecule first-in-patient trials: Learnings from a survey of FDA-approved drugs. Cancer Chemother. Pharmacol. 2021, 87, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Shord, S.S.; Zhu, H.; Liu, J.; Rahman, A.; Booth, B.; Zineh, I. US Food and Drug Administration embraces using innovation to identify optimized dosages for patients with cancer. CPT Pharmacomet. Syst. Pharmacol. 2023, 12, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhou, H.; Liu, S. Statistical and practical considerations in planning and conduct of dose-optimization trials. Clin. Trials 2024, 21, 17407745231207085. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. A flexible dose-response modeling framework based on continuous toxicity outcomes in phase I cancer clinical trials. Trials 2023, 24, 745. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, D.; Lin, R.; Huang, B.; Yuan, Y. Design and Sample Size Determination for Multiple-dose Randomized Phase II Trials for Dose Optimization. arXiv 2023, arXiv:2302.09612. [Google Scholar] [CrossRef]
- Guo, B.; Yuan, Y. DROID: Dose-ranging approach to optimizing dose in oncology drug development. Biometrics 2023, 79, 2907–2919. [Google Scholar] [CrossRef]
- Liu, S.Y.; Dong, X.R.; Wang, Z.; Du, Y.; Cui, J.W.; Chu, Q.; Xu, B.F.; Zheng, M.Y.; Deng, J.Y.; Lu, C.; et al. Efficacy, safety and dose selection of AZD3759 in patients with untreated EGFR-mutated non-small-cell lung cancer and central nervous system metastases in China (CTONG1702-Arm 8): A multi-center, single-arm, phase 2 trial. EClinicalMedicine 2023, 64, 102238. [Google Scholar] [CrossRef]
- Ahn, M.J.; Kim, D.W.; Cho, B.C.; Kim, S.W.; Lee, J.S.; Ahn, J.S.; Kim, T.M.; Lin, C.C.; Kim, H.R.; John, T.; et al. Activity and safety of AZD3759 in EGFR-mutant non-small-cell lung cancer with CNS metastases (BLOOM): A phase 1, open-label, dose-escalation and dose-expansion study. Lancet Respir. Med. 2017, 5, 891–902. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca. Protocol: A Phase I, Open-label, Multicentre Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Anti-Tumour Activity of AZD3759 or AZD9291 in Patients with EGFR Mutation Positive Advanced Stage Non Small Cell Lung Cancer (NSCLC). Available online: http://filehosting.pharmacm.com/DownloadService.ashx?client=CTR_MED_7111&studyid=2978&filename=BLOOM%20CSP_Redacted%20new.pdf (accessed on 27 April 2024).
- Simon, R. Optimal two-stage designs for phase II clinical trials. Control Clin. Trials 1989, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Alpha Biopharma. Press Release: Alpha Biopharma Submits New Drug Application for Zorifertinib, a Next-Generation EGFR-TKI to Treat EGFR-Mutated NSCLC Patients with CNS Metastases. 29 January 2023. Available online: https://www.prnewswire.com/news-releases/alpha-biopharma-submits-new-drug-application-for-zorifertinib-a-next-generation-egfr-tki-to-treat-egfr-mutated-nsclc-patients-with-cns-metastases-301733086.html (accessed on 27 April 2024).
- Wu, Y.L.; Zhou, Q.; Wang, J.; Yu, Y.; Xing, L.; Wang, Y.; Cheng, Y.; Pan, Y.; Fan, Y.; Shi, J.; et al. Randomized phase 3 study of first-line AZD3759 (zorifertinib) versus gefitinib or erlotinib in EGFR-mutant (EGFRm+) non–small-cell lung cancer (NSCLC) with central nervous system (CNS) metastasis. J. Clin. Oncol. 2023, 41, 9001. [Google Scholar] [CrossRef]
- Food and Drug Administration. Exposure-Response Relationships—Study Design, Data Analysis, and Regulatory Applications. Guidance for Industry. 2003. Available online: https://www.fda.gov/media/71277/download (accessed on 27 April 2024).
- Food and Drug Administration. Prescribing Information for Dasatinib. 2023. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/021986s027lbl.pdf (accessed on 27 April 2024).
- Lankheet, N.A.G.; Desar, I.M.E.; Mulder, S.F.; Burger, D.M.; Kweekel, D.M.; van Herpen, C.M.L.; van der Graaf, W.T.A.; van Erp, N.P. Optimizing the dose in cancer patients treated with imatinib, sunitinib and pazopanib. Br. J. Clin. Pharmacol. 2017, 83, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Reyner, E.; Lum, B.; Jing, J.; Kagedal, M.; Ware, J.A.; Dickmann, L.J. Intrinsic and Extrinsic Pharmacokinetic Variability of Small Molecule Targeted Cancer Therapy. Clin. Transl. Sci. 2020, 13, 410–418. [Google Scholar] [CrossRef]
- Maués, J.; Loeser, A.; Cowden, J.; Johnson, S.; Carlson, M.; Lee, S. The patient perspective on dose optimization for anticancer treatments: A new era of cancer drug dosing-Challenging the “more is better” dogma. Clin. Trials 2024, 21, 17407745241232428. [Google Scholar] [CrossRef]
- Food and Drug Administration. OCE-Funded Active Extramural Research Projects. Available online: https://www.fda.gov/about-fda/oncology-center-excellence/oce-funded-active-extramural-research-projects (accessed on 27 April 2024).
- American Society of Clinical Oncology. Press Release: ASCO Awarded $11M to Study Oral Drug Dosing Strategies in Older Adults with Metastatic Breast Cancer Receiving CDK4/6 Inhibitors. 18 July 2023. Available online: https://society.asco.org/about-asco/press-center/news-releases/asco-awarded-11m-study-oral-drug-dosing-strategies-older (accessed on 27 April 2024).



{kind=link}
{kind=link}
{kind=link}
| Drug | Approved Dose | Was the Approved Dose the MTD (or Maximum Studied Dose) in the Phase 1 Dose Escalation Study? | Design of Phase 1 Dose Escalation Study | Was Further Dose Optimization Undertaken (beyond the Phase 1 Dose Escalation Study) Prior to Approval? | Dose Investigated Post-Marketing | Impact of Post-Marketing Dose Exploration |
|---|---|---|---|---|---|---|
| Pazopanib | 800 mg without food | No | 3+3 | No | 600 mg with food | None known |
| Regorafenib | 160 mg | Yes | 3+3 | No | Dose escalation from 80 mg to 120 mg to 160 mg over the first cycle | Change to NCCN guidelines |
| Niraparib | 300 mg | Yes | Accelerated titration, 3+3 | No | 200 mg in a subset of patients | Change to prescribing information |
| Futibatinib | 20 mg | Yes | 3+3 | No | 16 mg | N/A, study is still ongoing |
| Melphanan flufenamide | 40 mg | Yes | 3+3 | No | N/A (drug withdrawn from market) | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zettler, M.E. Dose Optimization of Targeted Therapies for Oncologic Indications. Cancers 2024, 16, 2180. https://doi.org/10.3390/cancers16122180
Zettler ME. Dose Optimization of Targeted Therapies for Oncologic Indications. Cancers. 2024; 16(12):2180. https://doi.org/10.3390/cancers16122180
Chicago/Turabian StyleZettler, Marjorie E. 2024. "Dose Optimization of Targeted Therapies for Oncologic Indications" Cancers 16, no. 12: 2180. https://doi.org/10.3390/cancers16122180
APA StyleZettler, M. E. (2024). Dose Optimization of Targeted Therapies for Oncologic Indications. Cancers, 16(12), 2180. https://doi.org/10.3390/cancers16122180