

Galactosylceramide Upregulates the Expression of the BCL2 Gene and Downregulates the Expression of TNFRSF1B and TNFRSF9 Genes, Acting as an Anti-Apoptotic Molecule in Breast Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Vector Construction, Virus Production, and Transductions
2.3. Quantitative PCR (qPCR) Assay and Apoptosis Gene Expression Profiling with RT-qPCR Array
2.4. Western Blotting
2.5. Purification of Neutral GSLs
2.6. Thin-Layer Chromatogram (TLC)-Binding Assay
2.7. Cell Survival Assay
2.8. Apoptotic Assay
2.9. In-Vivo Tumor Growth Assay
2.10. Cloning of Human TNFRSF1B, TNFRSF9, and BCL2 Gene Promoters and Determination of Their Activity Using Luciferase Reporter Vectors
2.11. mRNA Stability Assay
2.12. siRNA Transfections
2.13. Statistical Analysis
3. Results
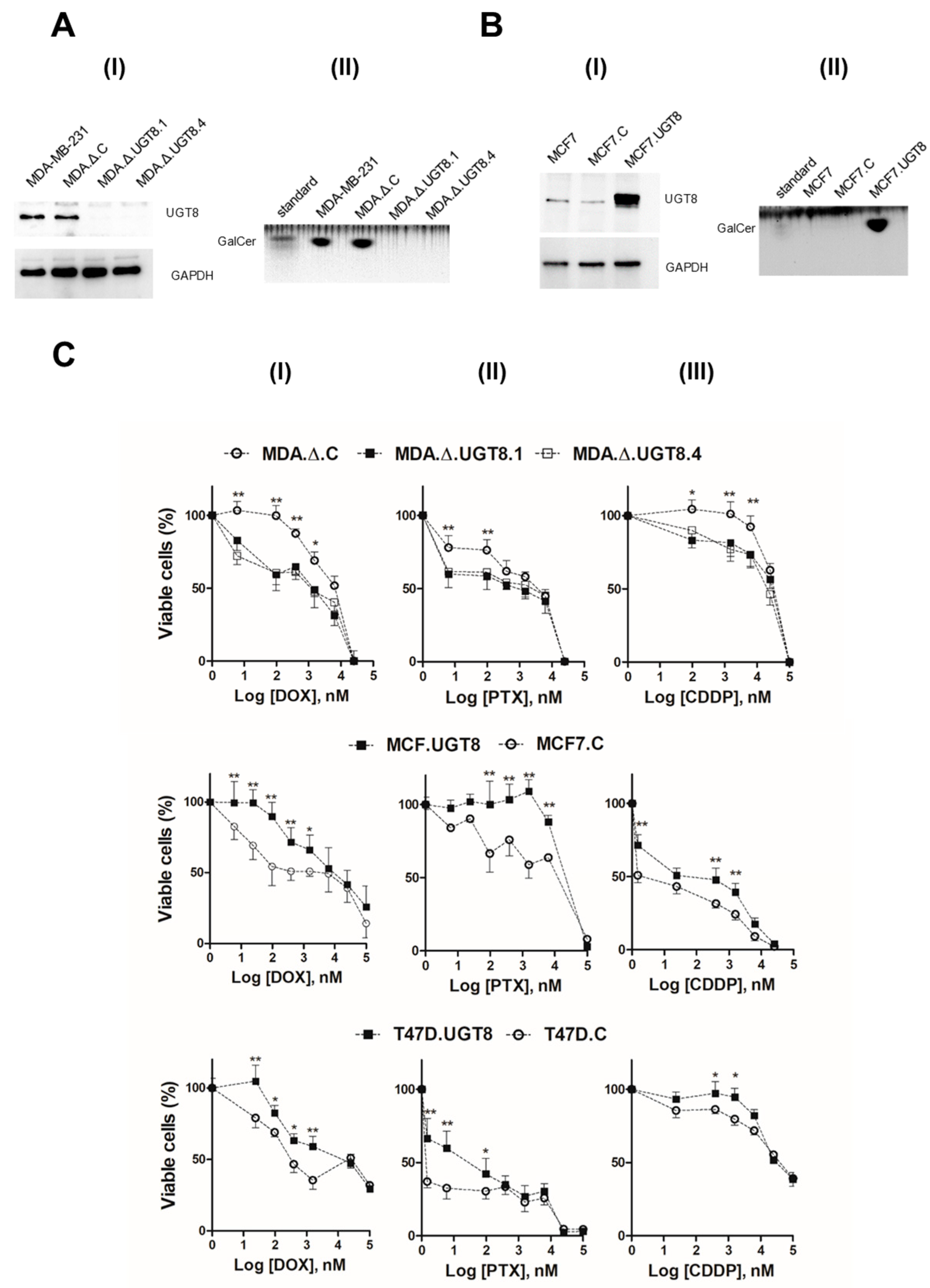
3.1. DR of BC Cells Depends on Expression of UGT8 and Synthesis of GalCer
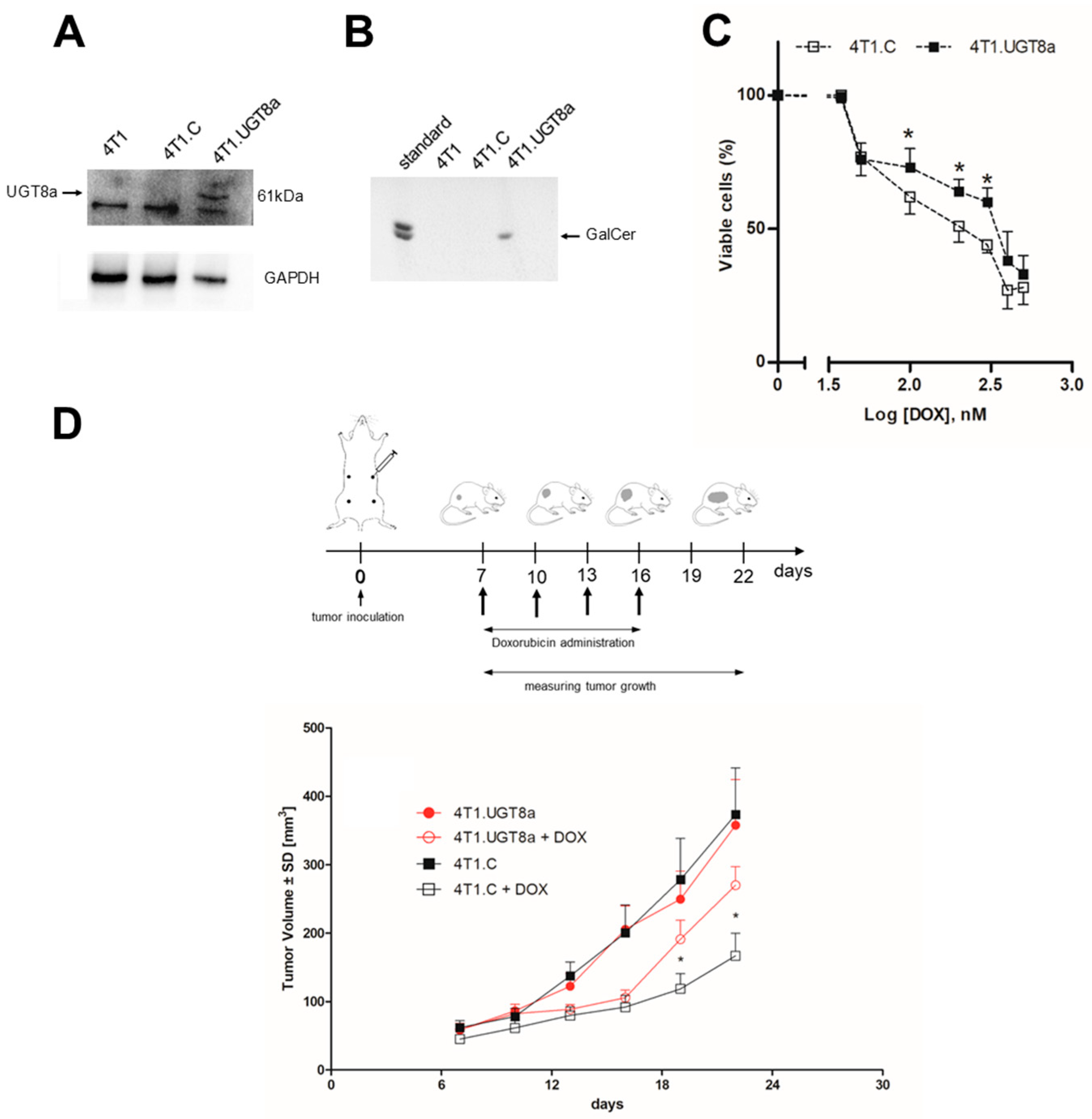
3.2. Resistance of Murine 4T1 Mammary Tumours to DOX Treatment Is Dependent on the Presence of GalCer
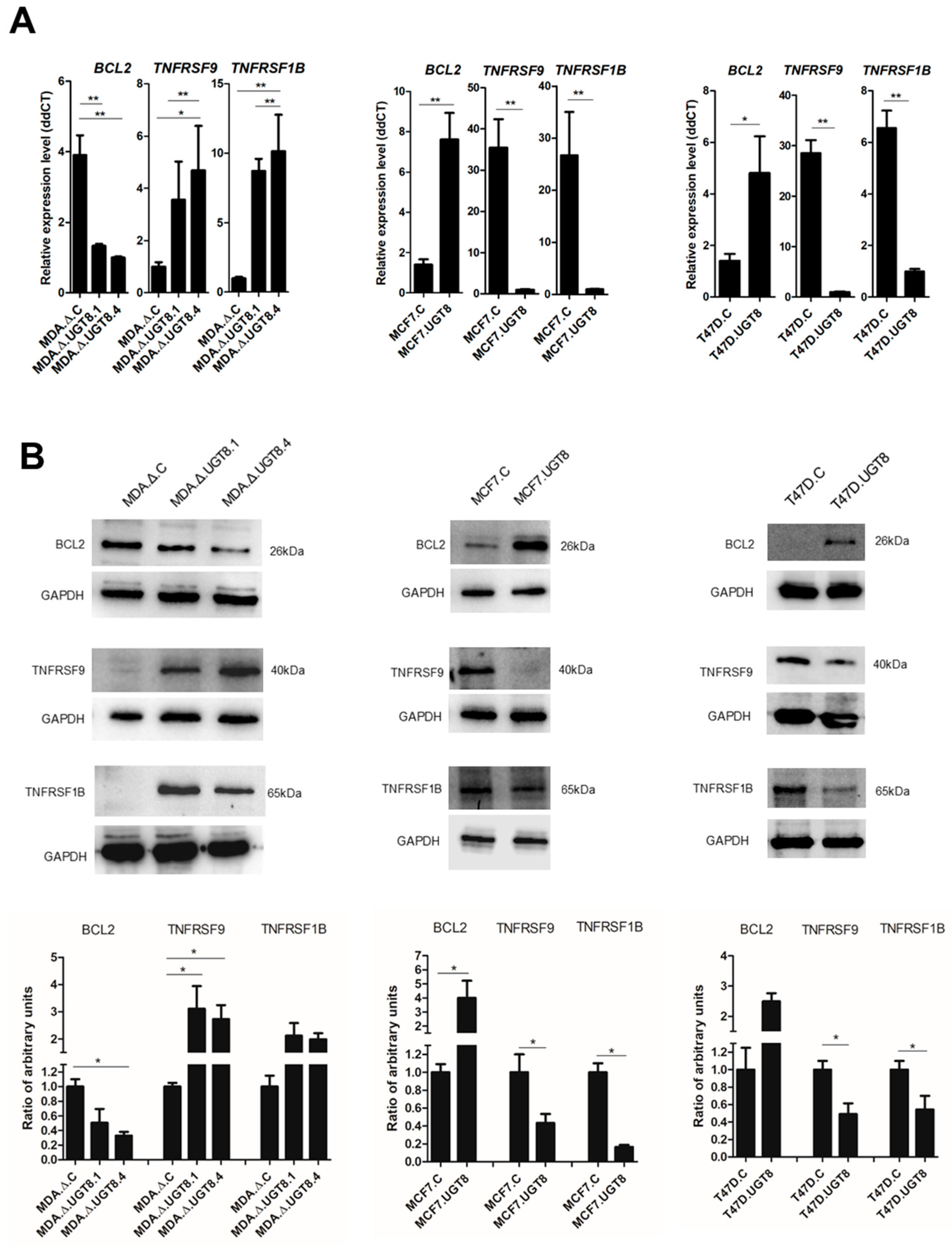
3.3. GalCer Is Responsible for Changes in the Expression of Specific Apoptotic Genes
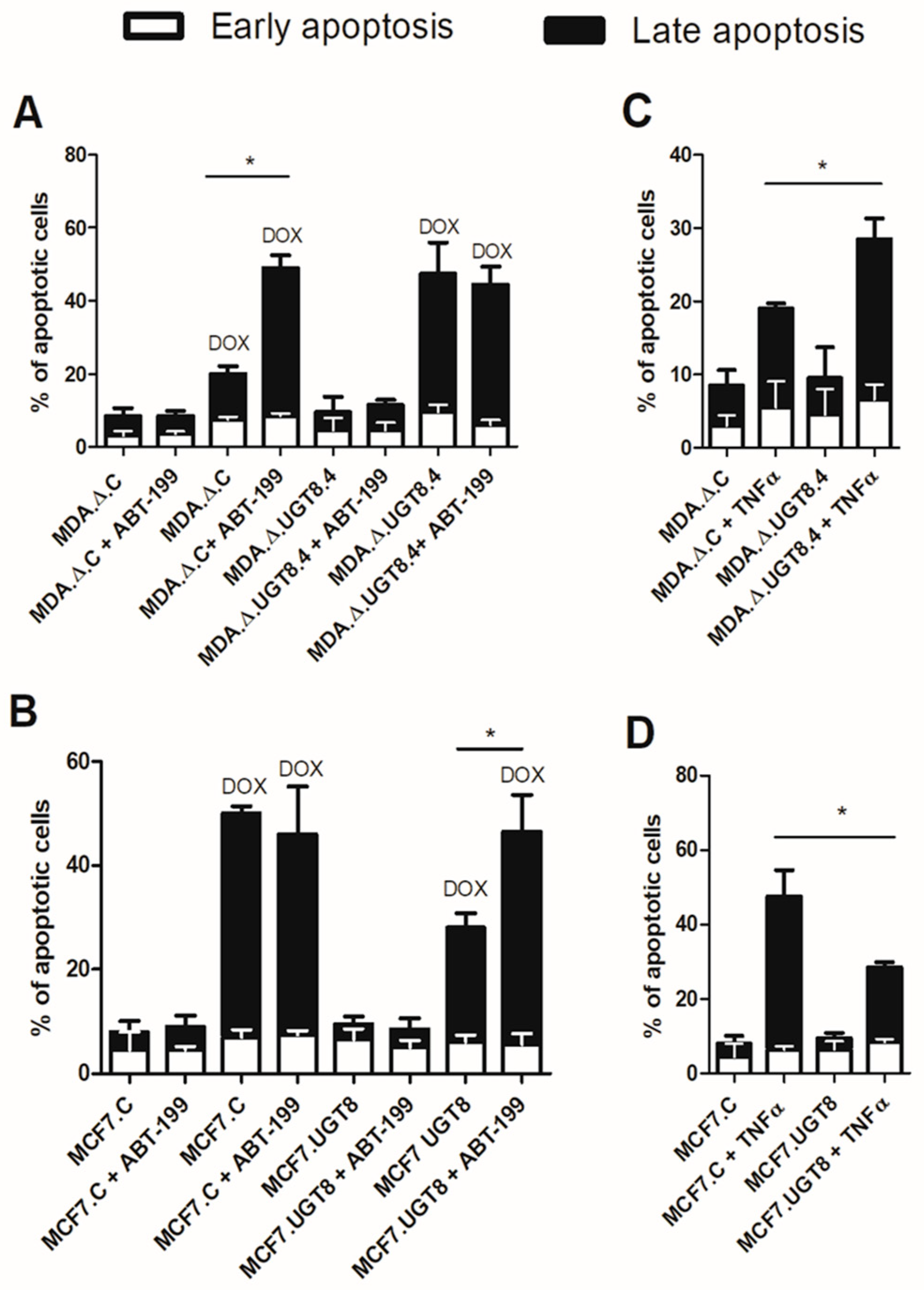
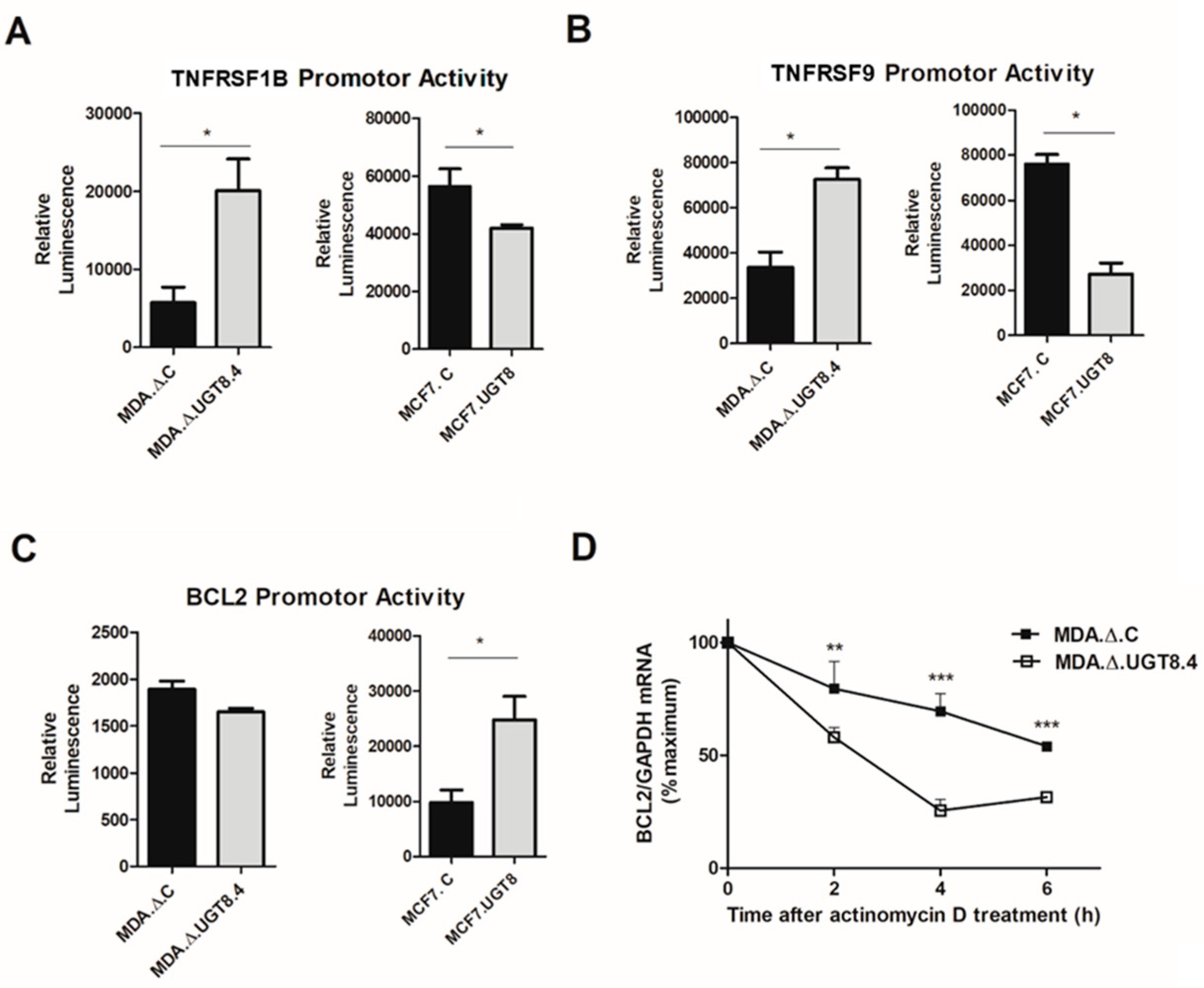
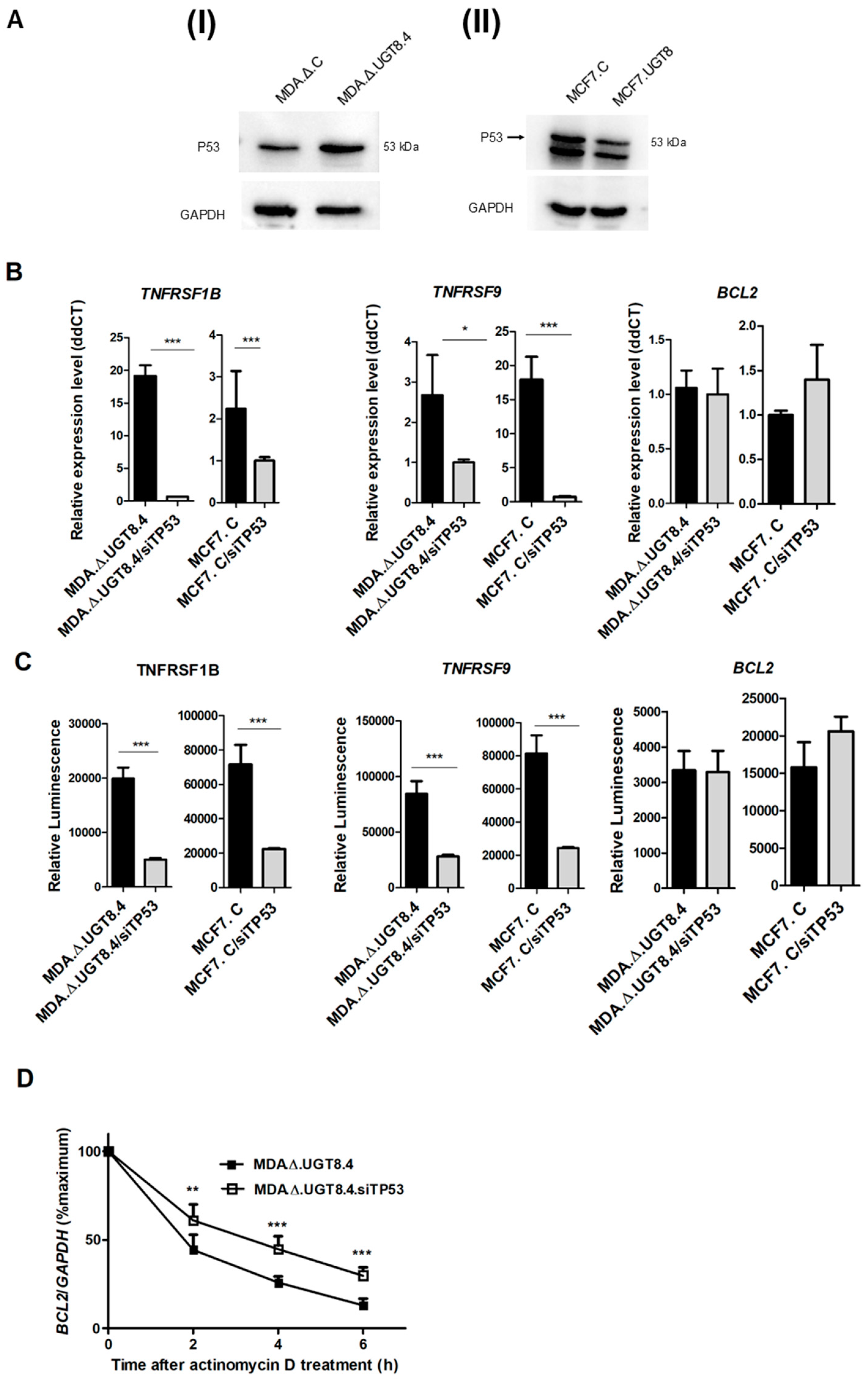
3.4. GalCer Regulates the Expression of TNFRSF1B, TNFRSF9, and Bcl-2 on the Level of Transcription and mRNA Stability
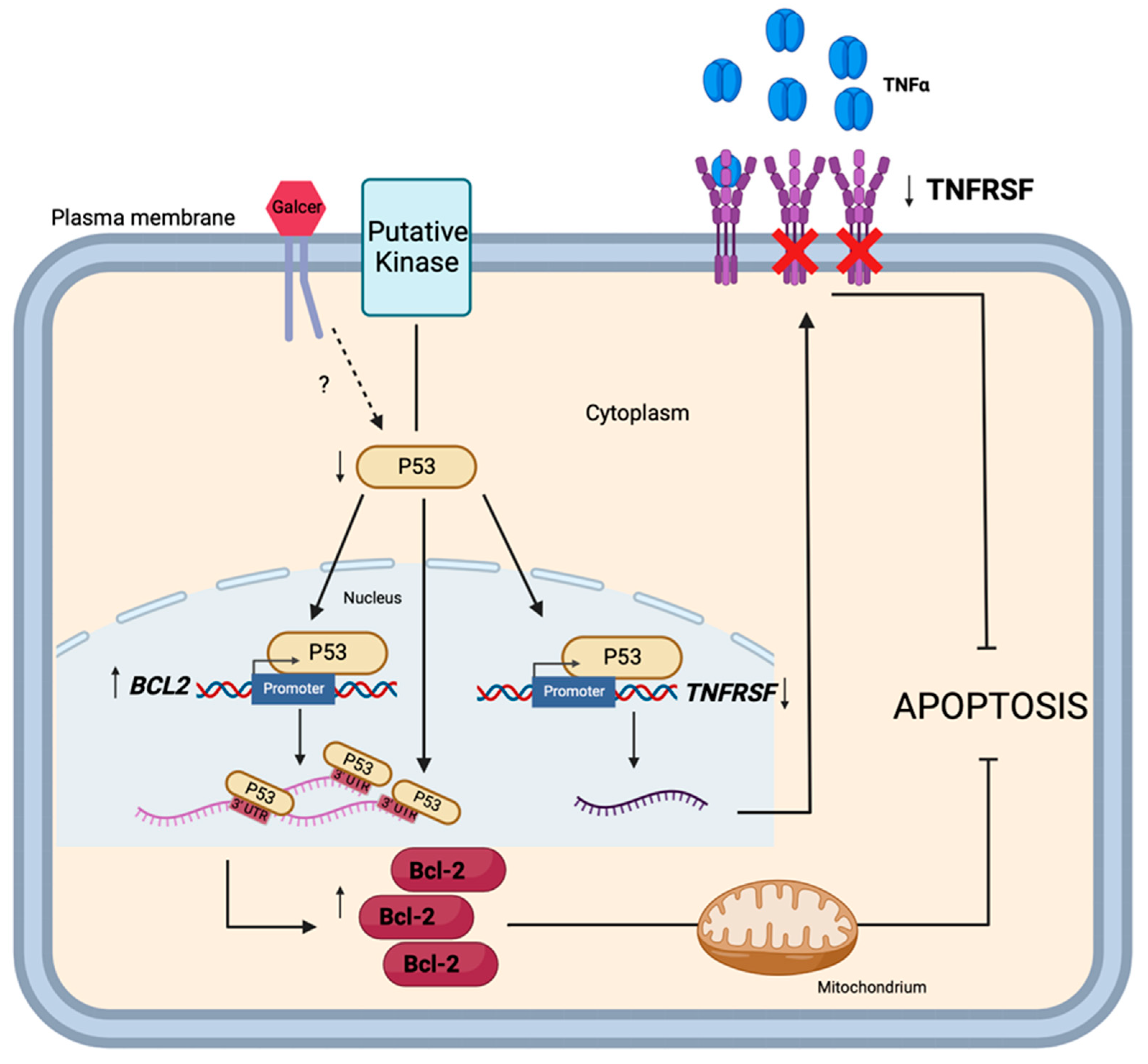
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kruh, G.D. Introduction to resistance to anticancer agents. Oncogene 2003, 22, 7262–7264. [Google Scholar] [CrossRef] [PubMed]
- Giussani, P.; Tringali, C.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance. Int. J. Mol. Sci. 2014, 15, 4356–4392. [Google Scholar] [CrossRef] [PubMed]
- Lavie, Y.; Cao, H.T.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar] [CrossRef] [PubMed]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Gouaze, V.; Yu, J.Y.; Bleicher, R.J.; Han, T.Y.; Liu, Y.Y.; Wang, H.T.; Gottesman, M.M.; Bitterman, A.; Giuliano, A.E.; Cabot, M.C. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol. Cancer Ther. 2004, 3, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Gupta, V.; Patwardhan, G.A.; Bhinge, K.; Zhao, Y.F.; Bao, J.X.; Mehendale, H.; Cabot, M.C.; Li, Y.T.; Jazwinski, S.M. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and beta-catenin signaling. Mol. Cancer 2010, 9, 145. [Google Scholar] [CrossRef]
- De Rosa, M.F.; Sillence, D.; Ackerley, C.; Lingwood, C. Role of multiple drug resistance protein 1 in neutral but not acidic glycosphingolipid biosynthesis. J. Biol. Chem. 2004, 279, 7867–7876. [Google Scholar] [CrossRef]
- De Rosa, M.F.; Ackerley, C.; Wang, B.; Ito, S.; Clarke, D.M.; Lingwood, C. Inhibition of multidrug resistance by AdamantylGb(3), a globotriaosylceramide analog. J. Biol. Chem. 2008, 283, 4501–4511. [Google Scholar] [CrossRef]
- Noguchi, M.; Kabayama, K.; Uemura, S.; Kang, B.W.; Saito, M.; Igarashi, Y.; Inokuchi, J. Endogenously produced ganglioside GM3 endows etoposide and doxorubicin resistance by up-regulating Bcl-2 expression in 3LL Lewis lung carcinoma cells. Glycobiology 2006, 16, 641–650. [Google Scholar] [CrossRef]
- Kiura, K.; Watarai, S.; Ueoka, H.; Tabata, M.; Gemba, K.I.; Aoe, K.; Yamane, H.; Yasuda, T.; Harada, M. An alteration of ganglioside composition in cisplatin-resistant lung cancer cell line. Anticancer Res. 1998, 18, 2957–2960. [Google Scholar]
- Tringali, C.; Lupo, B.; Cirillo, F.; Papini, N.; Anastasia, L.; Lamorte, G.; Colombi, P.; Bresciani, R.; Monti, E.; Tettamanti, G.; et al. Silencing of membrane-associated sialidase Neu3 diminishes apoptosis resistance and triggers megakaryocytic differentiation of chronic myeloid leukemic cells K562 through the increase of ganglioside GM3. Cell Death Differ. 2009, 16, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Schulte, S.; Stoffel, W. Ceramide UDPgalactosyltransferase from Myelinating Rat-Brain—Purification, Cloning, and Expression. Proc. Natl. Acad. Sci. USA 1993, 90, 10265–10269. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, D.; Yu, R.K. Cloning, characterization, and expression of human ceramide galactosyltransferase cDNA. Biochem. Biophys. Res. Commun. 1997, 232, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Marcus, J.; Popko, B. Galactolipids are molecular determinants of myelin development and axo-glial organization. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2002, 1573, 406–413. [Google Scholar] [CrossRef]
- Owczarek, T.B.; Suchanski, J.; Pula, B.; Kmiecik, A.M.; Chadalski, M.; Jethon, A.; Dziegiel, P.; Ugorski, M. Galactosylceramide Affects Tumorigenic and Metastatic Properties of Breast Cancer Cells as an Anti-Apoptotic Molecule. PLoS ONE 2013, 8, e84191. [Google Scholar] [CrossRef] [PubMed]
- Suchanski, J.; Grzegrzolka, J.; Owczarek, T.; Pasikowski, P.; Piotrowska, A.; Kocbach, B.; Nowak, A.; Dziegiel, P.; Wojnar, A.; Ugorski, M. Sulfatide decreases the resistance to stress-induced apoptosis and increases P-selectin-mediated adhesion: A two-edged sword in breast cancer progression. Breast Cancer Res. 2018, 20, 133. [Google Scholar] [CrossRef]
- Kok, J.W.; Veldman, R.J.; Klappe, K.; Koning, H.; Filipeanu, C.M.; Muller, M. Differential expression of sphingolipids in MRP1 overexpressing HT29 cells. Int. J. Cancer 2000, 87, 172–178. [Google Scholar] [CrossRef]
- Veldman, R.J.; Klappe, K.; Hinrichs, J.; Hummel, I.; van der Schaaf, G.; Sietsma, H.; Kok, J.W. Altered sphingolipid metabolism in multidrug-resistant ovarian cancer cells is due to uncoupling of glycolipid biosynthesis in the Golgi apparatus. FASEB J. 2002, 16, 1111–1113. [Google Scholar] [CrossRef]
- Grazide, S.; Terrisse, A.D.; Lerouge, S.; Laurent, G.; Jaffrezou, J.P. Cytoprotective effect of glucosylceramide synthase inhibition against daunorubicin-induced apoptosis in human leukemic cell lines. J. Biol. Chem. 2004, 279, 18256–18261. [Google Scholar] [CrossRef]
- Niimura, Y.; Nagai, K.I. Metabolic responses of sulfatide and related glycolipids in Madin-Darby canine kidney (MDCK) cells under osmotic stresses. Comp. Biochem. Phys. B 2008, 149, 161–167. [Google Scholar] [CrossRef]
- Niimura, Y.; Moue, T.; Takahashi, N.; Nagai, K.I. Modification of sphingoglycolipids and sulfolipids in kidney cell lines under heat stress: Activation of monohexosylceramide synthesis as a ceramide scavenger. Glycobiology 2010, 20, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E. Integration of glycosphingolipid metabolism and cell-fate decisions in cancer and stem cells: Review and hypothesis. Glycoconj. J. 2004, 21, 315–327. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Ugorski, M.; Pahlsson, P.; Dus, D.; Nilsson, B.; Radzikowski, C. Glycosphingolipids of Human Urothelial Cell-Lines with Different Grades of Transformation. Glycoconj. J. 1989, 6, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Lapucci, A.; Lulli, M.; Amedei, A.; Papucci, L.; Witort, E.; Di Gesualdo, F.; Bertolini, F.; Brewer, G.; Nicolin, A.; Bevilacqua, A.; et al. Zeta-Crystallin is a bcl-2 mRNA binding protein involved in bcl-2 overexpression in T-cell acute lymphocytic leukemia. FASEB J. 2010, 24, 1852–1865. [Google Scholar] [CrossRef]
- Ishimaru, D.; Zuraw, L.; Ramalingam, S.; Sengupta, T.K.; Bandyopadhyay, S.; Reuben, A.; Fernandes, D.J.; Spicer, E.K. Mechanism of Regulation of bcl-2 mRNA by Nucleolin and A plus U-rich Element-binding Factor 1 (AUF1). J. Biol. Chem. 2010, 285, 27182–27191. [Google Scholar] [CrossRef]
- Puimege, L.; Libert, C.; Van Hauwermeiren, F. Regulation and dysregulation of tumor necrosis factor receptor-1. Cytokine Growth Factor Rev. 2014, 25, 285–300. [Google Scholar] [CrossRef]
- Liu, X.G.; Yue, P.; Khuri, F.R.; Sun, S.Y. p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res. 2004, 64, 5078–5083. [Google Scholar] [CrossRef]
- Zhang, P.J.; Wang, J.; Lang, H.Y.; Wang, W.X.; Liu, X.H.; Liu, H.Y.; Tan, C.C.; Lia, X.T.; Zhao, Y.M.; Wu, X.H. Knockdown of CREB1 promotes apoptosis and decreases estradiol synthesis in mouse granulosa cells. Biomed. Pharmacother. 2018, 105, 1141–1146. [Google Scholar] [CrossRef]
- Heckman, C.A.; Mehew, J.W.; Boxer, L.M. NF-kappa B activates Bcl-2 expression in t(14;18) lymphoma cells. Oncogene 2002, 21, 3898–3908. [Google Scholar] [CrossRef] [PubMed]
- Bourgarel-Rey, V.; Savry, A.; Hua, G.Q.; Carre, M.; Bressin, C.; Chacon, C.; Imbert, J.; Braguer, D.; Barra, Y. Transcriptional down-regulation of Bcl-2 by vinorelbine: Identification of a novel binding site of p53 on Bcl-2 promoter. Biochem. Pharmacol. 2009, 78, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Mehew, J.W.; Heckman, C.A.; Arcinas, M.; Boxer, L.M. Negative regulation of bcl-2 expression by p53 in hematopoietic cells. Oncogene 2001, 20, 240–251. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-induced cell death—Where the cell cycle and apoptosis come together. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.T.; Lee, Y.C.; Kim, C.H. Overexpression of GD3 synthase induces apoptosis of ECV304 cells through downregulation of Bcl-2 (vol 568, pg 183, 2004). FEBS Lett. 2004, 576, 499. [Google Scholar] [CrossRef]
- Rippo, M.R.; Malisan, F.; Ravagnan, L.; Tomassini, B.; Condo, I.; Costantini, P.; Susin, S.A.; Rufini, A.; Todaro, M.; Kroemer, G.; et al. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 2000, 14, 2047–2054. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Petronilli, V.; Colonna, R.; Di Lisa, F.; Bernardi, P. Chloromethyltetramethylrosamine (Mitotracker Orange (TM)) induces the mitochondrial permeability transition and inhibits respiratory complex I—Implications for the mechanism of cytochrome c release. J. Biol. Chem. 1999, 274, 24657–24663. [Google Scholar] [CrossRef] [PubMed]
- Collel, A.; Garcia-Ruiz, C.; Roman, J.; Ballesta, A.; Fernandez-Checa, J.C. Ganglioside GD3 enhances apoptosis by suppressing the nuclear factor-κB-dependent survival pathway. FASEB J. 2001, 15, 1068–1070. [Google Scholar] [CrossRef]
- Paris, R.; Morales, A.; Coll, O.; Sanchez-Reyes, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Ganglioside GD3 sensitizes human hepatoma cells to cancer therapy. J. Biol. Chem. 2002, 277, 49870–49876. [Google Scholar] [CrossRef]
- Kakugawa, Y.; Wada, T.; Yamaguchi, K.; Yamanami, H.; Ouchi, K.; Sato, I.; Miyagi, T. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. USA 2002, 99, 10718–10723. [Google Scholar] [CrossRef]
- Li, Z.Q.; Xuan, Z.F.; Chen, J.; Song, W.F.; Zhang, S.Y.; Jin, C.; Zhou, M.Q.; Zheng, S.S.; Song, P.H. Inhibiting the NF-kappa B pathway enhances the antitumor effect of cabazitaxel by downregulating Bcl-2 in pancreatic cancer. Int. J. Oncol. 2020, 57, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Chand, H.S.; Montano, G.; Huang, X.S.; Randell, S.H.; Mebratu, Y.; Petersen, H.; Tesfaigzi, Y. A genetic variant of p53 restricts the mucous secretory phenotype by regulating SPDEF and Bcl-2 expression. Nat. Commun. 2014, 5, 5567. [Google Scholar] [CrossRef]
- Clore, G.M.; Omichinski, J.G.; Sakaguchi, K.; Zambrano, N.; Sakamoto, H.; Appella, E.; Gronenborn, A.M. High-Resolution Structure of the Oligomerization Domain of P53 by Multidimensional Nmr. Science 1994, 265, 386–391. [Google Scholar] [CrossRef]
- Lu, X.H.; Errington, J.; Curtin, N.J.; Lunec, J.; Newell, D.R. The impact of status on cellular sensitivity to antifolate drugs. Clin. Cancer Res. 2001, 7, 2114–2123. [Google Scholar] [PubMed]
- Katayose, D.; Gudas, J.; Nguyen, H.; Srivastava, S.; Cowan, K.H.; Seth, P. Cytotoxic effects of adenovirus-mediated wild-type p53 protein expression in normal and tumor mammary epithelial cells. Clin. Cancer Res. 1995, 1, 889–897. [Google Scholar]
- Jordan, J.J.; Inga, A.; Conway, K.; Edmiston, S.; Carey, L.A.; Wu, L.; Resnick, M.A. Altered-Function p53 Missense Mutations Identified in Breast Cancers Can Have Subtle Effects on Transactivation. Mol. Cancer Res. 2010, 8, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Dziegiel, P.; Owczarek, T.; Plazuk, E.; Gomulkiewicz, A.; Majchrzak, M.; Podhorska-Okolow, M.; Driouch, K.; Lidereau, R.; Ugorski, M. Ceramide galactosyltransferase (UGT8) is a molecular marker of breast cancer malignancy and lung metastases. Br. J. Cancer 2010, 103, 524–531. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suchanski, J.; Reza, S.; Urbaniak, A.; Woldanska, W.; Kocbach, B.; Ugorski, M. Galactosylceramide Upregulates the Expression of the BCL2 Gene and Downregulates the Expression of TNFRSF1B and TNFRSF9 Genes, Acting as an Anti-Apoptotic Molecule in Breast Cancer Cells. Cancers 2024, 16, 389. https://doi.org/10.3390/cancers16020389
Suchanski J, Reza S, Urbaniak A, Woldanska W, Kocbach B, Ugorski M. Galactosylceramide Upregulates the Expression of the BCL2 Gene and Downregulates the Expression of TNFRSF1B and TNFRSF9 Genes, Acting as an Anti-Apoptotic Molecule in Breast Cancer Cells. Cancers. 2024; 16(2):389. https://doi.org/10.3390/cancers16020389
Chicago/Turabian StyleSuchanski, Jaroslaw, Safoura Reza, Anna Urbaniak, Weronika Woldanska, Bartlomiej Kocbach, and Maciej Ugorski. 2024. "Galactosylceramide Upregulates the Expression of the BCL2 Gene and Downregulates the Expression of TNFRSF1B and TNFRSF9 Genes, Acting as an Anti-Apoptotic Molecule in Breast Cancer Cells" Cancers 16, no. 2: 389. https://doi.org/10.3390/cancers16020389
APA StyleSuchanski, J., Reza, S., Urbaniak, A., Woldanska, W., Kocbach, B., & Ugorski, M. (2024). Galactosylceramide Upregulates the Expression of the BCL2 Gene and Downregulates the Expression of TNFRSF1B and TNFRSF9 Genes, Acting as an Anti-Apoptotic Molecule in Breast Cancer Cells. Cancers, 16(2), 389. https://doi.org/10.3390/cancers16020389