
Current Treatment Concepts for Extra-Abdominal Desmoid-Type Fibromatosis: A Narrative Review


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Inclusion and Exclusion Criteria
2.3. Study Categories
3. Results
3.1. Study Selection Process
3.2. Active Surveillance
3.3. Surgery and Radiotherapy
3.4. Thermal Ablative Therapy
3.5. Antiestrogens and Nonsteroidal Anti-Inflammatory Drugs
3.6. Cytotoxic Chemotherapy
3.7. Tyrosine Kinase Inhibitors
3.8. γ-Secretase Inhibitors
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kasper, B.; Ströbel, P.; Hohenberger, P. Desmoid tumors: Clinical features and treatment options for advanced disease. Oncologist 2011, 16, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Kasper, B.; Raut, C.P.; Gronchi, A. Desmoid tumors: To treat or not to treat, that is the question. Cancer 2020, 126, 5213–5221. [Google Scholar] [CrossRef]
- Penel, N.; Bonvalot, S.; Le Deley, M.C.; Italiano, A.; Tlemsani, C.; Pannier, D.; Leguillette, C.; Kurtz, J.E.; Toulmonde, M.; Thery, J.; et al. Pain in desmoid-type fibromatosis: Prevalence, determinants and prognosis value. Int. J. Cancer 2023, 153, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Berri, R.N.; Baumann, D.P.; Madewell, J.E.; Lazar, A.; Pollock, R.E. Desmoid tumor: Current multidisciplinary approaches. Ann. Plast. Surg. 2011, 67, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.Y.; Bui, N.Q.; Charville, G.W.; Ghanouni, P.; Ganjoo, K.N. Current management and recent progress in desmoid tumors. Cancer Treat. Res. Commun. 2022, 31, 100562. [Google Scholar] [CrossRef]
- Mullen, J.T.; Delaney, T.F.; Kobayashi, W.K.; Szymonifka, J.; Yeap, B.Y.; Chen, Y.L.; Rosenberg, A.E.; Harmon, D.C.; Choy, E.; Yoon, S.S.; et al. Desmoid tumor: Analysis of prognostic factors and outcomes in a surgical series. Ann. Surg. Oncol. 2012, 19, 4028–4035. [Google Scholar] [CrossRef]
- Penel, N.; Chibon, F.; Salas, S. Adult desmoid tumors: Biology, management and ongoing trials. Curr. Opin. Oncol. 2017, 29, 268–274. [Google Scholar] [CrossRef]
- Houdek, M.T.; Rose, P.S.; Kakar, S. Desmoid tumors of the upper extremity. J. Hand. Surg. Am. 2014, 39, 1761–1765. [Google Scholar] [CrossRef]
- Shido, Y.; Nishida, Y.; Nakashima, H.; Katagiri, H.; Sugiura, H.; Yamada, Y.; Ishiguro, N. Surgical treatment for local control of extremity and trunk desmoid tumors. Arch. Orthop. Trauma Surg. 2009, 129, 929–933. [Google Scholar] [CrossRef]
- Crago, A.M.; Denton, B.; Salas, S.; Dufresne, A.; Mezhir, J.J.; Hameed, M.; Gonen, M.; Singer, S.; Brennan, M.F. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann. Surg. 2013, 258, 347–353. [Google Scholar] [CrossRef]
- Alman, B.; Attia, S.; Baumgarten, C.; Benson, C.; Blay, J.Y.; Bonvalot, S.; Breuing, J.; Cardona, K.; Casali, P.G.; van Coevorden, F.; et al. The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients. Eur. J. Cancer 2020, 127, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Testa, S.; Bui, N.Q.; Charville, G.W.; Avedian, R.S.; Steffner, R.; Ghanouni, P.; Mohler, D.G.; Ganjoo, K.N. Management of Patients with Newly Diagnosed Desmoid Tumors in a First-Line Setting. Cancers 2022, 14, 3907. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, P.; Scoccianti, G.; Schiavo, A.; Tortolini, V.; Wigley, C.; Muratori, F.; Matera, D.; Kukushkina, M.; Funovics, P.T.; Lingitz, M.T.; et al. Extra-abdominal desmoid tumor fibromatosis: A multicenter EMSOS study. BMC Cancer 2021, 21, 437. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Takahama, T.; Mavrogenis, A.F.; Tanaka, Y.; Tanaka, Y.; Errani, C. Clinical outcomes of medical treatments for progressive desmoid tumors following active surveillance: A systematic review. Musculoskelet. Surg. 2023, 107, 7–18. [Google Scholar] [CrossRef]
- Kasper, B. New treatments for desmoid tumors. Curr. Opin. Oncol. 2023, 35, 292–295. [Google Scholar] [CrossRef]
- Gounder, M.; Ratan, R.; Alcindor, T.; Schöffski, P.; van der Graaf, W.T.; Wilky, B.A.; Riedel, R.F.; Lim, A.; Smith, L.M.; Moody, S.; et al. Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. N. Engl. J. Med. 2023, 388, 898–912. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, J.E.; Buy, X.; Deschamps, F.; Sauleau, E.; Bouhamama, A.; Toulmonde, M.; Honoré, C.; Bertucci, F.; Brahmi, M.; Chevreau, C.; et al. CRYODESMO-O1: A prospective, open phase II study of cryoablation in desmoid tumour patients progressing after medical treatment. Eur. J. Cancer 2021, 143, 78–87. [Google Scholar] [CrossRef]
- Timbergen, M.J.M.; Schut, A.W.; Grünhagen, D.J.; Sleijfer, S.; Verhoef, C. Active surveillance in desmoid-type fibromatosis: A systematic literature review. Eur. J. Cancer 2020, 137, 18–29. [Google Scholar] [CrossRef]
- van Broekhoven, D.L.; Grünhagen, D.J.; den Bakker, M.A.; van Dalen, T.; Verhoef, C. Time trends in the incidence and treatment of extra-abdominal and abdominal aggressive fibromatosis: A population-based study. Ann. Surg. Oncol. 2015, 22, 2817–2823. [Google Scholar] [CrossRef]
- Kasper, B.; Baumgarten, C.; Garcia, J.; Bonvalot, S.; Haas, R.; Haller, F.; Hohenberger, P.; Penel, N.; Messiou, C.; van der Graaf, W.T.; et al. An update on the management of sporadic desmoid-type fibromatosis: A European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann. Oncol. 2017, 28, 2399–2408. [Google Scholar] [CrossRef]
- von Mehren, M.; Kane, J.M.; Agulnik, M.; Bui, M.M.; Carr-Ascher, J.; Choy, E.; Connelly, M.; Dry, S.; Ganjoo, K.N.; Gonzalez, R.J.; et al. Soft Tissue Sarcoma, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2022, 20, 815–833. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Matsumoto, S.; Ae, K.; Tanizawa, T.; Hayakawa, K.; Saito, M.; Kurosawa, N. The Clinical Features of Multicentric Extra-abdominal Desmoid Tumors. Cancer Diagn. Progn. 2021, 1, 339–343. [Google Scholar] [CrossRef]
- van Houdt, W.J.; Husson, O.; Patel, A.; Jones, R.L.; Smith, M.J.F.; Miah, A.B.; Messiou, C.; Moskovic, E.; Al-Muderis, O.; Benson, C.; et al. Outcome of Primary Desmoid Tumors at All Anatomic Locations Initially Managed with Active Surveillance. Ann. Surg. Oncol. 2019, 26, 4699–4706. [Google Scholar] [CrossRef] [PubMed]
- Bonvalot, S.; Ternès, N.; Fiore, M.; Bitsakou, G.; Colombo, C.; Honoré, C.; Marrari, A.; Le Cesne, A.; Perrone, F.; Dunant, A.; et al. Spontaneous regression of primary abdominal wall desmoid tumors: More common than previously thought. Ann. Surg. Oncol. 2013, 20, 4096–4102. [Google Scholar] [CrossRef]
- Park, J.S.; Nakache, Y.P.; Katz, J.; Boutin, R.D.; Steffner, R.J.; Monjazeb, A.M.; Canter, R.J. Conservative management of desmoid tumors is safe and effective. J. Surg. Res. 2016, 205, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Ruspi, L.; Cananzi, F.C.M.; Sicoli, F.; Samà, L.; Renne, S.L.; Marrari, A.; Gennaro, N.; Colombo, P.; Cozzaglio, L.; Politi, L.S.; et al. Event-free survival in Desmoid-Type fibromatosis (DTF): A pre-post comparison of upfront surgery versus wait-and-see approach. Eur. J. Surg. Oncol. 2021, 47, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Fiore, M.; Grignani, G.; Tolomeo, F.; Merlini, A.; Palassini, E.; Collini, P.; Stacchiotti, S.; Casali, P.G.; Perrone, F.; et al. A Prospective Observational Study of Active Surveillance in Primary Desmoid Fibromatosis. Clin. Cancer Res. 2022, 28, 4027–4032. [Google Scholar] [CrossRef]
- Schut, A.W.; Timbergen, M.J.M.; van Broekhoven, D.L.M.; van Dalen, T.; van Houdt, W.J.; Bonenkamp, J.J.; Sleijfer, S.; Grunhagen, D.J.; Verhoef, C. A Nationwide Prospective Clinical Trial on Active Surveillance in Patients with Non-intraabdominal Desmoid-type Fibromatosis: The GRAFITI Trial. Ann. Surg. 2023, 277, 689–696. [Google Scholar] [CrossRef]
- Penel, N.; Le Cesne, A.; Bonvalot, S.; Giraud, A.; Bompas, E.; Rios, M.; Salas, S.; Isambert, N.; Boudou-Rouquette, P.; Honore, C.; et al. Surgical versus non-surgical approach in primary desmoid-type fibromatosis patients: A nationwide prospective cohort from the French Sarcoma Group. Eur. J. Cancer 2017, 83, 125–131. [Google Scholar] [CrossRef]
- Cates, J.M.; Stricker, T.P. Surgical resection margins in desmoid-type fibromatosis: A critical reassessment. Am. J. Surg. Pathol. 2014, 38, 1707–1714. [Google Scholar] [CrossRef]
- Sobczuk, P.; Agnieszczak, I.M.; Grycuk, W.; Czarnecka, A.M.; Świtaj, T.; Koseła-Paterczyk, H.; Morysiński, T.; Zdzienicki, M.; Rutkowski, P. What is the best front-line approach in patients with desmoid fibromatosis?—A retrospective analysis from a reference center. Eur. J. Surg. Oncol. 2021, 47, 2602–2608. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.D.; Hyder, O.; Mavros, M.N.; Turley, R.; Groeschl, R.; Firoozmand, A.; Lidsky, M.; Herman, J.M.; Choti, M.; Ahuja, N.; et al. Management and recurrence patterns of desmoids tumors: A multi-institutional analysis of 211 patients. Ann. Surg. Oncol. 2012, 19, 4036–4042. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Miceli, R.; Le Péchoux, C.; Palassini, E.; Honoré, C.; Stacchiotti, S.; Mir, O.; Casali, P.G.; Dômont, J.; Fiore, M.; et al. Sporadic extra abdominal wall desmoid-type fibromatosis: Surgical resection can be safely limited to a minority of patients. Eur. J. Cancer 2015, 51, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Orbach, D.; Brennan, B.; Bisogno, G.; Van Noesel, M.; Minard-Colin, V.; Daragjati, J.; Casanova, M.; Corradini, N.; Zanetti, I.; De Salvo, G.L.; et al. The EpSSG NRSTS 2005 treatment protocol for desmoid-type fibromatosis in children: An international prospective case series. Lancet Child. Adolesc. Health 2017, 1, 284–292. [Google Scholar] [CrossRef] [PubMed]
- van Broekhoven, D.L.; Verhoef, C.; Elias, S.G.; Witkamp, A.J.; van Gorp, J.M.; van Geel, B.A.; Wijrdeman, H.K.; van Dalen, T. Local recurrence after surgery for primary extra-abdominal desmoid-type fibromatosis. Br. J. Surg. 2013, 100, 1214–1219. [Google Scholar] [CrossRef]
- Janssen, M.L.; van Broekhoven, D.L.; Cates, J.M.; Bramer, W.M.; Nuyttens, J.J.; Gronchi, A.; Salas, S.; Bonvalot, S.; Grünhagen, D.J.; Verhoef, C. Meta-analysis of the influence of surgical margin and adjuvant radiotherapy on local recurrence after resection of sporadic desmoid-type fibromatosis. Br. J. Surg. 2017, 104, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Kito, M.; Ogose, A.; Yoshida, M.; Nishida, Y. Usefulness of surgical treatment for asymptomatic patients with extra-peritoneal desmoid-type fibromatosis: A systematic review and meta-analysis. Jpn. J. Clin. Oncol. 2020, 50, 574–580. [Google Scholar] [CrossRef]
- Spolverato, G.; Capelli, G.; Kasper, B.; Gounder, M. Management of Desmoid Tumors. Surg. Oncol. Clin. N. Am. 2022, 31, 447–458. [Google Scholar] [CrossRef]
- Santti, K.; Beule, A.; Tuomikoski, L.; Rönty, M.; Jääskeläinen, A.S.; Saarilahti, K.; Ihalainen, H.; Tarkkanen, M.; Blomqvist, C. Radiotherapy in desmoid tumors: Treatment response, local control, and analysis of local failures. Strahlenther. Onkol. 2017, 193, 269–275. [Google Scholar] [CrossRef]
- Gluck, I.; Griffith, K.A.; Biermann, J.S.; Feng, F.Y.; Lucas, D.R.; Ben-Josef, E. Role of radiotherapy in the management of desmoid tumors. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 787–792. [Google Scholar] [CrossRef]
- Niu, X.; Jiang, R.; Hu, C. Radiotherapy in the treatment of primary or recurrent unresectable desmoid tumors of the neck. Cancer Investig. 2019, 37, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.M.; Jones, D.; Boyle, R.; Stalley, P. Radiation Therapy as an Alternative Treatment for Desmoid Fibromatosis. Clin. Oncol. (R. Coll. Radiol.) 2018, 30, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.J.; Landry, J.P.; Roland, C.L.; Ratan, R.; Feig, B.W.; Moon, B.S.; Zarzour, M.A.; Wang, W.L.; Lazar, A.J.; Lewis, V.O.; et al. Certain risk factors for patients with desmoid tumors warrant reconsideration of local therapy strategies. Cancer 2020, 126, 3265–3273. [Google Scholar] [CrossRef]
- Keus, R.B.; Nout, R.A.; Blay, J.Y.; de Jong, J.M.; Hennig, I.; Saran, F.; Hartmann, J.T.; Sunyach, M.P.; Gwyther, S.J.; Ouali, M.; et al. Results of a phase II pilot study of moderate dose radiotherapy for inoperable desmoid-type fibromatosis—An EORTC STBSG and ROG study (EORTC 62991-22998). Ann. Oncol. 2013, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed]
- Colak, C.; Hull, C.; Simpfendorfer, C.; Ilaslan, H.; Forney, M. Extra-abdominal desmoid fibromatosis: Cryoablation versus traditional therapies. Clin. Imaging 2022, 88, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.E.; Kim, D.; Yarmohammadi, H.; Ziv, E.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Whiting, K.; Qin, L.X.; Singer, S.; et al. Percutaneous Cryoablation Provides Disease Control for Extra-Abdominal Desmoid-Type Fibromatosis Comparable with Surgical Resection. Ann. Surg. Oncol. 2022, 29, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Bouhamama, A.; Lame, F.; Mastier, C.; Cuinet, M.; Thibaut, A.; Beji, H.; Ricoeur, A.; Blay, J.Y.; Pilleul, F. Local Control and Analgesic Efficacy of Percutaneous Cryoablation for Desmoid Tumors. CardioVascular Interv. Radiol. 2020, 43, 110–119. [Google Scholar] [CrossRef]
- Bocale, D.; Rotelli, M.T.; Cavallini, A.; Altomare, D.F. Anti-oestrogen therapy in the treatment of desmoid tumours: A systematic review. Color. Dis. 2011, 13, e388–e395. [Google Scholar] [CrossRef]
- Quast, D.R.; Schneider, R.; Burdzik, E.; Hoppe, S.; Möslein, G. Long-term outcome of sporadic and FAP-associated desmoid tumors treated with high-dose selective estrogen receptor modulators and sulindac: A single-center long-term observational study in 134 patients. Fam. Cancer 2016, 15, 31–40. [Google Scholar] [CrossRef]
- Fiore, M.; Colombo, C.; Radaelli, S.; Callegaro, D.; Palassini, E.; Barisella, M.; Morosi, C.; Baldi, G.G.; Stacchiotti, S.; Casali, P.G.; et al. Hormonal manipulation with toremifene in sporadic desmoid-type fibromatosis. Eur. J. Cancer 2015, 51, 2800–2807. [Google Scholar] [CrossRef]
- Libertini, M.; Mitra, I.; van der Graaf, W.T.A.; Miah, A.B.; Judson, I.; Jones, R.L.; Thomas, K.; Moskovic, E.; Szucs, Z.; Benson, C.; et al. Aggressive fibromatosis response to tamoxifen: Lack of correlation between MRI and symptomatic response. Clin. Sarcoma Res. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Anderson, J.R.; Hill, D.A.; Henry, D.; Spunt, S.L.; Meyer, W.; Kao, S.; Hoffer, F.A.; Grier, H.E.; Hawkins, D.S.; et al. Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: Results of a Children’s Oncology Group (COG) phase II study. Pediatr. Blood Cancer 2013, 60, 1108–1112. [Google Scholar] [CrossRef]
- Mignemi, N.A.; Itani, D.M.; Fasig, J.H.; Keedy, V.L.; Hande, K.R.; Whited, B.W.; Homlar, K.C.; Correa, H.; Coffin, C.M.; Black, J.O.; et al. Signal transduction pathway analysis in desmoid-type fibromatosis: Transforming growth factor-β, COX2 and sex steroid receptors. Cancer Sci. 2012, 103, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Eastley, N.C.; Hennig, I.M.; Esler, C.P.; Ashford, R.U. Nationwide trends in the current management of desmoid (aggressive) fibromatosis. Clin. Oncol. (R. Coll. Radiol.) 2015, 27, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Urakawa, H.; Kozawa, E.; Futamura, N.; Ikuta, K.; Shimoyama, Y.; Nakamura, S.; Ishiguro, N.; Nishida, Y. Nuclear expression of β-catenin predicts the efficacy of meloxicam treatment for patients with sporadic desmoid tumors. Tumour Biol. 2014, 35, 4561–4566. [Google Scholar] [CrossRef]
- Li, S.; Fan, Z.; Fang, Z.; Liu, J.; Bai, C.; Xue, R.; Zhang, L.; Gao, T. Efficacy of vinorelbine combined with low-dose methotrexate for treatment of inoperable desmoid tumor and prognostic factor analysis. Chin. J. Cancer Res. 2017, 29, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Crombé, A.; Kind, M.; Ray-Coquard, I.; Isambert, N.; Chevreau, C.; André, T.; Lebbe, C.; Cesne, A.L.; Bompas, E.; Piperno-Neumann, S.; et al. Progressive Desmoid Tumor: Radiomics Compared with Conventional Response Criteria for Predicting Progression During Systemic Therapy-A Multicenter Study by the French Sarcoma Group. AJR Am. J. Roentgenol. 2020, 215, 1539–1548. [Google Scholar] [CrossRef]
- Nishida, Y.; Hamada, S.; Urakawa, H.; Ikuta, K.; Sakai, T.; Koike, H.; Ito, K.; Emoto, R.; Ando, Y.; Matsui, S. Desmoid with biweekly methotrexate and vinblastine shows similar effects to weekly administration: A phase II clinical trial. Cancer Sci. 2020, 111, 4187–4194. [Google Scholar] [CrossRef]
- Palassini, E.; Frezza, A.M.; Mariani, L.; Lalli, L.; Colombo, C.; Fiore, M.; Messina, A.; Casale, A.; Morosi, C.; Collini, P.; et al. Long-term Efficacy of Methotrexate Plus Vinblastine/Vinorelbine in a Large Series of Patients Affected by Desmoid-Type Fibromatosis. Cancer J. 2017, 23, 86–91. [Google Scholar] [CrossRef]
- Ingley, K.M.; Burtenshaw, S.M.; Theobalds, N.C.; White, L.M.; Blackstein, M.E.; Gladdy, R.A.; Thipphavong, S.; Gupta, A.A. Clinical benefit of methotrexate plus vinorelbine chemotherapy for desmoid fibromatosis (DF) and correlation of treatment response with MRI. Cancer Med. 2019, 8, 5047–5057. [Google Scholar] [CrossRef]
- Chugh, R.; Wathen, J.K.; Patel, S.R.; Maki, R.G.; Meyers, P.A.; Schuetze, S.M.; Priebat, D.A.; Thomas, D.G.; Jacobson, J.A.; Samuels, B.L.; et al. Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin. Cancer Res. 2010, 16, 4884–4891. [Google Scholar] [CrossRef] [PubMed]
- Penel, N.; Le Cesne, A.; Bui, B.N.; Perol, D.; Brain, E.G.; Ray-Coquard, I.; Guillemet, C.; Chevreau, C.; Cupissol, D.; Chabaud, S.; et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): An FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann. Oncol. 2011, 22, 452–457. [Google Scholar] [CrossRef]
- Kasper, B.; Gruenwald, V.; Reichardt, P.; Bauer, S.; Rauch, G.; Limprecht, R.; Sommer, M.; Dimitrakopoulou-Strauss, A.; Pilz, L.; Haller, F.; et al. Imatinib induces sustained progression arrest in RECIST progressive desmoid tumours: Final results of a phase II study of the German Interdisciplinary Sarcoma Group (GISG). Eur. J. Cancer 2017, 76, 60–67. [Google Scholar] [CrossRef]
- Toulmonde, M.; Pulido, M.; Ray-Coquard, I.; Andre, T.; Isambert, N.; Chevreau, C.; Penel, N.; Bompas, E.; Saada, E.; Bertucci, F.; et al. Pazopanib or methotrexate-vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): A non-comparative, randomised, open-label, multicentre, phase 2 study. Lancet Oncol. 2019, 20, 1263–1272. [Google Scholar] [CrossRef]
- Jo, J.C.; Hong, Y.S.; Kim, K.P.; Lee, J.L.; Lee, J.; Park, Y.S.; Kim, S.Y.; Ryu, J.S.; Lee, J.S.; Kim, T.W. A prospective multicenter phase II study of sunitinib in patients with advanced aggressive fibromatosis. Investig. New Drugs 2014, 32, 369–376. [Google Scholar] [CrossRef]
- Gounder, M.M.; Mahoney, M.R.; Van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N. Engl. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Gangadharaiah, B.B.; Rastogi, S.; Upadhyay, A.; Barwad, A.; Dhamija, E.; Gamangatti, S. Efficacy and tolerability of sorafenib in desmoid-type fibromatosis: A need to review dose. Eur. J. Cancer 2023, 186, 142–150. [Google Scholar] [CrossRef]
- McCaw, T.R.; Inga, E.; Chen, H.; Jaskula-Sztul, R.; Dudeja, V.; Bibb, J.A.; Ren, B.; Rose, J.B. Gamma Secretase Inhibitors in Cancer: A Current Perspective on Clinical Performance. Oncologist 2021, 26, e608–e621. [Google Scholar] [CrossRef]
- Messersmith, W.A.; Shapiro, G.I.; Cleary, J.M.; Jimeno, A.; Dasari, A.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin. Cancer Res. 2015, 21, 60–67. [Google Scholar] [CrossRef]
- Villalobos, V.M.; Hall, F.; Jimeno, A.; Gore, L.; Kern, K.; Cesari, R.; Huang, B.; Schowinsky, J.T.; Blatchford, P.J.; Hoffner, B.; et al. Long-Term Follow-Up of Desmoid Fibromatosis Treated with PF-03084014, an Oral Gamma Secretase Inhibitor. Ann. Surg. Oncol. 2018, 25, 768–775. [Google Scholar] [CrossRef]
- Kummar, S.; O’Sullivan Coyne, G.; Do, K.T.; Turkbey, B.; Meltzer, P.S.; Polley, E.; Choyke, P.L.; Meehan, R.; Vilimas, R.; Horneffer, Y.; et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults with Desmoid Tumors (Aggressive Fibromatosis). J. Clin. Oncol. 2017, 35, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Jones, R.L.; Yovell, J.; Gordon, G.; Kasper, B. Abstract CT070: Double-blind placebo-controlled trial of AL102 for treatment of progressing desmoid tumors: The RINGSIDE phase 3 study design. Cancer Res. 2023, 83, CT070. [Google Scholar] [CrossRef]
- Whittle, S.B.; Pressey, J.G.; Liu, X.; Minard, C.G.; Denic, K.Z.; Reid, J.M.; Berg, S.L.; Fox, E.; Weigel, B.J. Abstract CT090: PEPN2011: A phase 1/2 study of tegavivint in children, adolescents, and young adults with recurrent or refractory solid tumors, including lymphomas and desmoid tumors: A report from the pediatric early phase clinical trials network. Cancer Res. 2023, 83, CT090. [Google Scholar] [CrossRef]
- Riedel, R.F.; Agulnik, M. Evolving strategies for management of desmoid tumor. Cancer 2022, 128, 3027–3040. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Desai, J.; Lazarakis, S.; Gyorki, D. Systematic Review of Clinical Outcomes Following Various Treatment Options for Patients with Extraabdominal Desmoid Tumors. Ann. Surg. Oncol. 2018, 25, 1544–1554. [Google Scholar] [CrossRef]
- Tsagozis, P.; Stevenson, J.D.; Grimer, R.; Carter, S. Outcome of surgery for primary and recurrent desmoid-type fibromatosis. A retrospective case series of 174 patients. Ann. Med. Surg. 2017, 17, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.H.; Wirth, C.; Lenze, U.; Kettelhack, C.; Coslovsky, M.; Baumhoer, D.; Klenke, F.M.; Siebenrock, K.A.; Exner, G.U.; Bode-Lesniewska, B.; et al. Extra-abdominal desmoid tumours—Further evidence for the watchful waiting policy. Swiss Med. Wkly. 2019, 149, w20107. [Google Scholar] [CrossRef]
- Ratan, R.; Roland, C.L.; Bishop, A.J. Desmoid Fibromatosis: Management in an Era of Increasing Options. Curr. Oncol. Rep. 2021, 23, 41. [Google Scholar] [CrossRef]
- Seinen, J.M.; Niebling, M.G.; Bastiaannet, E.; Pras, B.; Hoekstra, H.J. Four different treatment strategies in aggressive fibromatosis: A systematic review. Clin. Transl. Radiat. Oncol. 2018, 12, 1–7. [Google Scholar] [CrossRef]
- Okuda, M.; Yoshida, K.; Kobayashi, S.; Gabata, T. Desmoid-type fibromatosis: Imaging features and course. Skelet. Radiol. 2023, 52, 1293–1303. [Google Scholar] [CrossRef]
- Nakayama, T.; Tsuboyama, T.; Toguchida, J.; Hosaka, T.; Nakamura, T. Natural course of desmoid-type fibromatosis. J. Orthop. Sci. 2008, 13, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shi, J.; Yang, T.; Liu, T.; Zhang, K. Management of aggressive fibromatosis. Oncol. Lett. 2021, 21, 43. [Google Scholar] [PubMed]
- Shang, H.; Braggio, D.; Lee, Y.J.; Al Sannaa, G.A.; Creighton, C.J.; Bolshakov, S.; Lazar, A.J.; Lev, D.; Pollock, R.E. Targeting the Notch pathway: A potential therapeutic approach for desmoid tumors. Cancer 2015, 121, 4088–4096. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Ishida, H.; Ueno, H.; Kobayashi, H.; Yamaguchi, T.; Konishi, T.; Tomita, N.; Matsubara, N.; Ishida, F.; Hinoi, T.; et al. The treatment of desmoid tumors associated with familial adenomatous polyposis: The results of a Japanese multicenter observational study. Surg. Today 2017, 47, 1259–1267. [Google Scholar] [CrossRef]
- Walter, T.; Zhenzhen Wang, C.; Guillaud, O.; Cotte, E.; Pasquer, A.; Vinet, O.; Poncet, G.; Ponchon, T.; Saurin, J.C. Management of desmoid tumours: A large national database of familial adenomatous patients shows a link to colectomy modalities and low efficacy of medical treatments. United Eur. Gastroenterol. J. 2017, 5, 735–741. [Google Scholar] [CrossRef]
- Nieuwenhuis, M.H.; Mathus-Vliegen, E.M.; Baeten, C.G.; Nagengast, F.M.; van der Bijl, J.; van Dalsen, A.D.; Kleibeuker, J.H.; Dekker, E.; Langers, A.M.; Vecht, J.; et al. Evaluation of management of desmoid tumours associated with familial adenomatous polyposis in Dutch patients. Br. J. Cancer 2011, 104, 37–42. [Google Scholar] [CrossRef]
- Yang, W.; Ding, P.R. Update on Familial Adenomatous Polyposis-Associated Desmoid Tumors. Clin. Colon Rectal Surg. 2023, 36, 400–405. [Google Scholar] [CrossRef]
- Kumamoto, K.; Ishida, H.; Tomita, N. Recent Advances and Current Management for Desmoid Tumor Associated with Familial Adenomatous Polyposis. J. Anus Rectum Colon 2023, 7, 38–51. [Google Scholar] [CrossRef]
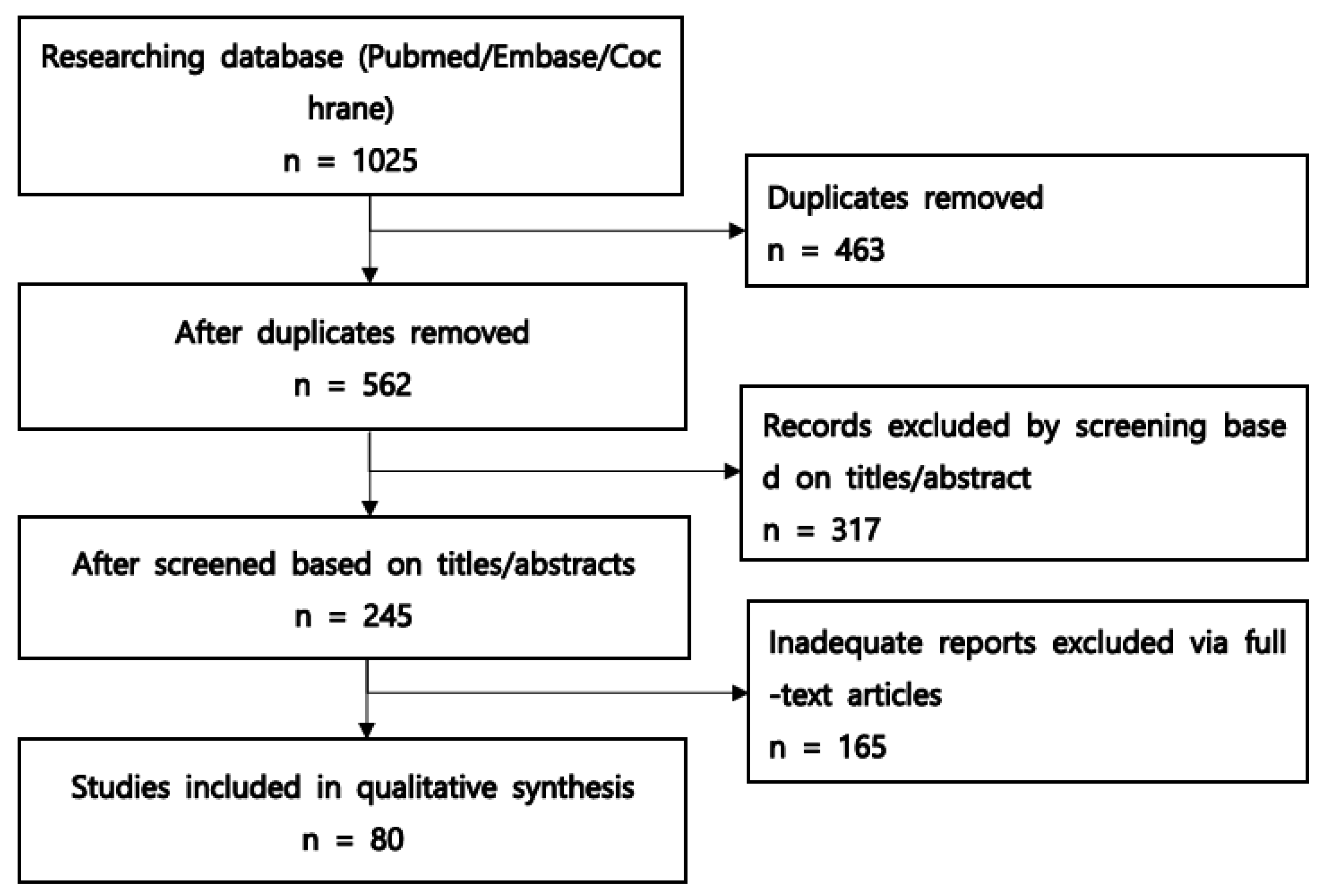

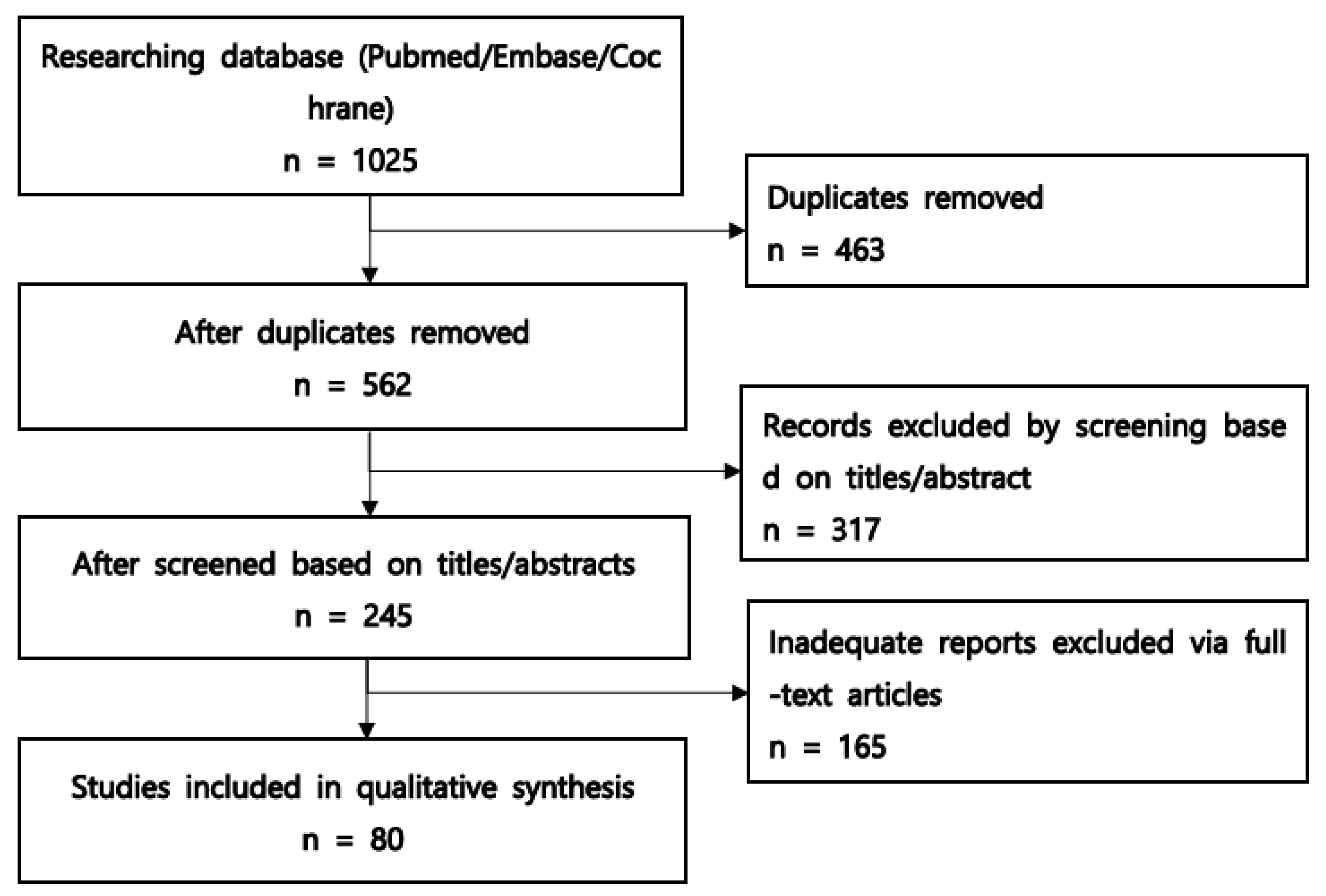{kind=link}
| Inclusion Criteria | Exclusion Criteria | |
|---|---|---|
| Population | Patients with extra-abdominal desmoid-type fibromatosis | Patients with intra-abdominal desmoid-type fibromatosis Patients with other neoplasms |
| Intervention and Comparators | Active surveillance Surgery Radiation Thermal ablative therapy Antiestrogens and anti-inflammatory drugs Cytotoxic chemotherapy Tyrosine kinase γ-secretase inhibitors | |
| Outcomes | Type of treatment modalities Rate of disease progression, stable, or regression Progression-free survival period Recurrence rate | Biochemical, molecular, or genetic outcomes |
| Study design | Case series studies (n ≥ 30) Prospective, observational studies Clinical trials (phase 2 or 3) Systematic reviews/meta-analyses Consensus or practice guidelines | Small sample size (n < 30) Clinical trials (phase 1) Guidelines or reviews before 2008 Letter, editorial, commentary |
| Treatment Option | Application | Outcome and Efficacy | Side Effects |
|---|---|---|---|
| Active surveillance | All guidelines recommend schedule for initial surveillance within 1–2 months of diagnosis and then at 3–6-month intervals [11,21,74]. | SD, 59%; PR, 19%; PD, 20% [18]. DC, 60–82% [75]. 2-year event-free survival, 58% [29]. SD, 36%; SR, 27% [23]. SR, 28.4% (follow-up, 32 months) [24]. SD, 65%; PR, 25%; SR, 5% (follow-up, 35.7 months) [25]. SR, 25%, PD, 39% (follow-up, 32.3 months) [27]. SD, 32%, SR, 28%, PD, 40% [28]. | |
| Surgery | Surgery is typically not the first-line treatment option, except under particular conditions approved by a multidisciplinary tumor board [21]. When considering surgical intervention, preserving function is the primary objective [2]. | Recurrence rates: positive margin, 32%; negative margin, 40% [30]. Recurrence rates: 14–47.2% [32,35,36,76,77]. Risk of local recurrence with microscopically positive margins: risk ratio, 1.78 [36]. | Surgical complications, the need for complex surgical reconstruction, and decreased quality of life [2]. |
| Radiotherapy | Postoperative RT or administered alone; 56–60 Gy in 28 fractions [44,78]. | DC: 55–92.3% [39,40,41,42]. 3-year DC: 81.5% (SD, 40.9%; PR, 36.4%; CR, 13.6%) [44]. 5-year DC with RT ± surgery: 77% and 65% [43]. | Fibrosis, fracture, and secondary malignancy [43,44]. |
| Cryoablation | Two 10 min freeze–thaw cycles [17,46]. | SD: 31%; PR: 26.2%; CR: 28.6% at 12 months [17]. 2-year DC: 85% [46]. 3-year DC: 42.2% [47]. | Nerve injury, rhadomyolysis, skin necrosis, bleeding, infection, and colo-cutaneous fistula: 2.4–30% [17]. |
| Antiestrogens | Lack of proof to regard antihormonal treatments, and current clinical guidelines have ceased to support hormone therapies as a standard recommendation [11,21]. | Wide range of DC (25–89.6%) [11,49,50,51]. Antihormonal therapy + NSAIDs: 36% of 2-year PFS [52]. No correlation between size and MRI signal changes and symptom release [51]. | |
| Anti-inflammatory drugs | Not considered as agents for disease management, and current guidelines recommend their use solely for the control of pain [11]. | Various response rates [54,55]. Prospective studies with meloxicam: PD, 35.5% [55]. | No life-threatening toxicity [52]. |
| Cytotoxic chemotherapy | MTX (30 mg/m2) and vinblastine (5 or 6 mg/m2) every 7–10 days. Weekly MTX (30 mg/m2) and vinorelbine (20 mg/m2); 40 to 50 cycles. | DC: 64–100% [56,57,59]. 1-year PFS: 79% [64]. SD, 17%; PR, 39%; CR, 42% [59]. | Hematologic toxicity: bone marrow suppression. Nonhematologic toxicity: nausea, vomiting [59]. |
| Tyrosine kinase inhibitors | 200 to 800 mg of oral Imatinib daily. 37.5 to 52 mg of Sunitinib daily dose. 400 mg of Sorafenib daily dose. 800 mg of Pazopanib daily dose. | CR + PR: 2–6% (between 3–6 months) [61,62,63]. 1-year PFS: 66% [63]. DC: 68.4% [65]. 2-year PFS: 74.7% [65]. 1-year PFS: 86.6–89% [66,67]. 6-month PFS: 83.7% [64]. | Hematologic toxicity: neutropenia [65]. Nonhematologic toxicity: fatigue, diarrhea, nausea, weight loss, hypertension, hand-foot skin reaction, rash, alopecia [64,65,66,67]. |
| γ-secretase inhibitors | 150 mg twice daily of Nirogacestat (NCT03785964; DeFi trial). 1.2–4 mg daily of AL 102 (RINGSIDE phase 2/3 trial). | 2-year event-free: 76% CR, 7% [16]. Endpoints: PFS, Overall Response Rate, Duration of Response, Quality-of-life measures [72]. | Diarrhea, nausea, fatigue, hypophosphatemia, maculopapular rash [16]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-S.; Joo, M.W.; Shin, S.-H.; Hong, S.; Chung, Y.-G. Current Treatment Concepts for Extra-Abdominal Desmoid-Type Fibromatosis: A Narrative Review. Cancers 2024, 16, 273. https://doi.org/10.3390/cancers16020273
Lee Y-S, Joo MW, Shin S-H, Hong S, Chung Y-G. Current Treatment Concepts for Extra-Abdominal Desmoid-Type Fibromatosis: A Narrative Review. Cancers. 2024; 16(2):273. https://doi.org/10.3390/cancers16020273
Chicago/Turabian StyleLee, Yong-Suk, Min Wook Joo, Seung-Han Shin, Sungan Hong, and Yang-Guk Chung. 2024. "Current Treatment Concepts for Extra-Abdominal Desmoid-Type Fibromatosis: A Narrative Review" Cancers 16, no. 2: 273. https://doi.org/10.3390/cancers16020273
APA StyleLee, Y.-S., Joo, M. W., Shin, S.-H., Hong, S., & Chung, Y.-G. (2024). Current Treatment Concepts for Extra-Abdominal Desmoid-Type Fibromatosis: A Narrative Review. Cancers, 16(2), 273. https://doi.org/10.3390/cancers16020273