
Unravelling the Complexity of HNSCC Using Single-Cell Transcriptomics
 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. HNSCC Etiology
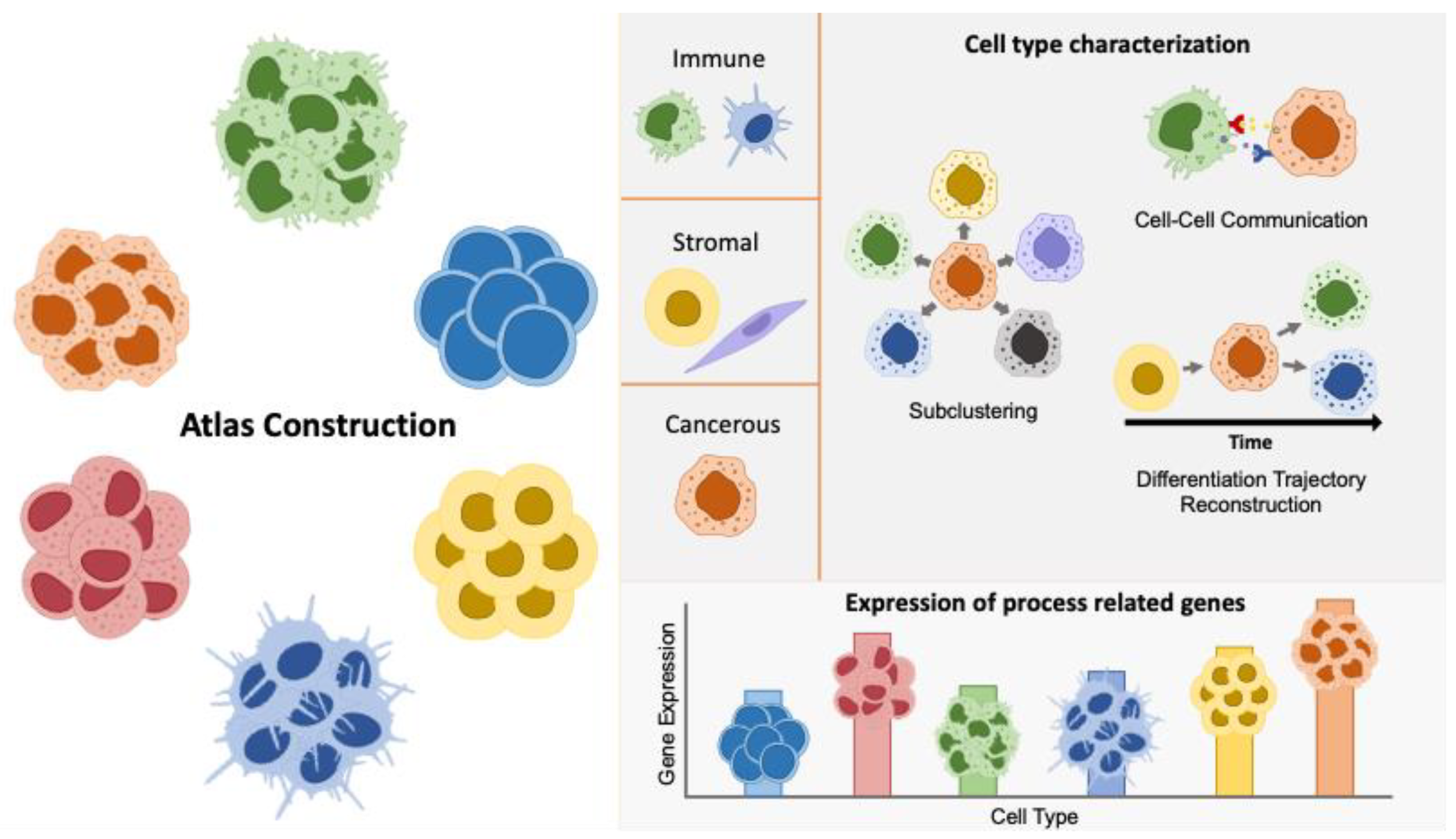
2.1. Atlas Construction
2.2. Cell Type-Specific Characterization
2.2.1. Tumor Cell Characterization and Crosstalk with Stromal Compartments
2.2.2. Immune Landscape
2.3. Expression of Process-Related Genes
3. HNSCC Diagnosis and Prognosis
4. HNSCC Treatment
5. Outlook
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| scRNAseq | Single-Cell RNA Sequencing |
| HNCs | Head and Neck Cancers |
| TME | Tumor Microenvironment |
| OS | Overall Survival |
| TCGA | The Cancer Genome Atlas |
| CPTAC | Clinical Proteomic Tumor Analysis Consortium |
| OPSCC | Oropharyngeal Squamous Cell Carcinoma |
| TCR | T Cell Receptor |
| TILs | Tumor-Infiltrating Lymphocytes |
| HPV | Human Papilloma Virus |
| TAMs | Tumor-Associated Macrophages |
| SC | Single Cell |
| GEO | Gene Expression Omnibus |
| CAFs | Cancer-Associated Fibroblasts |
| DE | Differential Expression |
| CCC | Cell–Cell Communication |
| DTR | Differentiation Trajectory Reconstruction |
| CNV | Copy Number Variation |
| GO | Gene Ontology |
| CSCs | Cancer Stem Cells |
| EMT | Epithelial–Mesenchymal Transition |
| TNF | Tumor Necrosis Factor |
| pDCs | Plasmacytoid Dendritic Cells |
| PNI | Perineural Invasion |
| MDSCs | Myeloid-Derived Suppressor Cells |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Cell Death Ligand 1 |
| MARGs | Methylation/Autophagy-Related Genes |
| FGFR | Fibroblast Growth Factor Receptor |
| pRS | Prognostic Risk Scoring System |
| MCs | Mast Cells |
| PFS | Progression-Free Survival |
| TFH | CD4+ T Follicular Helper |
| DRGs | Cell Differentiation-Related Genes |
| tsCAFs | T Cell-Stimulating Cancer-Associated Fibroblasts |
| ICT | Induction Chemotherapy |
| ICB | Immune Checkpoint Blockade |
| GC | Germinal Center |
| MOC | Murine Oral Carcinoma |
| KO | Knockout |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Jawa, Y.; Yadav, P.; Gupta, S.; Mathan, S.V.; Pandey, J.; Saxena, A.K.; Kateriya, S.; Tiku, A.B.; Mondal, N.; Bhattacharya, J.; et al. Current Insights and Advancements in Head and Neck Cancer: Emerging Biomarkers and Therapeutics with Cues from Single Cell and 3D Model Omics Profiling. Front. Oncol. 2021, 11, 676948. [Google Scholar] [CrossRef]
- Cai, Z.; Tang, B.; Chen, L.; Lei, W. Mast Cell Marker Gene Signature in Head and Neck Squamous Cell Carcinoma. BMC Cancer 2022, 22, 577. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- Stampe, H.; Jakobsen, K.K.; Bendtsen, S.K.; Grønhøj, C.; von Buchwald, C. Systematic Review on the Current Knowledge and Use of Single-Cell RNA Sequencing in Head and Neck Cancer. APMIS 2021, 129, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-D.; Liu, Z.-Z.; Liu, Y.-Y.; Fu, Y.-C.; Lin, L.-L.; Hu, C.; Gu, H.-Y.; Wei, R.-X. Molecular Subtypes Based on Cell Differentiation Trajectories in Head and Neck Squamous Cell Carcinoma: Differential Prognosis and Immunotherapeutic Responses. Front. Immunol. 2021, 12, 791621. [Google Scholar] [CrossRef]
- Qi, Z.; Barrett, T.; Parikh, A.S.; Tirosh, I.; Puram, S.V. Single-Cell Sequencing and Its Applications in Head and Neck Cancer. Oral Oncol. 2019, 99, 104441. [Google Scholar] [CrossRef]
- Cao, S.; Wang, J.R.; Ji, S.; Yang, P.; Dai, Y.; Guo, S.; Montierth, M.D.; Shen, J.P.; Zhao, X.; Chen, J.; et al. Estimation of Tumor Cell Total MRNA Expression in 15 Cancer Types Predicts Disease Progression. Nat. Biotechnol. 2022, 40, 1624–1633. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular Heterogeneity during Mouse Pancreatic Ductal Adenocarcinoma Progression at Single-Cell Resolution. JCI Insight 2019, 4, e129212. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA Sequencing in Cancer Research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Sahoo, S.; Brien, R.; Jung, S.; Humphries, B.; Lee, W.; Cheng, Y.-H.; Zhang, Z.; Luker, K.E.; Wicha, M.S.; et al. Single-Cell RNA-Sequencing of Migratory Breast Cancer Cells: Discovering Genes Associated with Cancer Metastasis. Analyst 2019, 144, 7296–7309. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-T.; Lee, H.W.; Lee, H.-O.; Song, H.J.; Jeong, D.E.; Shin, S.; Kim, H.; Shin, Y.; Nam, D.-H.; Jeong, B.C.; et al. Application of Single-Cell RNA Sequencing in Optimizing a Combinatorial Therapeutic Strategy in Metastatic Renal Cell Carcinoma. Genome Biol. 2016, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Lee, B.-S.; Jang, J.Y.; Lee, Y.S.; Kim, H.J.; Roh, J.; Shin, Y.S.; Woo, H.G.; Kim, C.-H. Single-Cell Transcriptome Profiling of the Stepwise Progression of Head and Neck Cancer. Nat. Commun. 2023, 14, 1055. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, Y.A.; Conejo-Garcia, J.R.; Chung, C.H.; Wang, X. Estimation of Immune Cell Content in Tumor Using Single-Cell RNA-seq Reference Data. BMC Cancer 2019, 19, 715. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Puram, S.V.; Mints, M.; Pal, A.; Qi, Z.; Reeb, A.; Gelev, K.; Barrett, T.F.; Gerndt, S.; Liu, P.; Parikh, A.S.; et al. Cellular States Are Coupled to Genomic and Viral Heterogeneity in HPV-Related Oropharyngeal Carcinoma. Nat. Genet. 2023, 55, 640–650. [Google Scholar] [CrossRef]
- Kürten, C.H.L.; Kulkarni, A.; Cillo, A.R.; Santos, P.M.; Roble, A.K.; Onkar, S.; Reeder, C.; Lang, S.; Chen, X.; Duvvuri, U.; et al. Investigating Immune and Non-Immune Cell Interactions in Head and Neck Tumors by Single-Cell RNA Sequencing. Nat. Commun. 2021, 12, 7338. [Google Scholar] [CrossRef]
- Song, L.; Zhang, S.; Yu, S.; Ma, F.; Wang, B.; Zhang, C.; Sun, J.; Mao, X.; Wei, L. Cellular Heterogeneity Landscape in Laryngeal Squamous Cell Carcinoma. Int. J. Cancer 2020, 147, 2879–2890. [Google Scholar] [CrossRef]
- Bill, R.; Wirapati, P.; Messemaker, M.; Roh, W.; Zitti, B.; Duval, F.; Kiss, M.; Park, J.C.; Saal, T.M.; Hoelzl, J.; et al. CXCL9:SPP1 Macrophage Polarity Identifies a Network of Cellular Programs That Control Human Cancers. Science 2023, 381, 515–524. [Google Scholar] [CrossRef]
- Cillo, A.R.; Kürten, C.H.L.; Tabib, T.; Qi, Z.; Onkar, S.; Wang, T.; Liu, A.; Duvvuri, U.; Kim, S.; Soose, R.J.; et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 2020, 52, 183–199.e9. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, A.; Graves, D.; Korrer, M.; Wang, Y.; Roy, S.; Naveed, A.; Xu, Y.; Luginbuhl, A.; Curry, J.; Gibson, M.; et al. Immunostimulatory Cancer-Associated Fibroblast Subpopulations Can Predict Immunotherapy Response in Head and Neck Cancer. Clin. Cancer Res. 2022, 28, 2094–2109. [Google Scholar] [CrossRef] [PubMed]
- Luoma, A.M.; Suo, S.; Wang, Y.; Gunasti, L.; Porter, C.B.M.; Nabilsi, N.; Tadros, J.; Ferretti, A.P.; Liao, S.; Gurer, C.; et al. Tissue-Resident Memory and Circulating T Cells Are Early Responders to Pre-Surgical Cancer Immunotherapy. Cell 2022, 185, 2918–2935.e29. [Google Scholar] [CrossRef] [PubMed]
- Wieland, A.; Patel, M.R.; Cardenas, M.A.; Eberhardt, C.S.; Hudson, W.H.; Obeng, R.C.; Griffith, C.C.; Wang, X.; Chen, Z.G.; Kissick, H.T.; et al. Defining HPV-Specific B Cell Responses in Patients with Head and Neck Cancer. Nature 2021, 597, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, C.S.; Kissick, H.T.; Patel, M.R.; Cardenas, M.A.; Prokhnevska, N.; Obeng, R.C.; Nasti, T.H.; Griffith, C.C.; Im, S.J.; Wang, X.; et al. Functional HPV-Specific PD-1+ Stem-like CD8 T Cells in Head and Neck Cancer. Nature 2021, 597, 279–284. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Wang, X.-L.; Peng, W.; Chen, Q.-Y.; Chi, D.-M.; Chen, J.-R.; Han, B.-W.; Lin, G.-W.; Li, Y.-Q.; et al. Tumour Heterogeneity and Intercellular Networks of Nasopharyngeal Carcinoma at Single Cell Resolution. Nat. Commun. 2021, 12, 741. [Google Scholar] [CrossRef]
- Lin, M.; Sade-Feldman, M.; Wirth, L.; Lawrence, M.S.; Faden, D.L. Single-Cell Transcriptomic Profiling for Inferring Tumor Origin and Mechanisms of Therapeutic Resistance. NPJ Precis Oncol. 2022, 6, 71. [Google Scholar] [CrossRef]
- Peng, Y.; Xiao, L.; Rong, H.; Ou, Z.; Cai, T.; Liu, N.; Li, B.; Zhang, L.; Wu, F.; Lan, T.; et al. Single-Cell Profiling of Tumor-Infiltrating TCF1/TCF7+ T Cells Reveals a T Lymphocyte Subset Associated with Tertiary Lymphoid Structures/Organs and a Superior Prognosis in Oral Cancer. Oral Oncol. 2021, 119, 105348. [Google Scholar] [CrossRef]
- Janjic, B.M.; Kulkarni, A.; Ferris, R.L.; Vujanovic, L.; Vujanovic, N.L. Human B Cells Mediate Innate Anti-Cancer Cytotoxicity Through Concurrent Engagement of Multiple TNF Superfamily Ligands. Front. Immunol. 2022, 13, 837842. [Google Scholar] [CrossRef]
- Huynh, N.C.-N.; Huang, T.-T.; Nguyen, C.T.-K.; Lin, F.-K. Comprehensive Integrated Single-Cell Whole Transcriptome Analysis Revealed the p-EMT Tumor Cells-CAFs Communication in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 6470. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, X.; Zhang, D.; Deng, S.; Zheng, A.; Du, F.; Shen, J.; Yue, L.; Yi, T.; Xiao, Z.; et al. Complex Interaction and Heterogeneity among Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma Revealed by Single-Cell Sequencing. Front. Immunol. 2022, 13, 1050951. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, J.; Li, H.; Yang, Z.; Zhang, X.; Li, X.; Wang, J.; Zhang, Y.; Chen, S.; Song, M. Single-Cell Transcriptomics Reveal the Intratumoral Landscape of Infiltrated T-Cell Subpopulations in Oral Squamous Cell Carcinoma. Mol. Oncol. 2021, 15, 866–886. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, J.; Baranovskii, A.; Ronen, J.; Uyar, B.; Franke, V.; Akalin, A. Identifying Tumor Cells at the Single-Cell Level Using Machine Learning. Genome Biol. 2022, 23, 123. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Z.; Zeng, T. Essential Gene Expression Pattern of Head and Neck Squamous Cell Carcinoma Revealed by Tumor-Specific Expression Rule Based on Single-Cell RNA Sequencing. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Y.; Xia, C.; Ding, L.; Pu, Y.; Hu, X.; Cai, H.; Hu, Q. Integrated Analysis of Single-Cell RNA-seq and Bulk RNA-seq Reveals Distinct Cancer-Associated Fibroblasts in Head and Neck Squamous Cell Carcinoma. Ann. Transl. Med. 2021, 9, 1017. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-Associated Fibroblasts in the Single-Cell Era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Meng, L.; Lu, H.; Li, Y.; Zhao, J.; He, S.; Wang, Z.; Shen, J.; Huang, H.; Xiao, J.; Sooranna, S.R.; et al. Human Papillomavirus Infection Can Alter the Level of Tumour Stemness and T Cell Infiltration in Patients with Head and Neck Squamous Cell Carcinoma. Front. Immunol. 2022, 13, 1013542. [Google Scholar] [CrossRef]
- Chen, S.M.Y.; Krinsky, A.L.; Woolaver, R.A.; Wang, X.; Chen, Z.; Wang, J.H. Tumor Immune Microenvironment in Head and Neck Cancers. Mol. Carcinog. 2020, 59, 766–774. [Google Scholar] [CrossRef]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52. [Google Scholar] [CrossRef]
- Gameiro, S.F.; Evans, A.M.; Mymryk, J.S. The Tumor Immune Microenvironments of HPV+ and HPV− Head and Neck Cancers. WIREs Mech. Dis. 2022, 14, e1539. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, H.; Liu, X.; Li, N.; Bai, H.; Guo, C.; Xu, T.; Zhu, L.; Liu, C.; Xiao, J. The Chemokines Initiating and Maintaining Immune Hot Phenotype Are Prognostic in ICB of HNSCC. Front. Genet. 2022, 13, 820065. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Y.; Sun, R.; Franceschi, D.; Pan, H.; Wei, C.; Ogbuehi, A.C.; Lethaus, B.; Savkovic, V.; Gaus, S.; et al. Single-Cell Transcriptome Analysis Reveals Different Immune Signatures in HPV- and HPV + Driven Human Head and Neck Squamous Cell Carcinoma. J. Immunol. Res. 2022, 2022, 2079389. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, S.; Tang, L.; Li, R.; Zhai, J.; Luo, S.; Peng, Y.; Chen, X.; Wei, L. Single-Cell RNA Sequencing Reveals TCR+ Macrophages in HPV-related Head and Neck Squamous Cell Carcinoma. Front. Immunol. 2022, 13, 1030222. [Google Scholar] [CrossRef]
- Parker, T.M.; Henriques, V.; Beltran, A.; Nakshatri, H.; Gogna, R. Cell Competition and Tumor Heterogeneity. Semin. Cancer Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Liu, F.; Tang, L.; Li, Q.; Chen, L.; Pan, Y.; Yin, Z.; He, J.; Tian, J. Single-Cell Transcriptomics Uncover the Key Ferroptosis Regulators Contribute to Cancer Progression in Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2022, 9, 962742. [Google Scholar] [CrossRef]
- Jiang, X.; Ke, J.; Jia, L.; An, X.; Ma, H.; Li, Z.; Yuan, W. A Novel Cuproptosis-Related Gene Signature of Prognosis and Immune Microenvironment in Head and Neck Squamous Cell Carcinoma Cancer. J. Cancer Res. Clin. Oncol. 2023, 149, 203–218. [Google Scholar] [CrossRef]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: A Cancer Single-Cell State Atlas. Nucleic Acids Res. 2019, 47, D900–D908. [Google Scholar] [CrossRef]
- Sun, D.; Wang, J.; Han, Y.; Dong, X.; Ge, J.; Zheng, R.; Shi, X.; Wang, B.; Li, Z.; Ren, P.; et al. TISCH: A Comprehensive Web Resource Enabling Interactive Single-Cell Transcriptome Visualization of Tumor Microenvironment. Nucleic Acids Res. 2021, 49, D1420–D1430. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, R.; Jin, R.; Fan, Y.; Li, T.; Shuai, Y.; Li, X.; Wang, X.; Luo, J. Integrating Clinical and Genetic Analysis of Perineural Invasion in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 434. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y. Potential Roles of Cornichon Family AMPA Receptor Auxiliary Protein 4 (CNIH4) in Head and Neck Squamous Cell Carcinoma. Cancer Biomark. 2022, 35, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Daimary, U.D.; Jose, S.; Sajeev, A.; Chinnathambi, A.; Alharbi, S.A.; Shakibaei, M.; Kunnumakkara, A.B. Differential Expression of Genes Regulating Store-Operated Calcium Entry in Conjunction with Mitochondrial Dynamics as Potential Biomarkers for Cancer: A Single-Cell RNA Analysis. Front. Genet. 2022, 13, 866473. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, C.; Obermayer, B.; Klinghammer, K.; Ochsenreither, S.; Treue, D.; Stenzinger, A.; Glimm, H.; Fröhling, S.; Kindler, T.; Brandts, C.H.; et al. Distinct Immune Evasion in APOBEC-enriched, HPV-negative HNSCC. Int. J. Cancer 2020, 147, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Fan, H.; Liu, H.; Tang, S.; Zheng, Y. YTHDC1 Promotes Stemness Maintenance and Malignant Progression in Head and Neck Squamous Cell Carcinoma. Stem Cells Int. 2022, 2022, 7494354. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Fan, J.; He, G.; Dong, C.; Zhou, S.; Zheng, Y. Signal Peptidase Complex Catalytic Subunit SEC11A Upregulation Is a Biomarker of Poor Prognosis in Patients with Head and Neck Squamous Cell Carcinoma. PLoS ONE 2022, 17, e0269166. [Google Scholar] [CrossRef] [PubMed]
- Carofino, B.L.; Dinshaw, K.M.; Ho, P.Y.; Cataisson, C.; Michalowski, A.M.; Ryscavage, A.; Alkhas, A.; Wong, N.W.; Koparde, V.; Yuspa, S.H. Head and Neck Squamous Cancer Progression Is Marked by CLIC4 Attenuation in Tumor Epithelium and Reciprocal Stromal Upregulation of MiR-142-3p, a Novel Post-Transcriptional Regulator of CLIC4. Oncotarget 2019, 10, 7251–7275. [Google Scholar] [CrossRef]
- Yorozu, A.; Sekiguchi, S.; Takasawa, A.; Okazaki, F.; Niinuma, T.; Kitajima, H.; Yamamoto, E.; Kai, M.; Toyota, M.; Hatanaka, Y.; et al. CXCL12 Is Expressed by Skeletal Muscle Cells in Tongue Oral Squamous Cell Carcinoma. Cancer Med. 2022, 12, 5953–5963. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, W.; Zhang, J. 8-Gene Signature Related to CD8+ T Cell Infiltration by Integrating Single-Cell and Bulk RNA-sequencing in Head and Neck Squamous Cell Carcinoma. Front. Genet. 2022, 13, 938611. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Kang, X.; Chen, Y.; Yi, B.; Yan, X.; Jiang, C.; Chen, B.; Lu, L.; Sun, Y.; Shi, R. An Integrative Microenvironment Approach for Laryngeal Carcinoma: The Role of Immune/Methylation/Autophagy Signatures on Disease Clinical Prognosis and Single-Cell Genotypes. J. Cancer 2021, 12, 4148–4171. [Google Scholar] [CrossRef]
- Chen, Q.; Chu, L.; Li, X.; Li, H.; Zhang, Y.; Cao, Q.; Zhuang, Q. Investigation of an FGFR-signaling-related Prognostic Model and Immune Landscape in Head and Neck Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 801715. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liang, Y.; Dong, Y.; Huang, L.; Li, A.; Du, R.; Huang, H. Novel Prognostic Matrisome-Related Gene Signature of Head and Neck Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2022, 10, 884590. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Liu, Y.; Mints, M.; Mullins, R.; Sample, R.; Law, T.; Barrett, T.; Mazul, A.L.; Jackson, R.S.; Kang, S.Y.; et al. Single-Cell Deconvolution of Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 1230. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zheng, S.; He, X.; Huang, Y.; Hu, L.; Qin, F.; Zhong, L.; Li, S.; Hu, W.; Zhu, J. Identification of Molecular Classification and Gene Signature for Predicting Prognosis and Immunotherapy Response in HNSCC Using Cell Differentiation Trajectories. Sci. Rep. 2022, 12, 20404. [Google Scholar] [CrossRef] [PubMed]
- Han, P.-Z.; Tan, L.-C.; Ouyang, Q.-S.; Yu, P.-C.; Shi, X.; Hu, J.-Q.; Wei, W.-J.; Lu, Z.-W.; Wang, Y.; Ji, Q.-H.; et al. Development and Validation of a Gene Model Predicting Lymph Node Metastasis and Prognosis of Oral Squamous Cell Carcinoma Based on Single-Cell and Bulk RNA-seq Analysis. J. Oral Pathol. Med. 2022, 52, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, H.-C.; Cao, C.; Hu, J.-D.; Qian, J.; Jiang, T.; Jiang, W.-B.; Zhou, S.; Qiu, X.-W.; Wang, H.-L. Identification and Validation of a Novel Signature Based on Cell-Cell Communication in Head and Neck Squamous Cell Carcinoma by Integrated Analysis of Single-Cell Transcriptome and Bulk RNA-Sequencing. Front. Oncol. 2023, 13, 1136729. [Google Scholar] [CrossRef]
- Caudell, J.J.; Gillison, M.L.; Maghami, E.; Spencer, S.; Pfister, D.G.; Adkins, D.; Birkeland, A.C.; Brizel, D.M.; Busse, P.M.; Cmelak, A.J.; et al. NCCN Guidelines® Insights: Head and Neck Cancers, Version 1.2022. J. Natl. Compr. Canc. Netw. 2022, 20, 224–234. [Google Scholar] [CrossRef]
- Pulte, D.; Brenner, H. Changes in Survival in Head and Neck Cancers in the Late 20th and Early 21st Century: A Period Analysis. Oncologist 2010, 15, 994–1001. [Google Scholar] [CrossRef]
- Davis-Marcisak, E.F.; Sherman, T.D.; Orugunta, P.; Stein-O’Brien, G.L.; Puram, S.V.; Roussos Torres, E.T.; Hopkins, A.C.; Jaffee, E.M.; Favorov, A.V.; Afsari, B.; et al. Differential Variation Analysis Enables Detection of Tumor Heterogeneity Using Single-Cell RNA-sequencing Data. Cancer Res. 2019, 79, 5102–5112. [Google Scholar] [CrossRef]
- Song, H.; Lou, C.; Ma, J.; Gong, Q.; Tian, Z.; You, Y.; Ren, G.; Guo, W.; Wang, Y.; He, K.; et al. Single-Cell Transcriptome Analysis Reveals Changes of Tumor Immune Microenvironment in Oral Squamous Cell Carcinoma after Chemotherapy. Front. Cell Dev. Biol. 2022, 10, 914120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Shen, S.; Miyauchi, S.; Sanders, P.D.; Franiak-Pietryga, I.; Mell, L.; Gutkind, J.S.; Cohen, E.E.W.; Califano, J.A.; Sharabi, A.B. B Cells Improve Overall Survival in HPV-Associated Squamous Cell Carcinomas and Are Activated by Radiation and PD-1 Blockade. Clin. Cancer Res. 2020, 26, 3345–3359. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Donnelly, C.R.; Heath, B.R.; Bellile, E.; Donnelly, L.A.; Taner, H.F.; Broses, L.; Brenner, J.C.; Chinn, S.B.; Ji, R.-R.; et al. Cancer-Specific Type-I Interferon Receptor Signaling Promotes Cancer Stemness and Effector CD8+ T-Cell Exhaustion. Oncoimmunology 2021, 10, 1997385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zeng, Z.; Egloff, A.M.; Zhang, F.; Guo, F.; Campbell, K.M.; Du, P.; Fu, J.; Zolkind, P.; Ma, X.; et al. Checkpoint Blockade-Induced CD8+ T Cell Differentiation in Head and Neck Cancer Responders. J. Immunother. Cancer 2022, 10, e004034. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, K.; Chen, J.; Wang, X.; Ling, R.; Cheng, M.; Chen, Z.; Chen, F.; He, Q.; Li, S.; et al. Aberrant Translation Regulated by METTL1/WDR4-Mediated TRNA N7-Methylguanosine Modification Drives Head and Neck Squamous Cell Carcinoma Progression. Cancer Commun. 2022, 42, 223–244. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, K.; Chen, Y.; Ge, X.; Wu, J.; Xu, P.; Yao, J. Progress of Single-Cell RNA Sequencing Combined with Spatial Transcriptomics in Tumour Microenvironment and Treatment of Pancreatic Cancer. J. Transl. Med. 2024, 22, 563. [Google Scholar] [CrossRef]
- Chen, J.; Song, Y.; Huang, J.; Wan, X.; Li, Y. Integrated Single-Cell RNA Sequencing and Spatial Transcriptomics Analysis Reveals the Tumour Microenvironment in Patients with Endometrial Cancer Responding to Anti-PD-1 Treatment. Clin. Transl. Med. 2024, 14, e1668. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Andreatta, M.; Berenstein, A.J.; Carmona, S.J. ScGate: Marker-Based Purification of Cell Types from Heterogeneous Single-Cell RNA-Seq Datasets. Bioinformatics 2022, 38, 2642–2644. [Google Scholar] [CrossRef]
- Zappia, L.; Theis, F.J. Over 1000 Tools Reveal Trends in the Single-Cell RNA-Seq Analysis Landscape. Genome Biol. 2021, 22, 301. [Google Scholar] [CrossRef]
- Heumos, L.; Schaar, A.C.; Lance, C.; Litinetskaya, A.; Drost, F.; Zappia, L.; Lücken, M.D.; Strobl, D.C.; Henao, J.; Curion, F.; et al. Best Practices for Single-Cell Analysis across Modalities. Nat. Rev. Genet. 2023, 24, 550–572. [Google Scholar] [CrossRef]
- Du, P.; Fan, R.; Zhang, N.; Wu, C.; Zhang, Y. Advances in Integrated Multi-Omics Analysis for Drug-Target Identification. Biomolecules 2024, 14, 692. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.E.; Skene, N.G. A Balanced Measure Shows Superior Performance of Pseudobulk Methods in Single-Cell RNA-Sequencing Analysis. Nat. Commun. 2022, 13, 7851. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, Sensitive and Accurate Integration of Single-Cell Data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Andreatta, M.; Hérault, L.; Gueguen, P.; Gfeller, D.; Berenstein, A.J.; Carmona, S.J. Semi-Supervised Integration of Single-Cell Transcriptomics Data. Nat. Commun. 2024, 15, 872. [Google Scholar] [CrossRef]
- Shapiro, G.K. HPV Vaccination: An Underused Strategy for the Prevention of Cancer. Curr. Oncol. 2022, 29, 3780–3792. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.C.; Pearse, R.; Young-Pearse, T.; Mostafavi, S. Mosaic Loss of Chromosome Y in Aged Human Microglia. Genome Res. 2022, 32, 1795–1807. [Google Scholar] [CrossRef]
- Hollows, R.; Wei, W.; Cazier, J.-B.; Mehanna, H.; Parry, G.; Halford, G.; Murray, P. Association between Loss of Y Chromosome and Poor Prognosis in Male Head and Neck Squamous Cell Carcinoma. Head Neck 2019, 41, 993–1006. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Accession Number | Year | Publication | Data Type | Tissue Types | Sample Number | Cell Number |
|---|---|---|---|---|---|---|
| GSE234933 | 2023 | Bill, R.; Wirapati, P.; Messemaker, M.; Roh, W.; et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 2023, 381, 515–524 [20] | scRNAseq | Primary tumor Local recurrence Distant metastasis | 52 | 87,399 |
| GSE182227 | 2022 | Puram, S.V.; Mints, M.; Pal, A.; Qi, Z.; et al. Cellular states are coupled to genomic and viral heterogeneity in HPV−related oropharyngeal carcinoma. Nat. Genet. 2023, 55, 640–650 [17] | scRNAseq | Primary tumor Normal tissue | 24 | 70,970 |
| GSE139324 | 2019 | Cillo, A.R.; Kürten, C.H.L.; Tabib, T.; Qi, Z.; et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 2020, 52, 183–199.e9 [21] | scRNAseq | Peripheral and intra-tumoral CD45+ populations | 63 | 131,224 |
| GSE164690 | 2021 | Kürten, C.H.L.; Kulkarni, A.; Cillo, A.R.; Santos, P.M.; et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat. Commun. 2021, 12, 7338 [18] | scRNAseq | Primary tumor Peripheral blood leucocytes | 51 | 134,606 |
| GSE103322 | 2017 | Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24 [16] | scRNAseq | Primary tumor | 18 | 5902 |
| GSE181919 | 2022 | Choi, J.H.; Lee, B.S.; Jang, J.Y.; Lee, Y.S;. et al. Single-cell transcriptome profiling of the stepwise progression of head and neck cancer. Nat. Commun. 2023, 14, 1055 [14] | scRNAseq | Primary tumor Normal tissue Leukoplakia Lymph node metastasis | 37 | 54,239 |
| GSE173647 | 2022 | — | scRNAseq | Primary tumor | 2 | 13,903 |
| GSE195832 | 2022 | Obradovic, A.; Graves, D.; Korrer, M.; Wang, Y.; et al. Immunostimulatory Cancer-Associated Fibroblast Subpopulations Can Predict Immunotherapy Response in Head and Neck Cancer. Clin. Cancer Res. 2022, 28, 2094–2109 [22] | scRNAseq | Primary tumor | 8 | 22,906 |
| GSE140042 | 2021 | — | scRNAseq | Primary tumor Lymph node metastasis | 9 | — |
| GSE200996 | 2022 | Luoma, A.M.; Suo, S.; Wang, Y.; Gunasti, L.; et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell 2022, 185, 2918–2935.e29 [23] | scRNAseq + scTCR | Peripheral and intra-tumoral CD45+ populations | 204 | 74,557 |
| GSE153559 | 2020 | Wieland, A.; Patel, M.R.; Cardenas, M.A.; Eberhardt, C.S.; et al. Defining HPV−specific B cell responses in patients with head and neck cancer. Nature 2021, 597, 274–278 [24] | scRNAseq | B cells Primary tumor Lymph node metastasis Peripheral tumor | 7 | 8271 |
| GSE180268 | 2021 | Eberhardt, C.S.; Kissick, H.T.; Patel, M.R.; Cardenas, M.A.; et al. Functional HPV−specific PD-1(+) stem-like CD8 T cells in head and neck cancer. Nature 2021, 597, 279–284 [25] | scRNAseq | TILs from primary tumor Lymph node metastasis | 39 | — |
| GSE162025 | 2020 | Liu, Y.; He, S.; Wang, X.L.; Peng, W.; et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 2021, 12, 741 [26] | scRNAseq + scTCR | Primary tumor Peripheral blood leucocytes | 40 | 176,447 |
| GSE150321 | 2020 | Song, L.; Zhang, S.; Yu, S.; Ma, F.; et al. Cellular heterogeneity landscape in laryngeal squamous cell carcinoma. Int. J. Cancer 2020, 147, 2879–2890 [19] | scRNAseq | Primary tumor | 2 | 12,985 |
| GSE213047 | 2022 | Lin, M.; Sade-Feldman, M.; Wirth, L.; Lawrence, M.S.; et al. Single-cell transcriptomic profiling for inferring tumor origin and mechanisms of therapeutic resistance. NPJ Precis. Oncol. 2022, 6, 71 [27] | scRNAseq | Primary tumor Normal tissue Lymph node metastasis | 3 | 11,470 |
| GSE172577 | 2021 | Peng, Y.; Xiao, L.; Rong, H.; Ou, Z.; et al. Single-cell profiling of tumor-infiltrating TCF1/TCF7(+) T cells reveals a T lymphocyte subset associated with tertiary lymphoid structures/organs and a superior prognosis in oral cancer. Oral Oncol. 2021, 119, 105348 [28] | scRNAseq | Primary tumor | 6 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conde-Lopez, C.; Marripati, D.; Elkabets, M.; Hess, J.; Kurth, I. Unravelling the Complexity of HNSCC Using Single-Cell Transcriptomics. Cancers 2024, 16, 3265. https://doi.org/10.3390/cancers16193265
Conde-Lopez C, Marripati D, Elkabets M, Hess J, Kurth I. Unravelling the Complexity of HNSCC Using Single-Cell Transcriptomics. Cancers. 2024; 16(19):3265. https://doi.org/10.3390/cancers16193265
Chicago/Turabian StyleConde-Lopez, Cristina, Divyasree Marripati, Moshe Elkabets, Jochen Hess, and Ina Kurth. 2024. "Unravelling the Complexity of HNSCC Using Single-Cell Transcriptomics" Cancers 16, no. 19: 3265. https://doi.org/10.3390/cancers16193265
APA StyleConde-Lopez, C., Marripati, D., Elkabets, M., Hess, J., & Kurth, I. (2024). Unravelling the Complexity of HNSCC Using Single-Cell Transcriptomics. Cancers, 16(19), 3265. https://doi.org/10.3390/cancers16193265