
Germline Polymorphisms Associated with Overall Survival in Lung Adenocarcinoma: Genome-Wide Analysis

 , , ,
, , ,  ,
,  , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Case Series and Research Ethics
2.2. Clinical Data and Biological Samples
2.3. Genome-Wide Genotyping
2.4. Statistical Analyses
2.5. In Silico Functional Analyses
3. Results
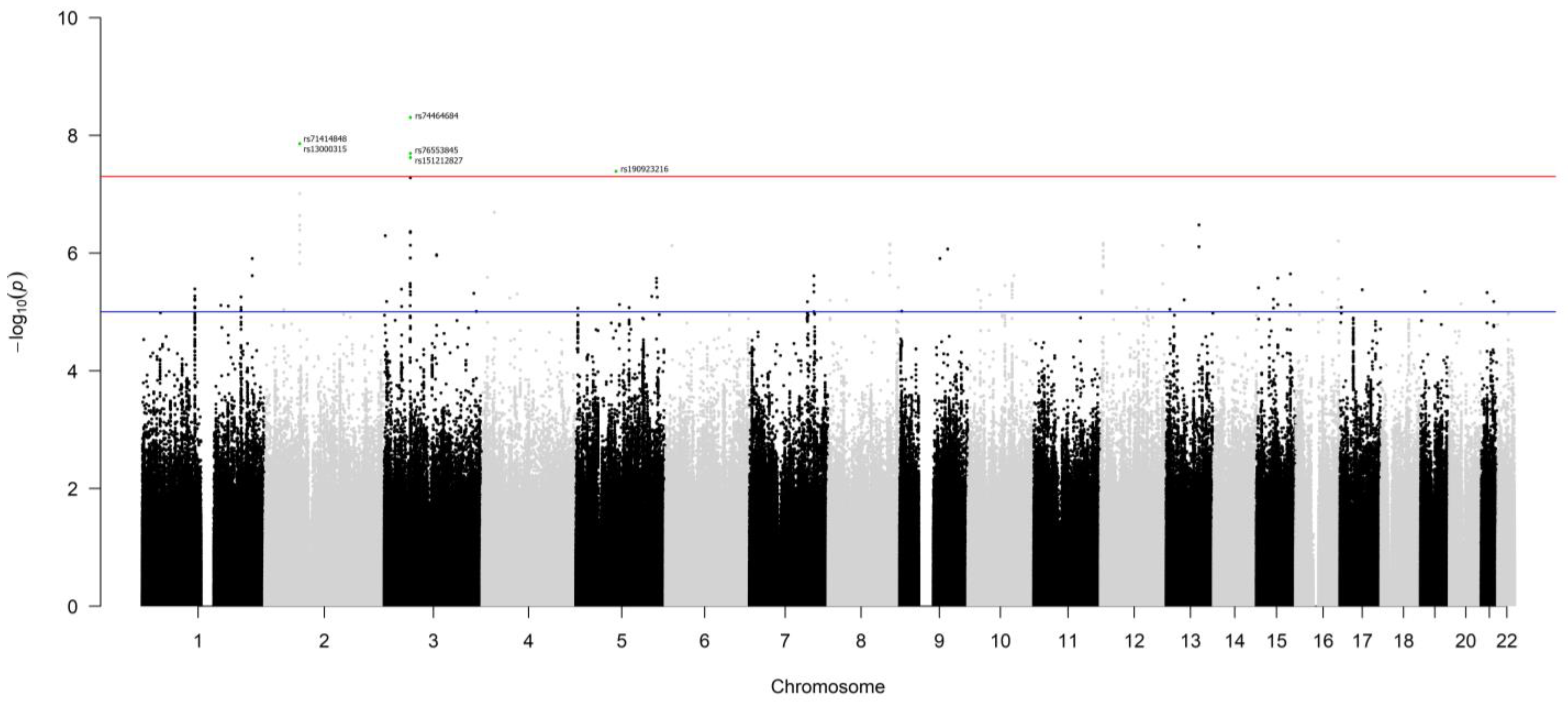
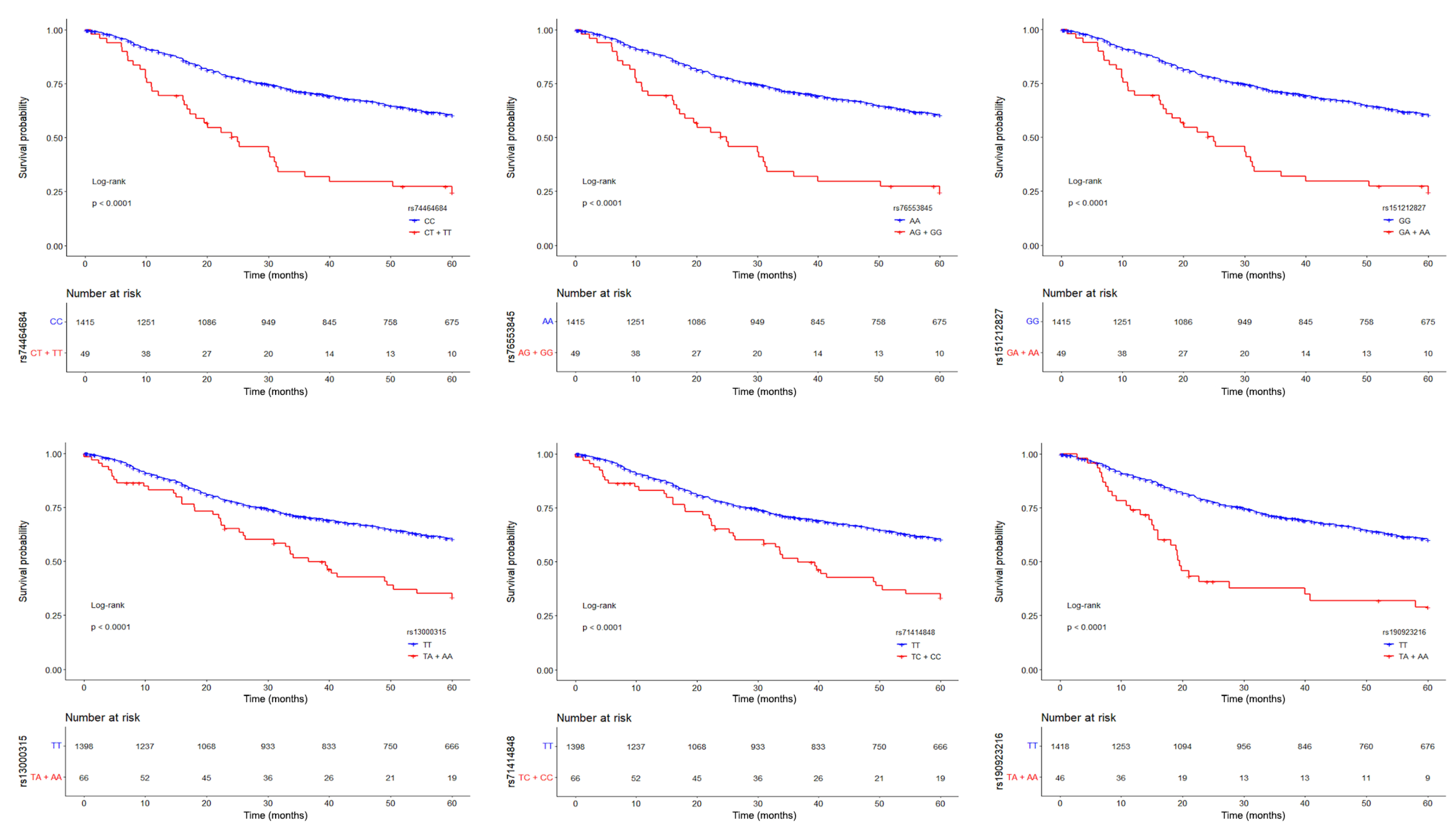
3.1. Germline Variants Associated with Overall Survival
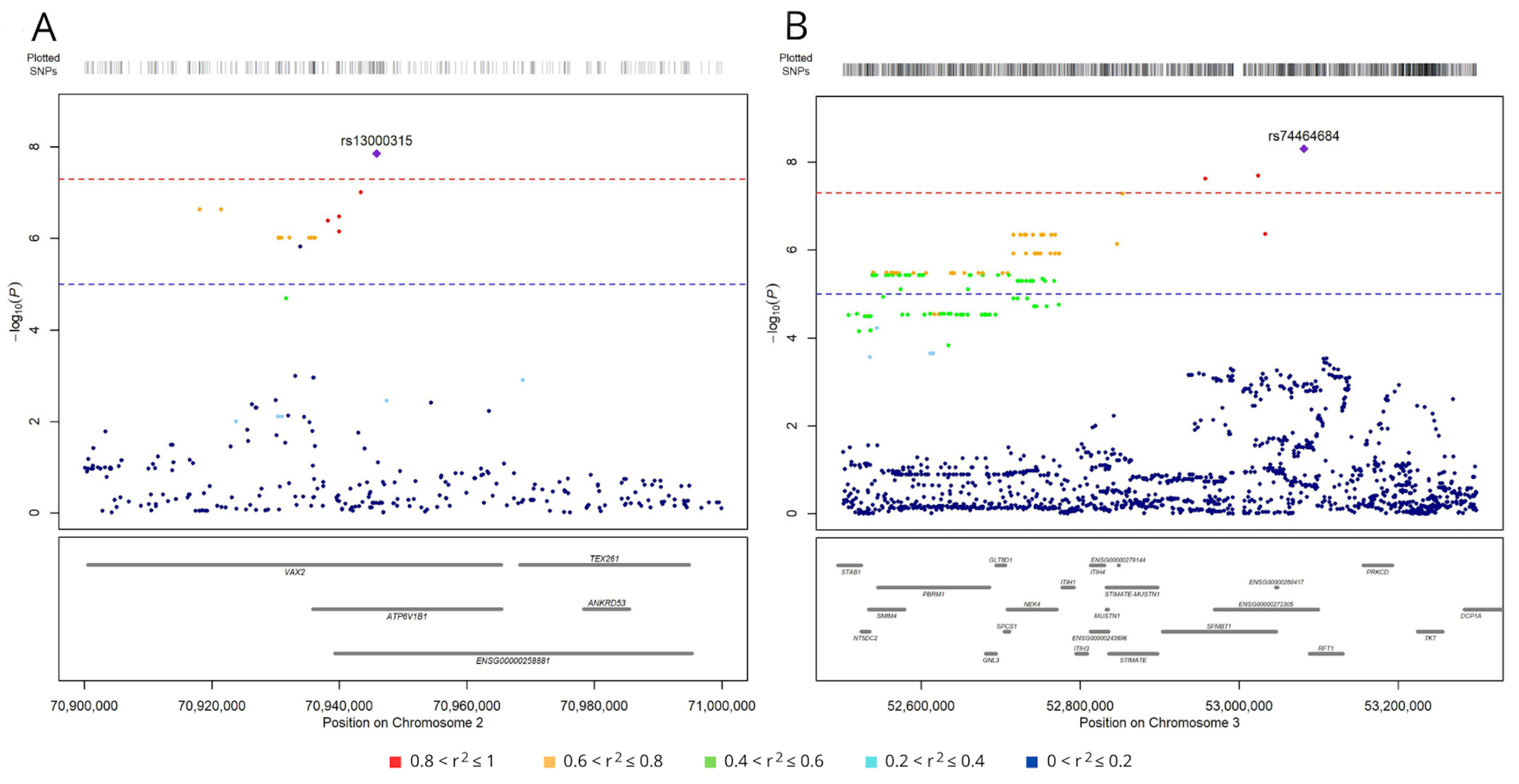
3.2. SNPs Associated with Lung Adenocarcinoma Survival Have Regulatory Roles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Potter, A.L.; Rosenstein, A.L.; Kiang, M.V.; Shah, S.A.; Gaissert, H.A.; Chang, D.C.; Fintelmann, F.J.; Yang, C.-F.J. Association of Computed Tomography Screening with Lung Cancer Stage Shift and Survival in the United States: Quasi-Experimental Study. BMJ 2022, 376, e069008. [Google Scholar] [CrossRef] [PubMed]
- Whitson, B.A.; Groth, S.S.; Duval, S.J.; Swanson, S.J.; Maddaus, M.A. Surgery for Early-Stage Non-Small Cell Lung Cancer: A Systematic Review of the Video-Assisted Thoracoscopic Surgery Versus Thoracotomy Approaches to Lobectomy. Ann. Thorac. Surg. 2008, 86, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Rami-Porta, R.; Call, S.; Dooms, C.; Obiols, C.; Sánchez, M.; Travis, W.D.; Vollmer, I. Lung Cancer Staging: A Concise Update. Eur. Respir. J. 2018, 51, 1800190. [Google Scholar] [CrossRef]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef]
- Garinet, S.; Wang, P.; Mansuet-Lupo, A.; Fournel, L.; Wislez, M.; Blons, H. Updated Prognostic Factors in Localized NSCLC. Cancers 2022, 14, 1400. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Chen, K.; Liu, H.; Liu, Z.; Luo, S.; Patz, E.F.; Moorman, P.G.; Su, L.; Shen, S.; Christiani, D.C.; Wei, Q. Genetic Variants in RUNX3, AMD1 and MSRA in the Methionine Metabolic Pathway and Survival in Nonsmall Cell Lung Cancer Patients. Int. J. Cancer 2019, 145, 621–631. [Google Scholar] [CrossRef]
- Du, H.; Liu, L.; Liu, H.; Luo, S.; Patz, E.F.; Glass, C.; Su, L.; Du, M.; Christiani, D.C.; Wei, Q. Genetic Variants of DOCK2, EPHB1 and VAV2 in the Natural Killer Cell-Related Pathway Are Associated with Non-Small Cell Lung Cancer Survival. Am. J. Cancer Res. 2021, 11, 2264–2277. [Google Scholar]
- Qian, D.; Liu, H.; Zhao, L.; Wang, X.; Luo, S.; Moorman, P.G.; Patz Jr, E.F.; Su, L.; Shen, S.; Christiani, D.C.; et al. Novel Genetic Variants in Genes of the Fc Gamma Receptor-Mediated Phagocytosis Pathway Predict Non-Small Cell Lung Cancer Survival. Transl. Lung Cancer Res. 2020, 9, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Guo, S.; Wang, Y.; Wang, H.; Lu, D.; Wang, J.; Jin, L.; Jiang, G.; Wu, J.; et al. Effect of ERCC2 Rs13181 and Rs1799793 Polymorphisms and Environmental Factors on the Prognosis of Patients with Lung Cancer. Am. J. Transl. Res. 2020, 12, 6941–6953. [Google Scholar] [PubMed]
- Pintarelli, G.; Cotroneo, C.E.; Noci, S.; Dugo, M.; Galvan, A.; Delli Carpini, S.; Citterio, L.; Manunta, P.; Incarbone, M.; Tosi, D.; et al. Genetic Susceptibility Variants for Lung Cancer: Replication Study and Assessment as Expression Quantitative Trait Loci. Sci. Rep. 2017, 7, 42185. [Google Scholar] [CrossRef]
- Du, H.; Mu, R.; Liu, L.; Liu, H.; Luo, S.; Patz, E.F.; Glass, C.; Su, L.; Du, M.; Christiani, D.C.; et al. Single Nucleotide Polymorphisms in FOXP1 and RORA of the Lymphocyte Activation-Related Pathway Affect Survival of Lung Cancer Patients. Transl. Lung Cancer Res. 2022, 11, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.S.; Liu, H.; Wu, Y.; Luo, S.; Patz, E.F.; Glass, C.; Su, L.; Du, M.; Christiani, D.C.; Wei, Q. Genetic Variants in DDO and PEX5L in Peroxisome-Related Pathways Predict Non-Small Cell Lung Cancer Survival. Mol. Carcinog. 2022, 61, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Tang, D.; Zhao, Y.C.; Liu, H.; Luo, S.; Stinchcombe, T.E.; Glass, C.; Su, L.; Shen, S.; Christiani, D.C.; et al. Potentially Functional Variants of ERAP1, PSMF1 and NCF2 in the MHC-I-Related Pathway Predict Non-Small Cell Lung Cancer Survival. Cancer Immunol. Immunother. 2021, 70, 2819–2833. [Google Scholar] [CrossRef]
- Galvan, A.; Colombo, F.; Frullanti, E.; Dassano, A.; Noci, S.; Wang, Y.; Eisen, T.; Matakidou, A.; Tomasello, L.; Vezzalini, M.; et al. Germline Polymorphisms and Survival of Lung Adenocarcinoma Patients: A Genome-Wide Study in Two European Patient Series. Int. J. Cancer 2015, 136, E262–E271. [Google Scholar] [CrossRef]
- Zhu, M.; Geng, L.; Shen, W.; Wang, Y.; Liu, J.; Cheng, Y.; Wang, C.; Dai, J.; Jin, G.; Hu, Z.; et al. Exome-Wide Association Study Identifies Low-Frequency Coding Variants in 2p23.2 and 7p11.2 Associated with Survival of Non–Small Cell Lung Cancer Patients. J. Thorac. Oncol. 2017, 12, 644–656. [Google Scholar] [CrossRef]
- Dragani, T.A.; Muley, T.; Schneider, M.A.; Kobinger, S.; Eichhorn, M.; Winter, H.; Hoffmann, H.; Kriegsmann, M.; Noci, S.; Incarbone, M.; et al. Lung Adenocarcinoma Diagnosed at a Younger Age Is Associated with Advanced Stage, Female Sex, and Ever-Smoker Status, in Patients Treated with Lung Resection. Cancers 2023, 15, 2395. [Google Scholar] [CrossRef]
- Sobin, L.; Christian, W. (Eds.) TNM Classification of Malignant Tumours; UICC International Union Against Cancer: Geneva, Switzerland, 2002. [Google Scholar]
- Sobin, L.H.; Gospodarowicz Mary, K.; Christian, W. (Eds.) TNM Classification of Malignant Tumours, 7th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; ISBN 978-1-4443-3241-4. [Google Scholar]
- Brierley James, D.; Gospodarowicz Mary, K.; Christian, W. (Eds.) TNM Classification of Malignant Tumours, 8th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2016; ISBN 978-1-119-26357-9. [Google Scholar]
- Maspero, D.; Dassano, A.; Pintarelli, G.; Noci, S.; de Cecco, L.; Incarbone, M.; Tosi, D.; Santambrogio, L.; Dragani, T.A.; Colombo, F. Read-through Transcripts in Lung: Germline Genetic Regulation and Correlation with the Expression of Other Genes. Carcinogenesis 2020, 41, 918–926. [Google Scholar] [CrossRef]
- Cotroneo, C.E.; Mangano, N.; Dragani, T.A.; Colombo, F. Lung Expression of Genes Putatively Involved in SARS-CoV-2 Infection Is Modulated in Cis by Germline Variants. Eur. J. Hum. Genet. 2021, 29, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Anderson, C.A.; Pettersson, F.H.; Clarke, G.M.; Cardon, L.R.; Morris, A.P.; Zondervan, K.T. Data Quality Control in Genetic Case-Control Association Studies. Nat. Protoc. 2010, 5, 1564–1573. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-Generation Genotype Imputation Service and Methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-Based Phasing Using the Haplotype Reference Consortium Panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Abecasis, G.R.; Hinds, D.A. Minimac2: Faster Genotype Imputation. Bioinformatics 2015, 31, 782–784. [Google Scholar] [CrossRef]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 Diverse Genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Verlouw, J.A.M.; Clemens, E.; de Vries, J.H.; Zolk, O.; Verkerk, A.J.M.H.; am Zehnhoff-Dinnesen, A.; Medina-Gomez, C.; Lanvers-Kaminsky, C.; Rivadeneira, F.; Langer, T.; et al. A Comparison of Genotyping Arrays. Eur. J. Human Genet. 2021, 29, 1611–1624. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Delaneau, O.; Marchini, J.; McVean, G.A.; Donnelly, P.; Lunter, G.; Marchini, J.L.; Myers, S.; Gupta-Hinch, A.; Iqbal, Z.; Mathieson, I.; et al. Integrating Sequence and Array Data to Create an Improved 1000 Genomes Project Haplotype Reference Panel. Nat Commun 2014, 5, 3934. [Google Scholar] [CrossRef]
- Clark, T.G.; Bradburn, M.J.; Love, S.B.; Altman, D.G. Survival Analysis Part I: Basic Concepts and First Analyses. Br. J. Cancer 2003, 89, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.T.; Patricia, M. Grambsch Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]
- Aulchenko, Y.S.; Ripke, S.; Isaacs, A.; van Duijn, C.M. GenABEL: An R Library for Genome-Wide Association Analysis. Bioinformatics 2007, 23, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-Scale Cis- and Trans-EQTL Analyses Identify Thousands of Genetic Loci and Polygenic Scores That Regulate Blood Gene Expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS ONE 2013, 8, e111842. [Google Scholar] [CrossRef]
- Kratzer, T.B.; Bandi, P.; Freedman, N.D.; Smith, R.A.; Travis, W.D.; Jemal, A.; Siegel, R.L. Lung Cancer Statistics, 2023. Cancer 2024, 130, 1330–1348. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takada, M.; Kubo, A.; Matsumura, A.; Fukai, S.; Tamura, A.; Saito, R.; Maruyama, Y.; Kawahara, M.; Ignatius Ou, S.-H. Performance Status and Smoking Status Are Independent Favorable Prognostic Factors for Survival in Non-Small Cell Lung Cancer: A Comprehensive Analysis of 26,957 Patients with NSCLC. J. Thorac. Oncol. 2010, 5, 620–630. [Google Scholar] [CrossRef]
- Sheikh, M.; Mukeriya, A.; Shangina, O.; Brennan, P.; Zaridze, D. Postdiagnosis Smoking Cessation and Reduced Risk for Lung Cancer Progression and Mortality: A Prospective Cohort Study. Ann. Intern. Med. 2021, 174, 1232–1239. [Google Scholar] [CrossRef]
- Schulze, A.B.; Kuntze, A.; Schmidt, L.H.; Mohr, M.; Marra, A.; Hillejan, L.; Schulz, C.; Görlich, D.; Hartmann, W.; Bleckmann, A.; et al. High Expression of NT5DC2 Is a Negative Prognostic Marker in Pulmonary Adenocarcinoma. Cancers 2022, 14, 1395. [Google Scholar] [CrossRef]
- Jin, X.; Liu, X.; Zhang, Z.; Xu, L. NT5DC2 Suppression Restrains Progression towards Metastasis of Non-Small-Cell Lung Cancer through Regulation P53 Signaling. Biochem. Biophys. Res. Commun. 2020, 533, 354–361. [Google Scholar] [CrossRef]
- Niu, C.; Qiu, W.; Li, X.; Li, H.; Zhou, J.; Zhu, H. Transketolase Serves as a Biomarker for Poor Prognosis in Human Lung Adenocarcinoma. J. Cancer 2022, 13, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zeng, X.; Lin, S.; Liang, M.; Huang, H. Identification of Seven-Gene Marker to Predict the Survival of Patients with Lung Adenocarcinoma Using Integrated Multi-Omics Data Analysis. J. Clin. Lab. Anal. 2022, 36, e24190. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Mesaros, C.; Izzo, L.; Affronti, H.; Noji, M.; Schaffer, B.E.; Tsang, T.; Sun, K.; Trefely, S.; Kruijning, S.; et al. Glutamine Deprivation Triggers NAGK-Dependent Hexosamine Salvage. eLife 2021, 10, e62644. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total (n = 1479) | Italian Series (n = 1049) | German Series (n = 430) | p | ||
|---|---|---|---|---|---|---|
| Age at surgery, years, median (range) | 65 (30–90) | 66 (30–85) | 63 (37–88) | <0.001 a | ||
| Age group, years, n (%) | <0.001 b | |||||
| <55 | 227 (15.3) | 139 (13.3) | 88 (20.5) | |||
| 55–64 | 496 (33.5) | 347 (33.1) | 149 (34.7) | |||
| 65–74 | 563 (38.1) | 413 (39.3) | 150 (34.9) | |||
| ≥75 | 193 (13.0) | 150 (14.3) | 43 (10.0) | |||
| Sex, n (%) | 0.004 b | |||||
| Male | 922 (62.4) | 679 (64.7) | 243 (56.5) | |||
| Female | 557 (37.6) | 370 (35.3) | 187 (43.5) | |||
| Smoking habit, n (%) | 0.67 b | |||||
| Never | 225 (15.3) | 160 (15.3) | 65 (15.1) | |||
| Ever | 1190 (80.4) | 826 (78.7) | 364 (84.7) | |||
| Missing | 64 (4.3) | 63 (6.0) | 1 (0.2) | |||
| Pathological stage, n (%) | <0.001 b | |||||
| I | 778 (52.6) | 612 (58.7) | 166 (38.6) | |||
| II | 257 (17.4) | 174 (16.3) | 83 (19.3) | |||
| III | 348 (23.6) | 195 (18.5) | 153 (35.6) | |||
| IV | 82 (5.5) | 54 (5.2) | 28 (6.5) | |||
| Missing | 14 (0.95) | 14 (1.3) | 0 (0) | |||
| Decade of surgery, n (%) | ||||||
| Before 2000 | 221 (14.9) | 221 (21.1) | 0 (0) | 0.82 b | ||
| 2001–2010 | 558 (37.8) | 370 (35.3) | 188 (43.7) | |||
| After 2010 | 699 (47.2) | 458 (43.6) | 241 (56.0) | |||
| Missing | 1 (0.1) | 0 (0) | 1 (0.2) | |||
| Follow-up, months, median (IQR) | 54 (21–60) | 50 (20–60) | 60 (23–60) | 0.011 a | ||
| Survival status at 60 months, n (%) | 0.002 b | |||||
| Alive | 928 (62.7) | 685 (65.3) | 243 (56.5) | |||
| Dead | 551 (37.3) | 364 (34.7) | 187 (43.5) | |||
| Genotyping array, n (%) | <0.001 b | |||||
| Infinium Omni2.5-8 | ||||||
| All tumors | 559 (37.7) | 559 (53.3) | 0 (0) | |||
| Stage I tumors * | 366 (65.5) | 366 (65.5) | 0 (0) | |||
| Axiom PMRA | ||||||
| All tumors | 920 (62.3) | 490 (46.7) | 430 (100) | |||
| Stage I tumors * | 412 (44.8) | 246 (50.2) | 166 (38.6) | |||
| Characteristic | Univariable Analyses a | Multivariable Analyses b | ||
|---|---|---|---|---|
| HR (95% CI) | Cox P | HR (95% CI) | Cox P | |
| Age, years | 1.00 (1.00–1.01) | 0.40 | 1.02 (1.01–1.03) | 2.93 × 10−5 |
| Age group, years | ||||
| <55 | 1.00 | 1.00 | ||
| 55–64 | 0.84 (0.66–1.08) | 0.18 | 1.06 (0.82–1.37) * | 0.62 * |
| 65–74 | 0.91 (0.71–1.16) | 0.45 | 1.33 (1.03–1.71) * | 0.028 * |
| ≥75 | 1.08 (0.80–1.46) | 0.62 | 1.82 (1.31–2.52) * | 2.98 × 10−4 * |
| Sex | ||||
| Male | 1.00 | 1.00 | ||
| Female | 0.61 (0.51–0.74) | 1.88 × 10−7 | 0.66 (0.54–0.81) | 6.53 × 10−5 |
| Smoking habit | ||||
| Never | 1.00 | 1.00 | ||
| Ever | 1.25 (0.98–1.60) | 0.078 | 1.05 (0.81–1.37) | 0.72 |
| Pathological stage | ||||
| I | 1.00 | 1.00 | ||
| II | 2.06 (1.61–2.64) | 9.01 × 10−9 | 2.05 (1.60–2.65) | 2.38 × 10−8 |
| III | 4.04 (3.30–4.94) | <2.00 × 10−16 | 4.14 (3.35–5.12) | <2.00 × 10−16 |
| IV | 5.95 (4.45–7.97) | <2.00 × 10−16 | 5.59 (4.12–7.57) | <2.00 × 10−16 |
| Decade of surgery | ||||
| Before 2000 | 1.00 | 1.00 | ||
| 2001–2010 | 0.77 (0.62–0.95) | 0.017 | 0.68 (0.53–0.86) | 0.0014 |
| After 2010 | 0.45 (0.36–0.57) | 2.5 × 10−11 | 0.48 (0.37–0.63) | 8.62 × 10−8 |
| Country | ||||
| Italy | 0.85 (0.71–1.01) | 0.063 | 0.96 (0.77–1.21) | 0.73 |
| Germany | 1.00 | 1.00 | ||
| Genotyping array | ||||
| Infinium Omni2.5-8 | 0.79 (0.66–0.94) | 0.0075 | 0.84 (0.68–1.04) | 0.11 |
| Axiom PRMA | 1.00 | 1.00 | ||
| Variable | HR (95% CI) | Cox P |
|---|---|---|
| Age | 1.02 (1.01–1.03) | 2.8 × 10−6 |
| Sex | ||
| Male | 1.00 | |
| Female | 0.68 (0.56–0.82) | 8.0 × 10−5 |
| Pathological stage | ||
| I | 1.00 | |
| II | 2.00 (1.56–2.57) | 4.8 × 10−8 |
| III | 4.34 (3.53–5.34) | <2.0 × 10−16 |
| IV | 6.24 (4.62–8.41) | <2.0 × 10−16 |
| Decade of surgery | ||
| Before 2000 | 1.00 | |
| 2001–2010 | 0.70 (0.56–0.88) | 2.4 × 10−3 |
| After 2010 | 0.49 (0.39–0.63) | 2.4 × 10−8 |
| Genomic variant a | ||
| rs13000315 | 2.62 (1.92–3.56) | 9.6 × 10−10 |
| rs151212827 | 2.32 (1.63–3.29) | 2.8 × 10−6 |
| rs190923216 | 2.58 (1.75–3.79) | 1.5 × 10−6 |
| SNP | Chr. | Minor Allele | Major Allele | Regulated Gene | p-Value | Z-Score | FDR |
|---|---|---|---|---|---|---|---|
| rs13000315 | 2 | A | T | CLEC4F | 5.08 × 10−53 | 15.3 | 0 |
| A | T | NAGK | 4.30 × 10−10 | 6.24 | 0 | ||
| A | T | MCEE | 3.74 × 10−6 | 4.63 | 0.0100 | ||
| A | T | CD207 | 1.50 × 10−5 | 4.33 | 0.0387 | ||
| rs71414848 | 2 | C | T | CLEC4F | 3.16 × 10−53 | 15.4 | 0 |
| C | T | NAGK | 3.74 × 10−10 | 6.26 | 0 | ||
| C | T | MCEE | 4.81 × 10−6 | 4.57 | 0.0130 | ||
| C | T | CD207 | 1.88 × 10−5 | 4.28 | 0.0470 | ||
| rs74464684 | 3 | T | C | NT5DC2 | 6.54 × 10−49 | 14.7 | 0 |
| T | C | TKT | 8.97 × 10−16 | −8.04 | 0 | ||
| T | C | UQCC5 | 4.80 × 10−7 | −5.03 | 0.00147 | ||
| rs76553845 | 3 | G | A | NT5DC2 | 4.40 × 10−45 | 14.1 | 0 |
| G | A | TKT | 2.18 × 10−12 | −7.02 | 0 | ||
| G | A | UQCC5 | 4.12 × 10−7 | −5.06 | 0.00122 | ||
| rs151212827 | 3 | A | G | NT5DC2 | 1.57 × 10−47 | 14.5 | 0 |
| A | G | TKT | 1.08 × 10−11 | −6.80 | 0 | ||
| A | G | UQCC5 | 2.76 × 10−8 | −5.56 | 0.000121 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minnai, F.; Noci, S.; Esposito, M.; Schneider, M.A.; Kobinger, S.; Eichhorn, M.; Winter, H.; Hoffmann, H.; Kriegsmann, M.; Incarbone, M.A.; et al. Germline Polymorphisms Associated with Overall Survival in Lung Adenocarcinoma: Genome-Wide Analysis. Cancers 2024, 16, 3264. https://doi.org/10.3390/cancers16193264
Minnai F, Noci S, Esposito M, Schneider MA, Kobinger S, Eichhorn M, Winter H, Hoffmann H, Kriegsmann M, Incarbone MA, et al. Germline Polymorphisms Associated with Overall Survival in Lung Adenocarcinoma: Genome-Wide Analysis. Cancers. 2024; 16(19):3264. https://doi.org/10.3390/cancers16193264
Chicago/Turabian StyleMinnai, Francesca, Sara Noci, Martina Esposito, Marc A. Schneider, Sonja Kobinger, Martin Eichhorn, Hauke Winter, Hans Hoffmann, Mark Kriegsmann, Matteo A. Incarbone, and et al. 2024. "Germline Polymorphisms Associated with Overall Survival in Lung Adenocarcinoma: Genome-Wide Analysis" Cancers 16, no. 19: 3264. https://doi.org/10.3390/cancers16193264
APA StyleMinnai, F., Noci, S., Esposito, M., Schneider, M. A., Kobinger, S., Eichhorn, M., Winter, H., Hoffmann, H., Kriegsmann, M., Incarbone, M. A., Mattioni, G., Tosi, D., Muley, T., Dragani, T. A., & Colombo, F. (2024). Germline Polymorphisms Associated with Overall Survival in Lung Adenocarcinoma: Genome-Wide Analysis. Cancers, 16(19), 3264. https://doi.org/10.3390/cancers16193264