
Generation of Hydrogen Peroxide in Cancer Cells: Advancing Therapeutic Approaches for Cancer Treatment

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
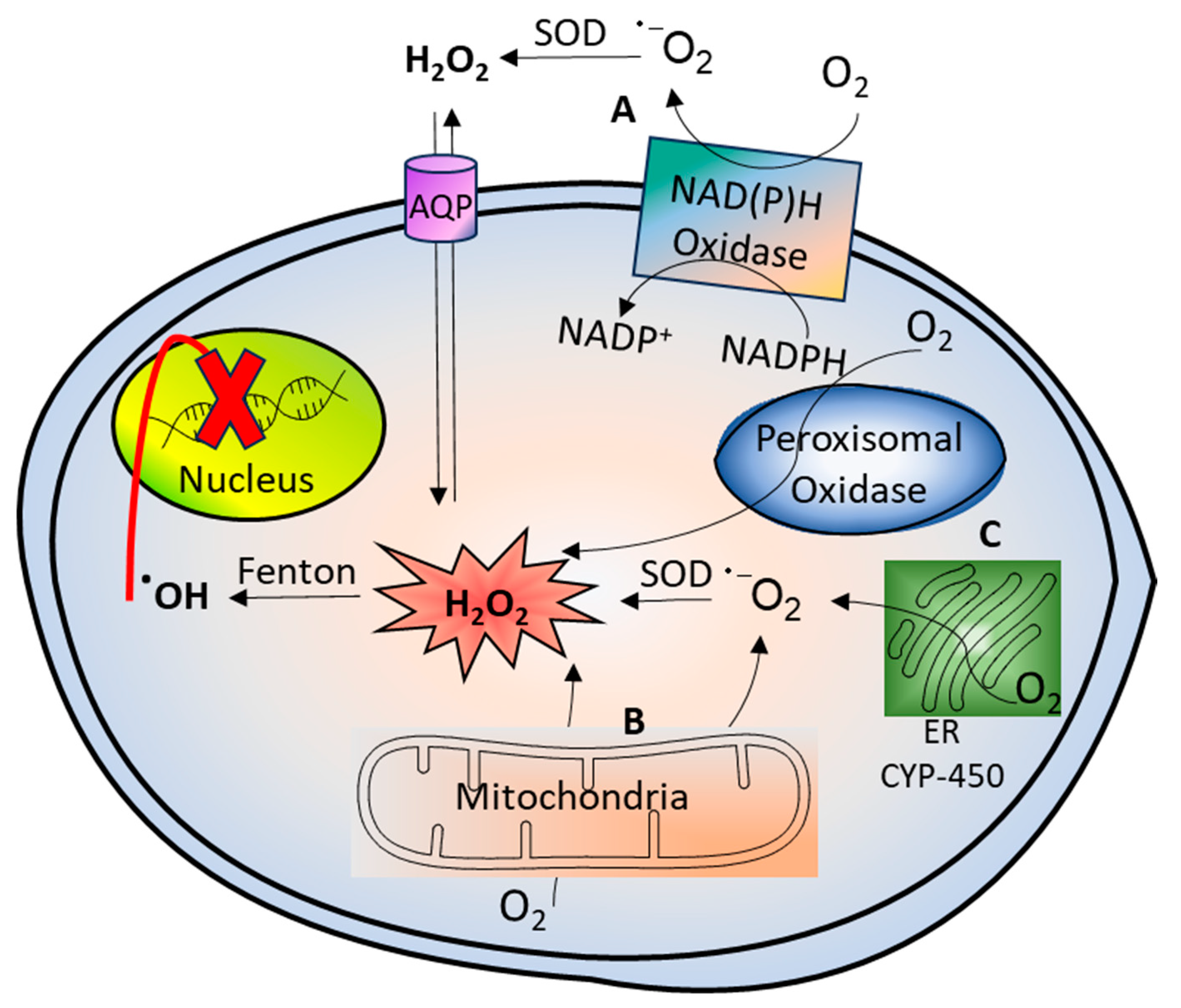
2. Hydrogen Peroxide Generation and Its Reactivity with Biomolecules
3. Concentration-Dependent Selection between Apoptosis and Necrosis Induced by H2O2
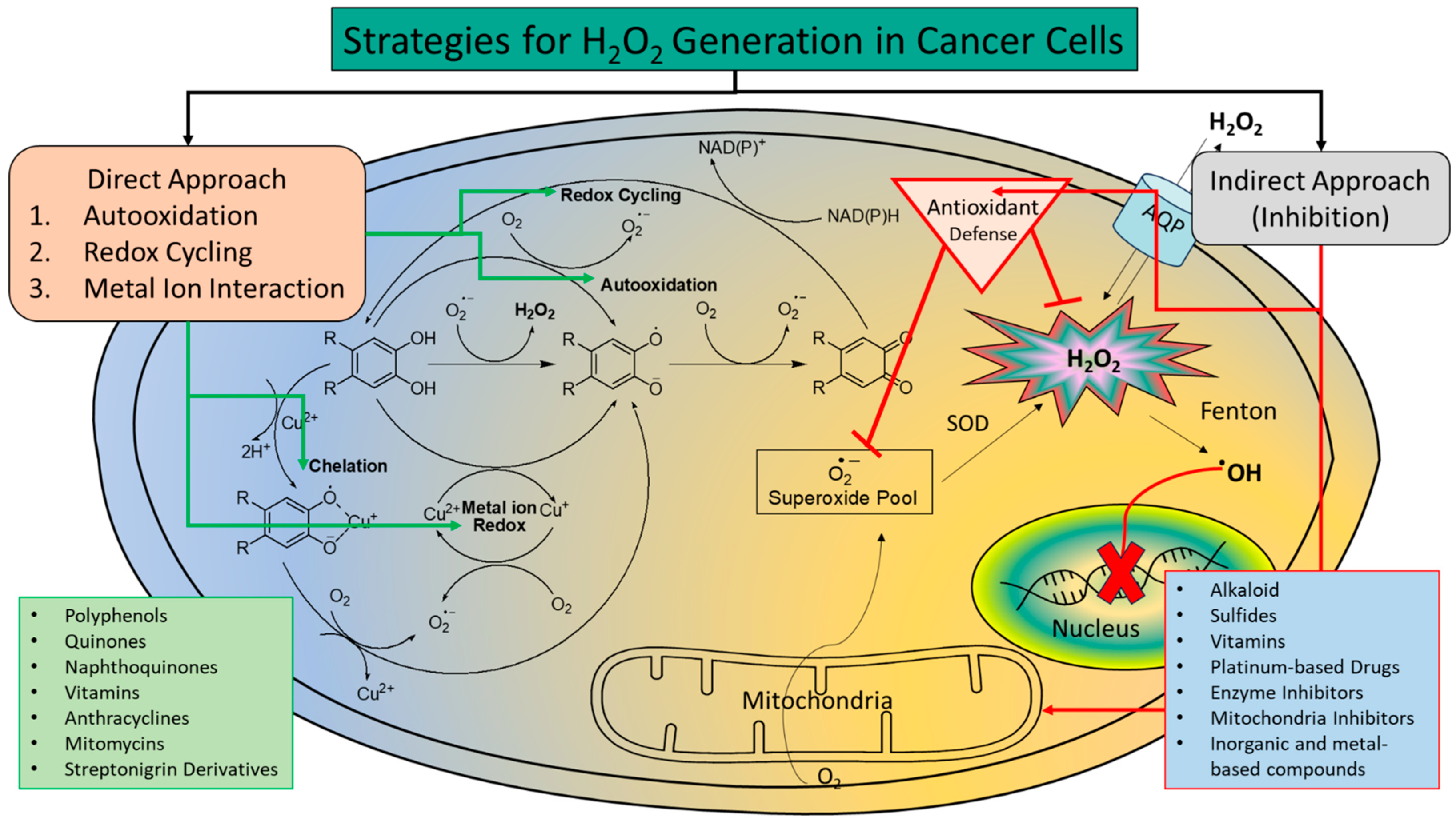
4. Increasing H2O2 Level as an Anticancer Therapy
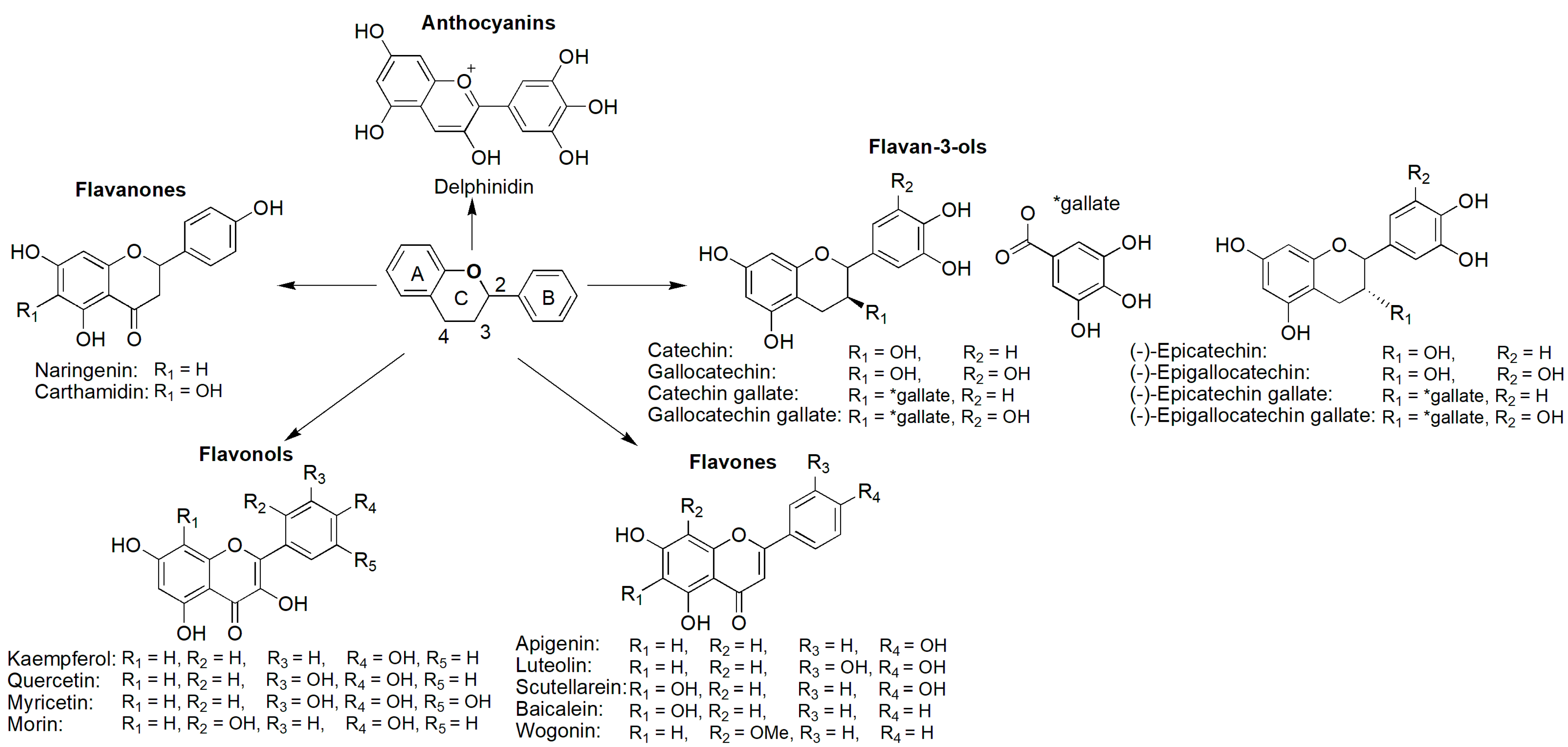
4.1. Phenol and Polyphenol Analogues
4.1.1. Flavonoids
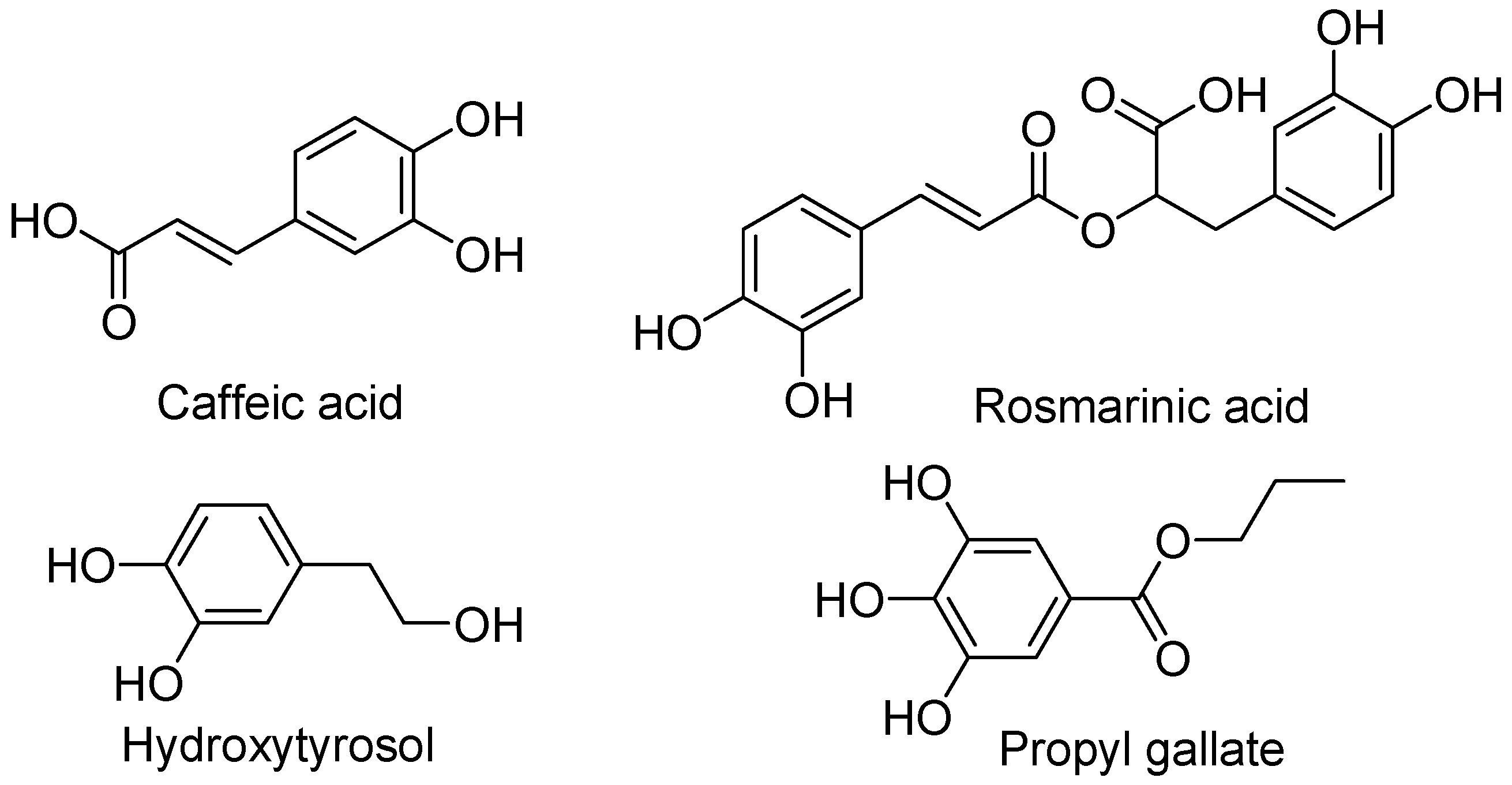
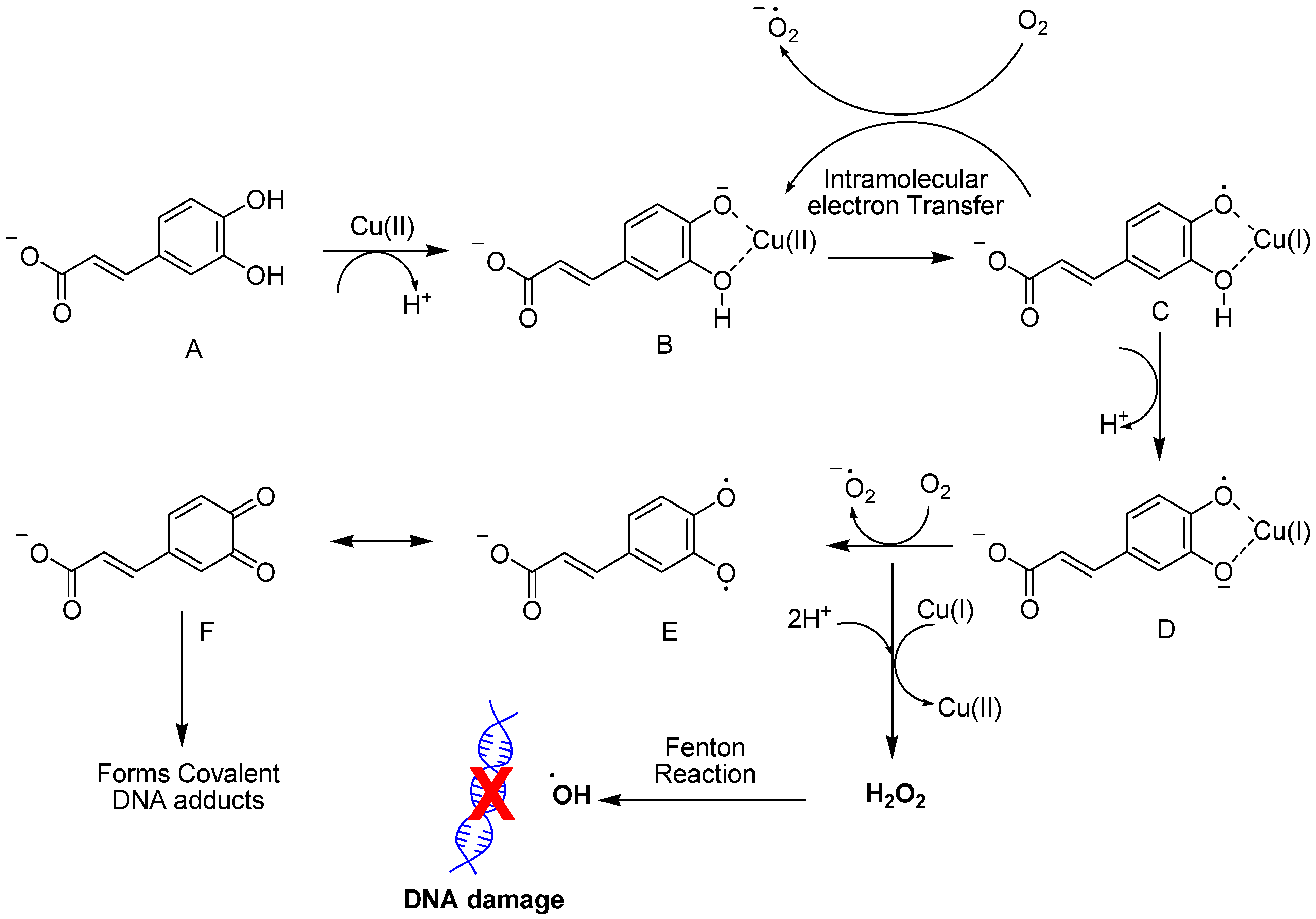
4.1.2. Hydroxycinnamic Acid
4.1.3. Hydroxytyrosol
4.1.4. Propyl Gallate
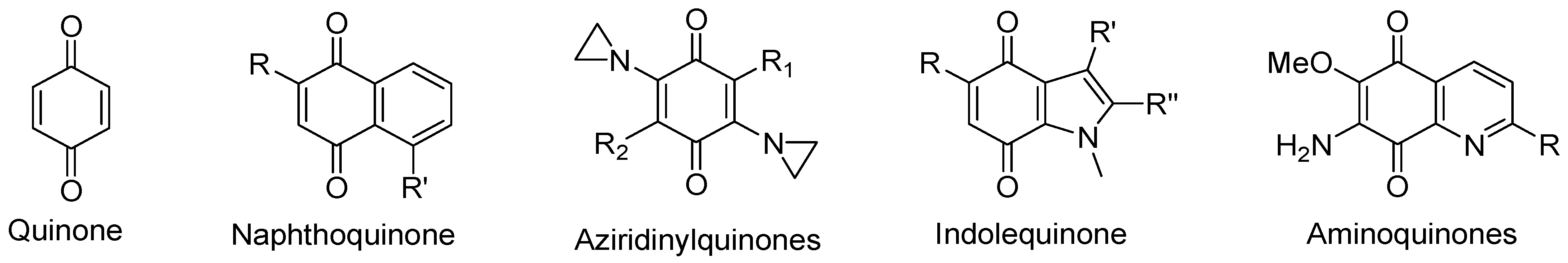
4.2. Compounds with Quinone Moieties
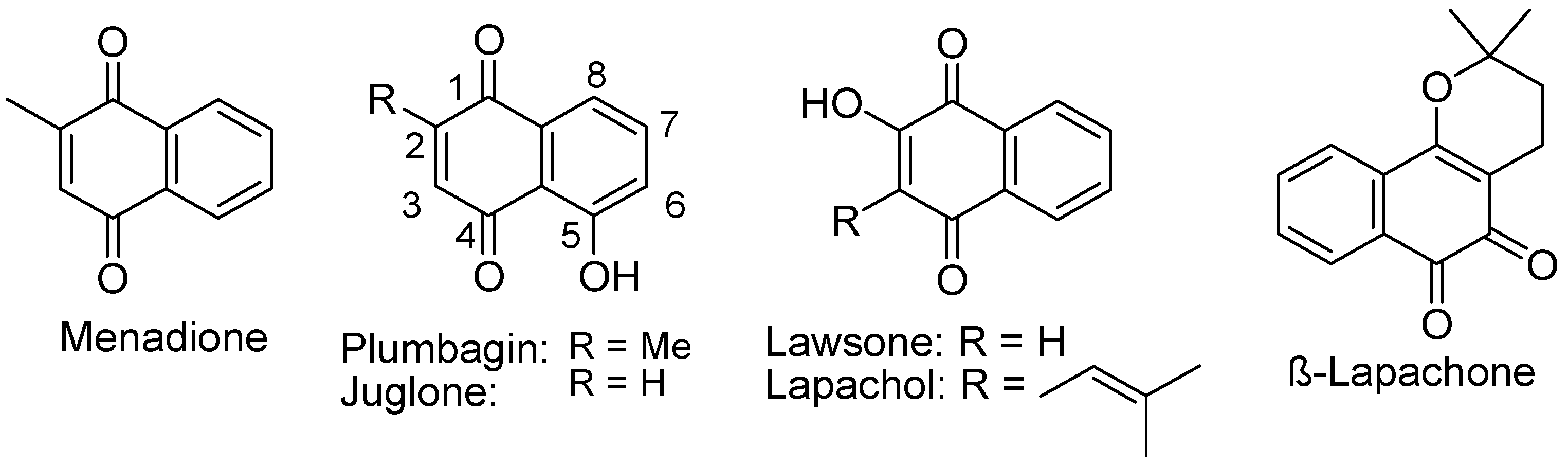
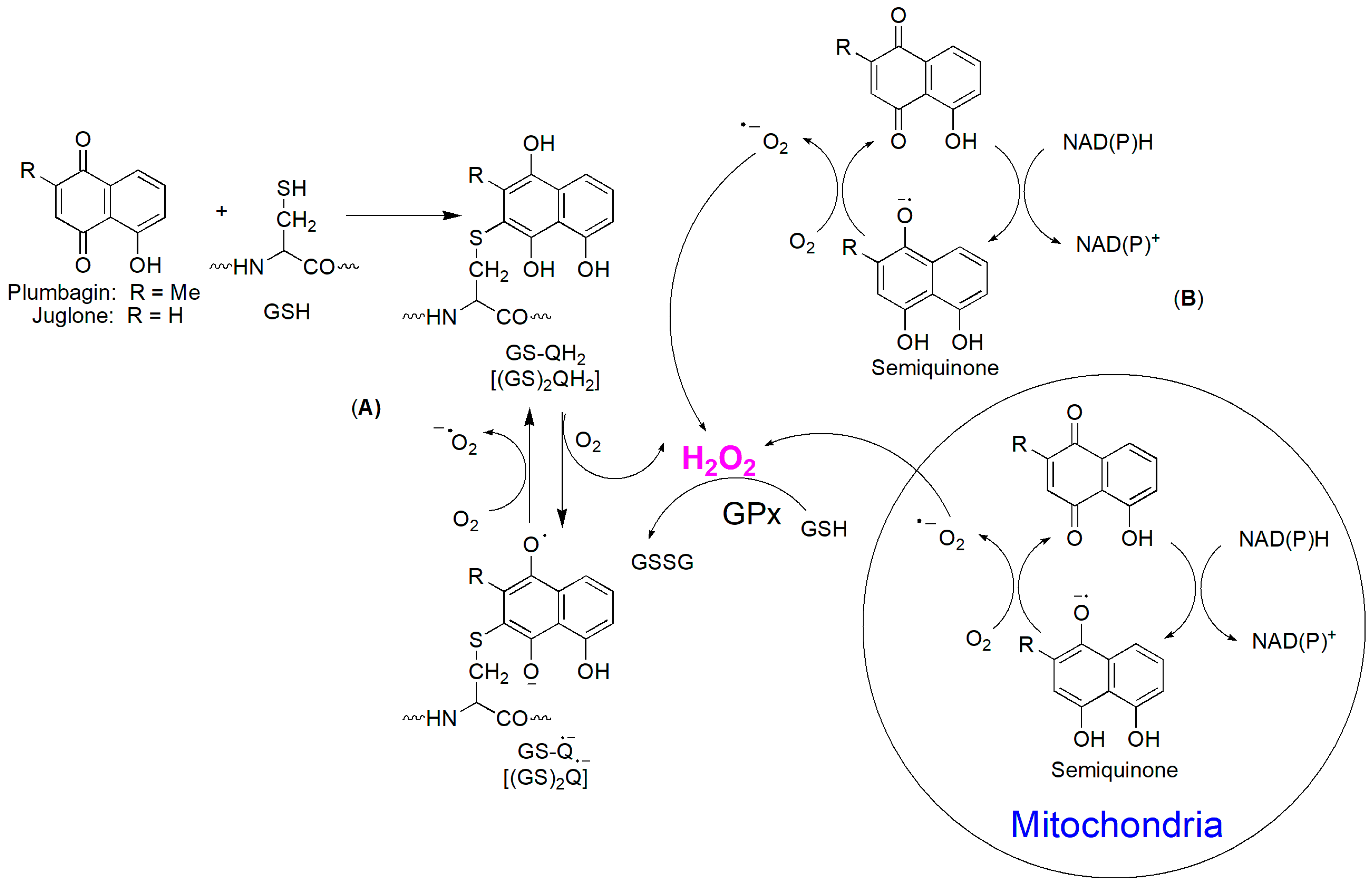
4.2.1. Naphthoquinones
4.2.2. Hydroxyl Naphthoquinone
4.2.3. 1,2-Naphthoquinone
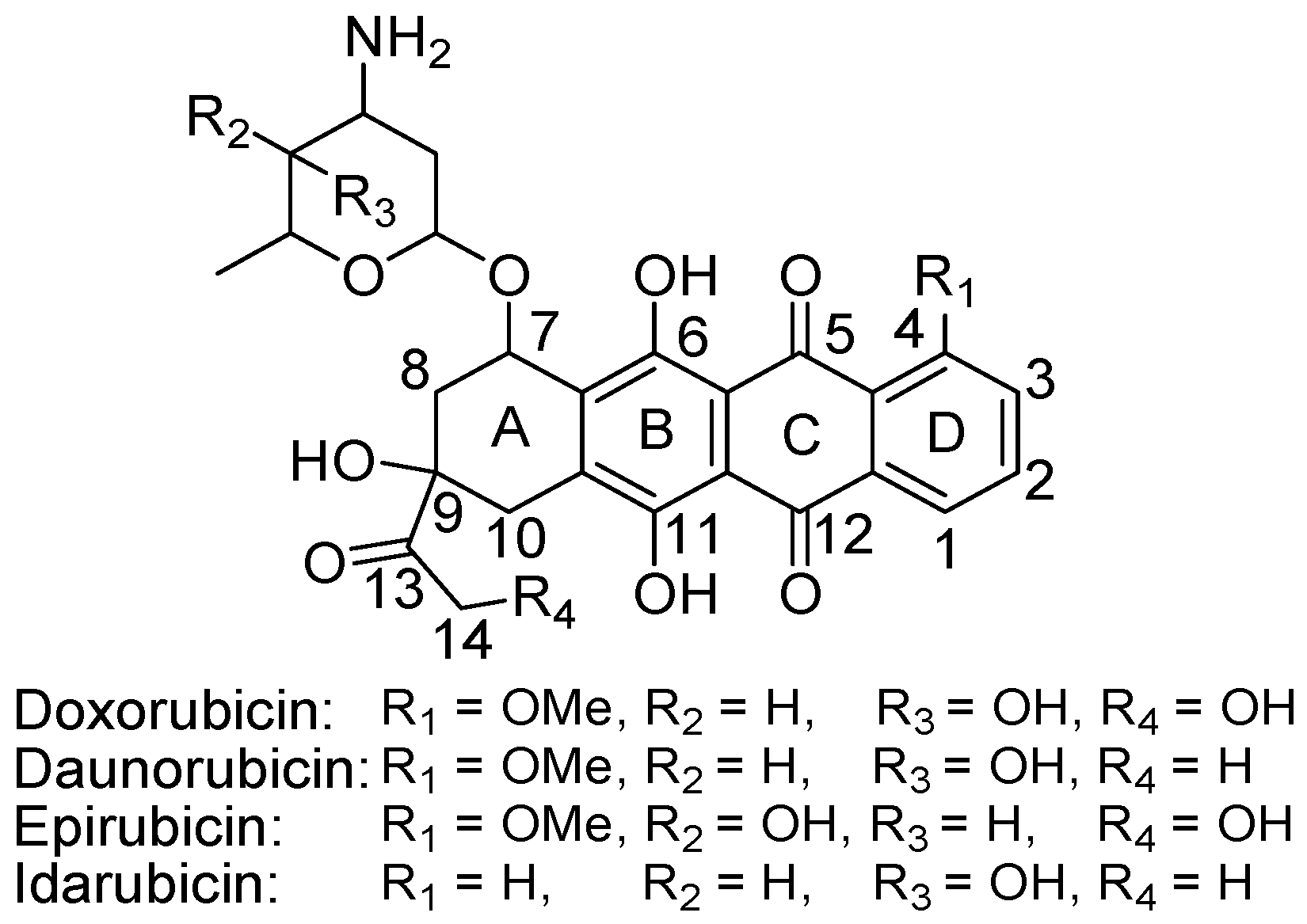
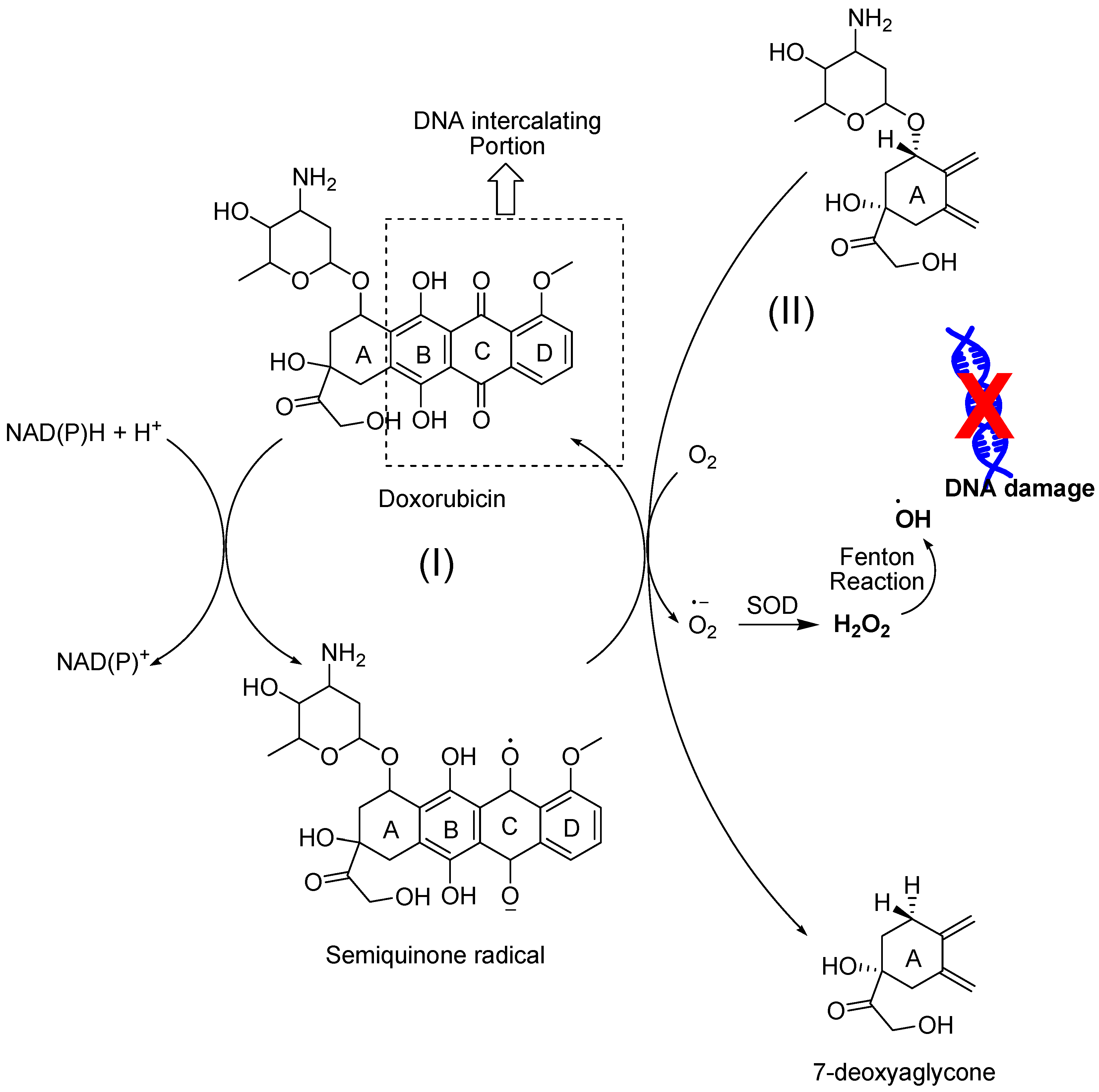
4.2.4. Anthracyclines
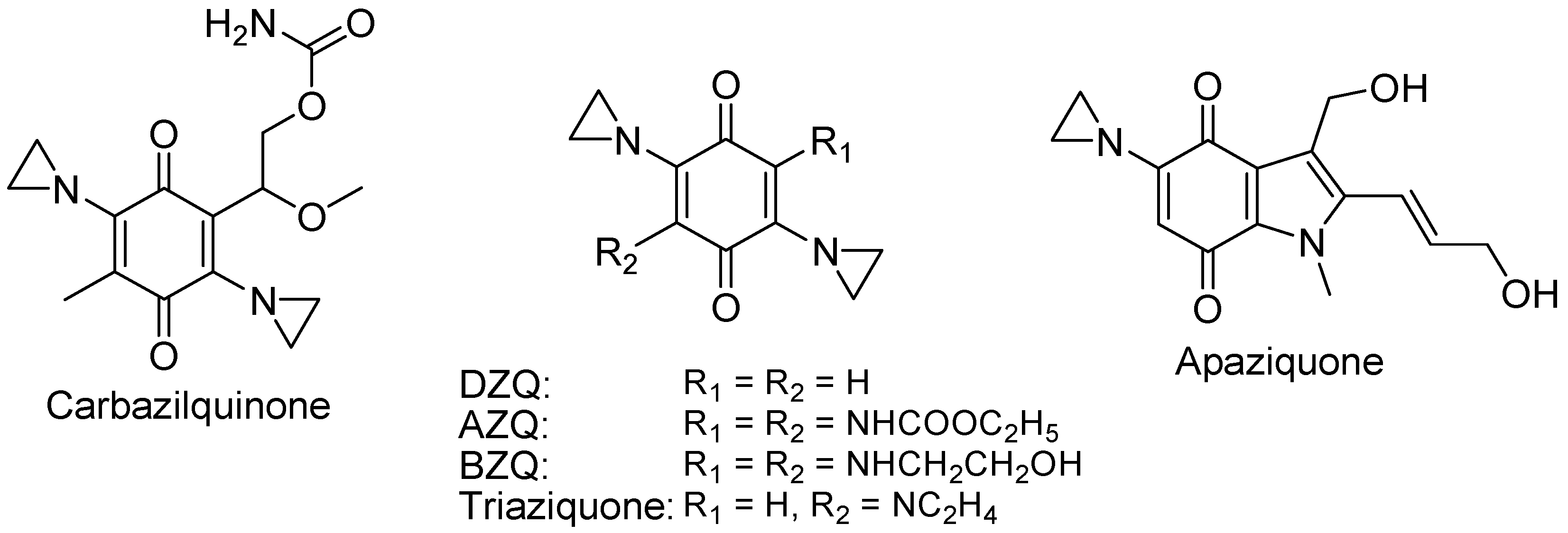
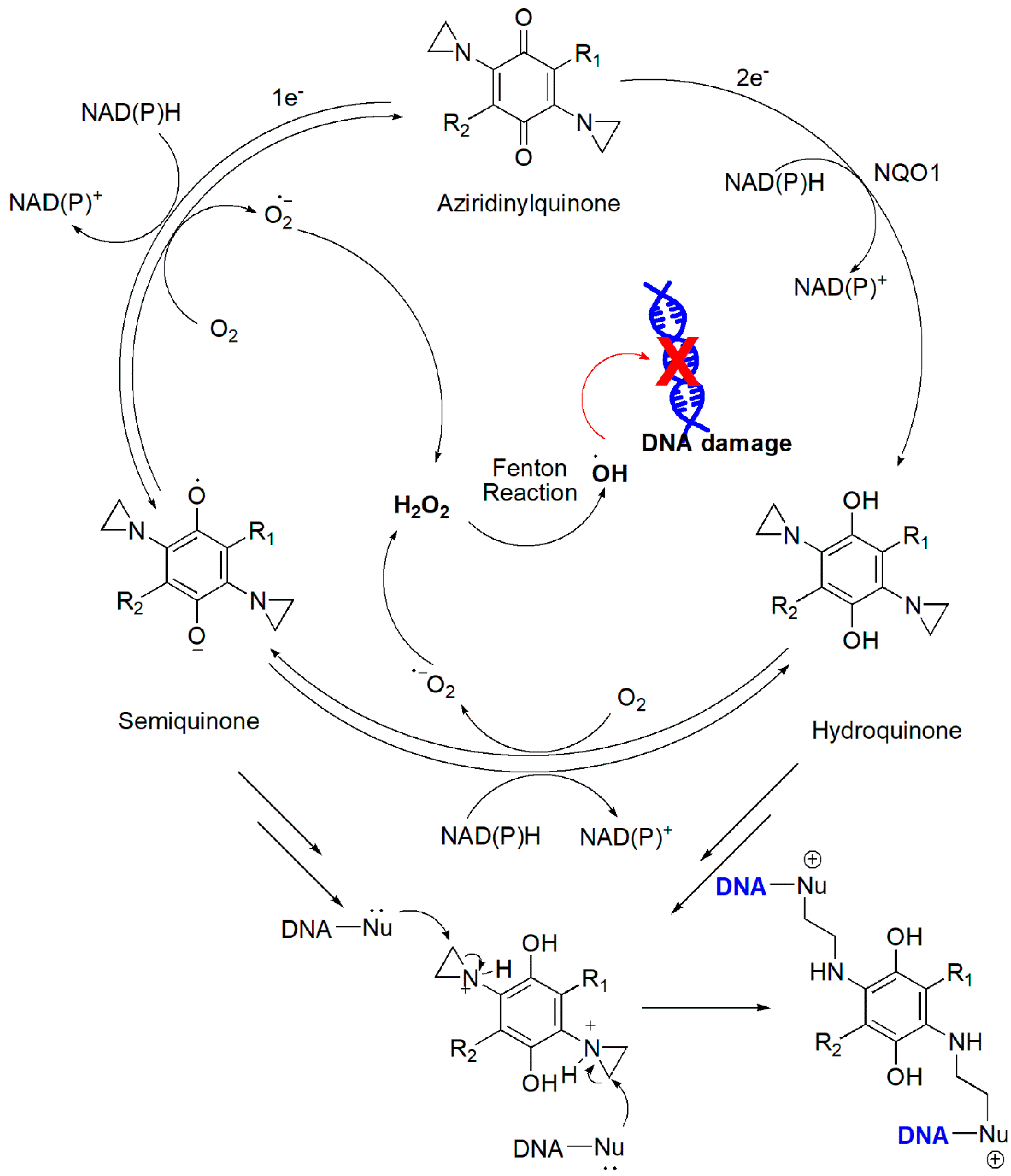
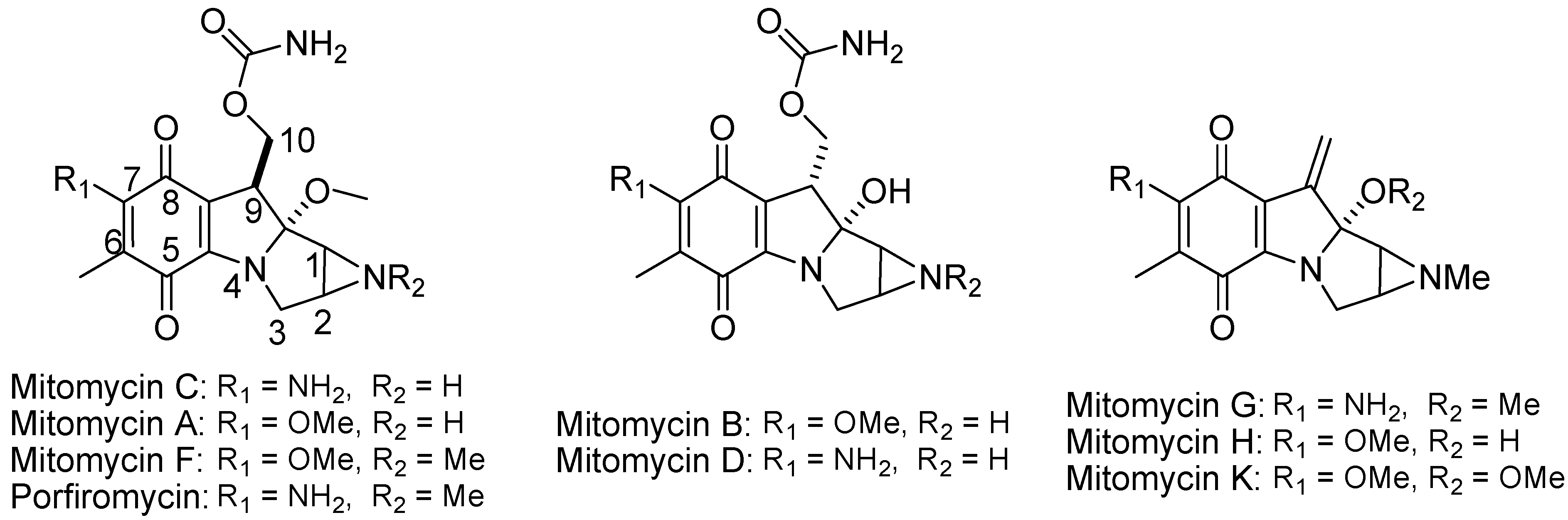
4.2.5. Aziridinylquinones
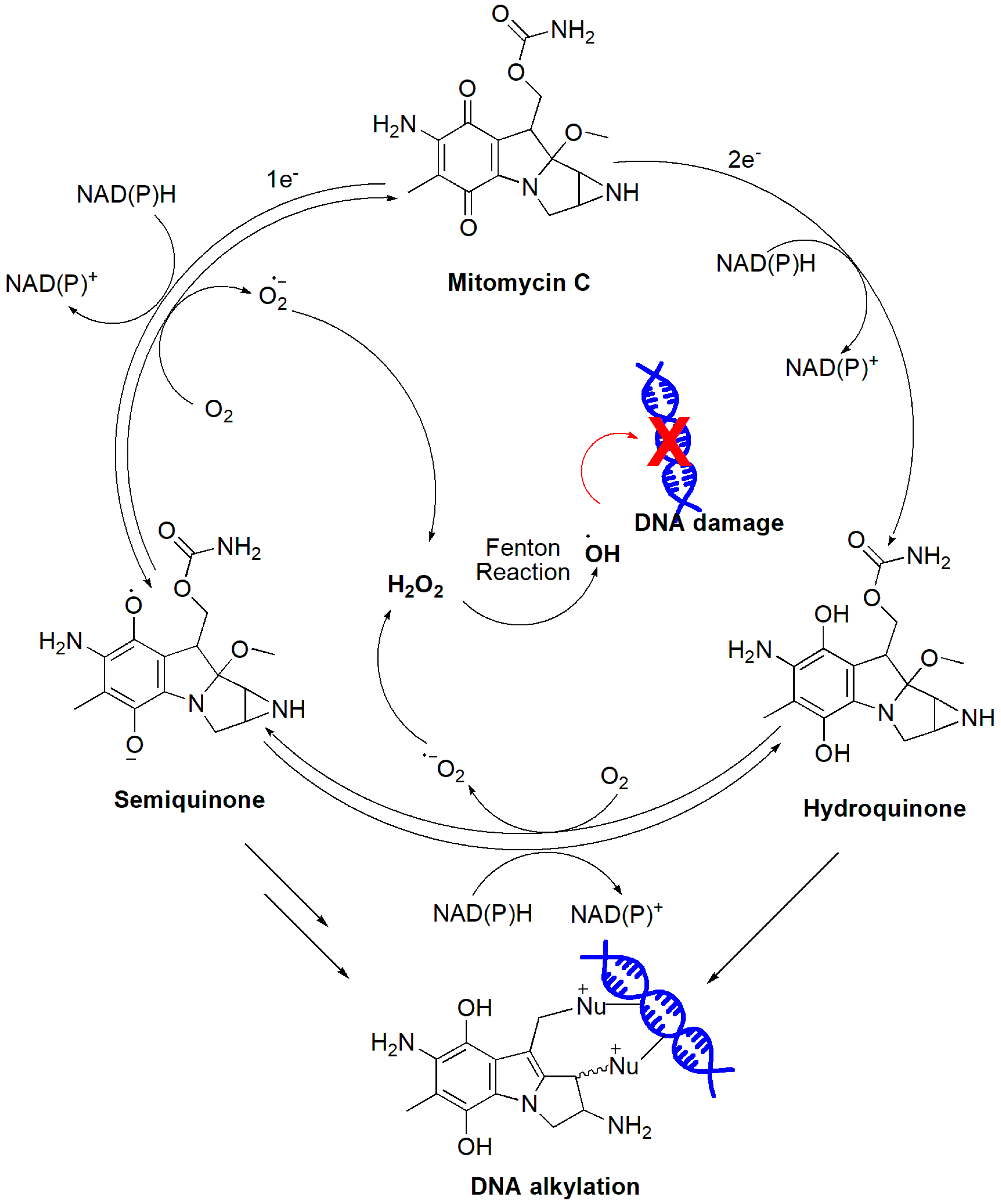
4.2.6. Indolequinones
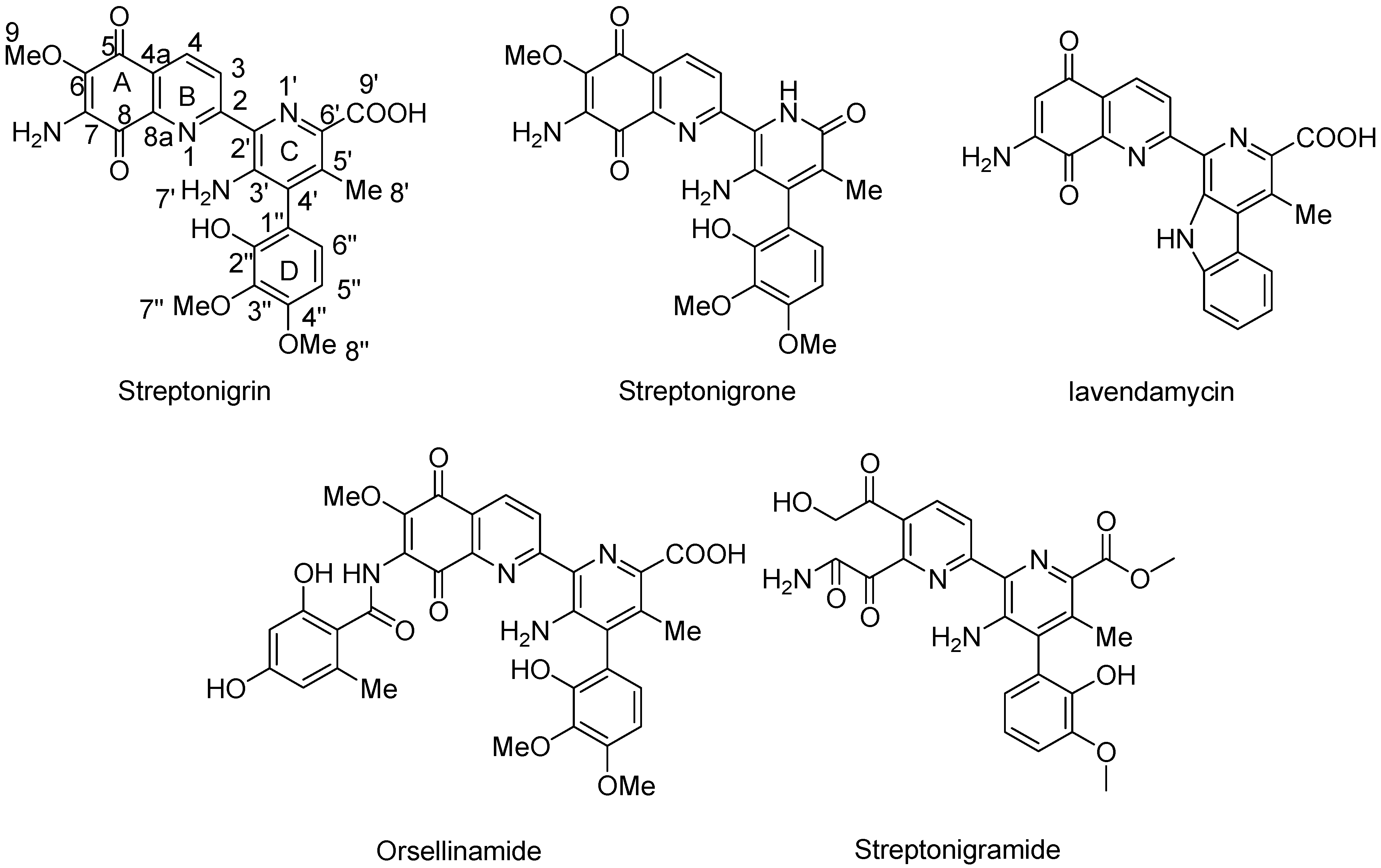
4.2.7. Aminoquinones
4.3. Vitamin C
4.4. Metal, Metal Oxides, and Metal Peroxides
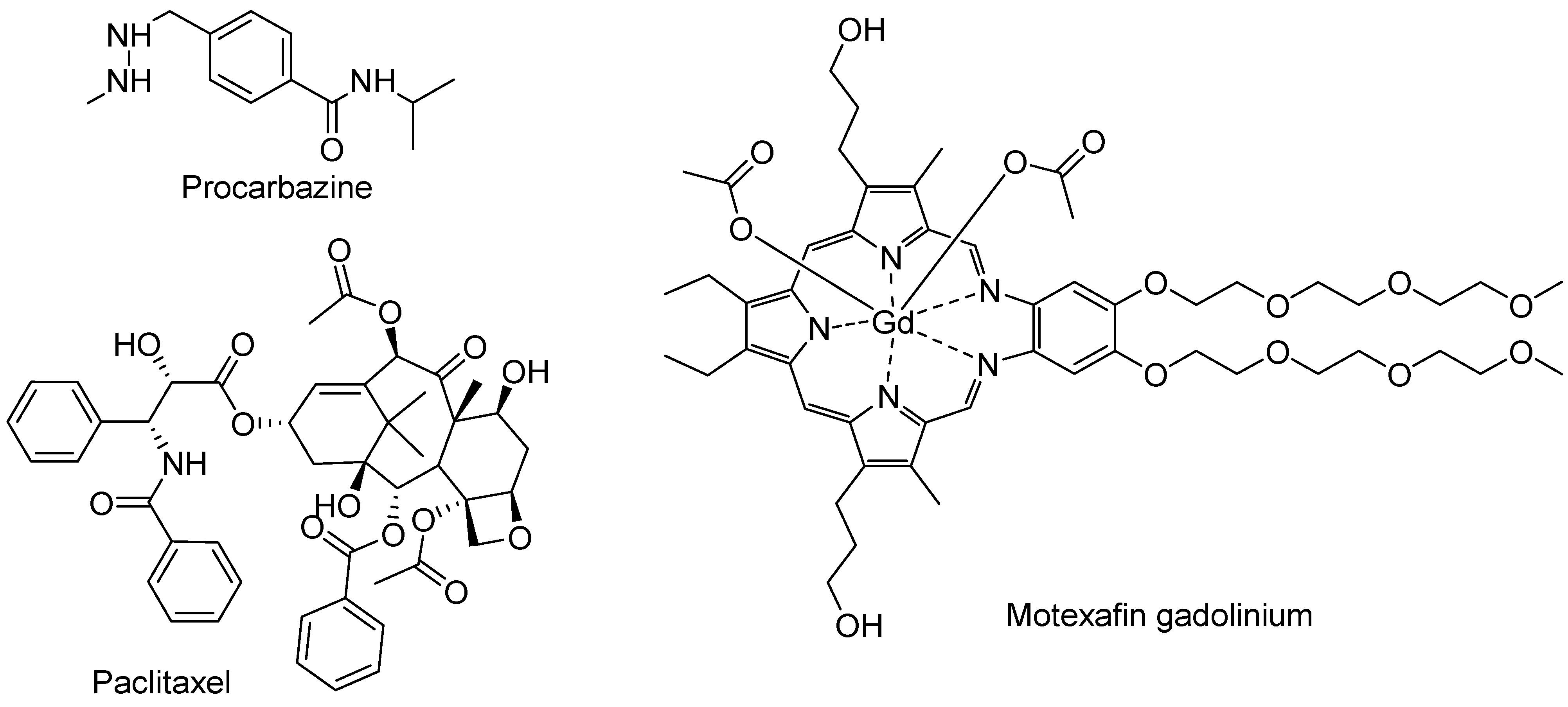
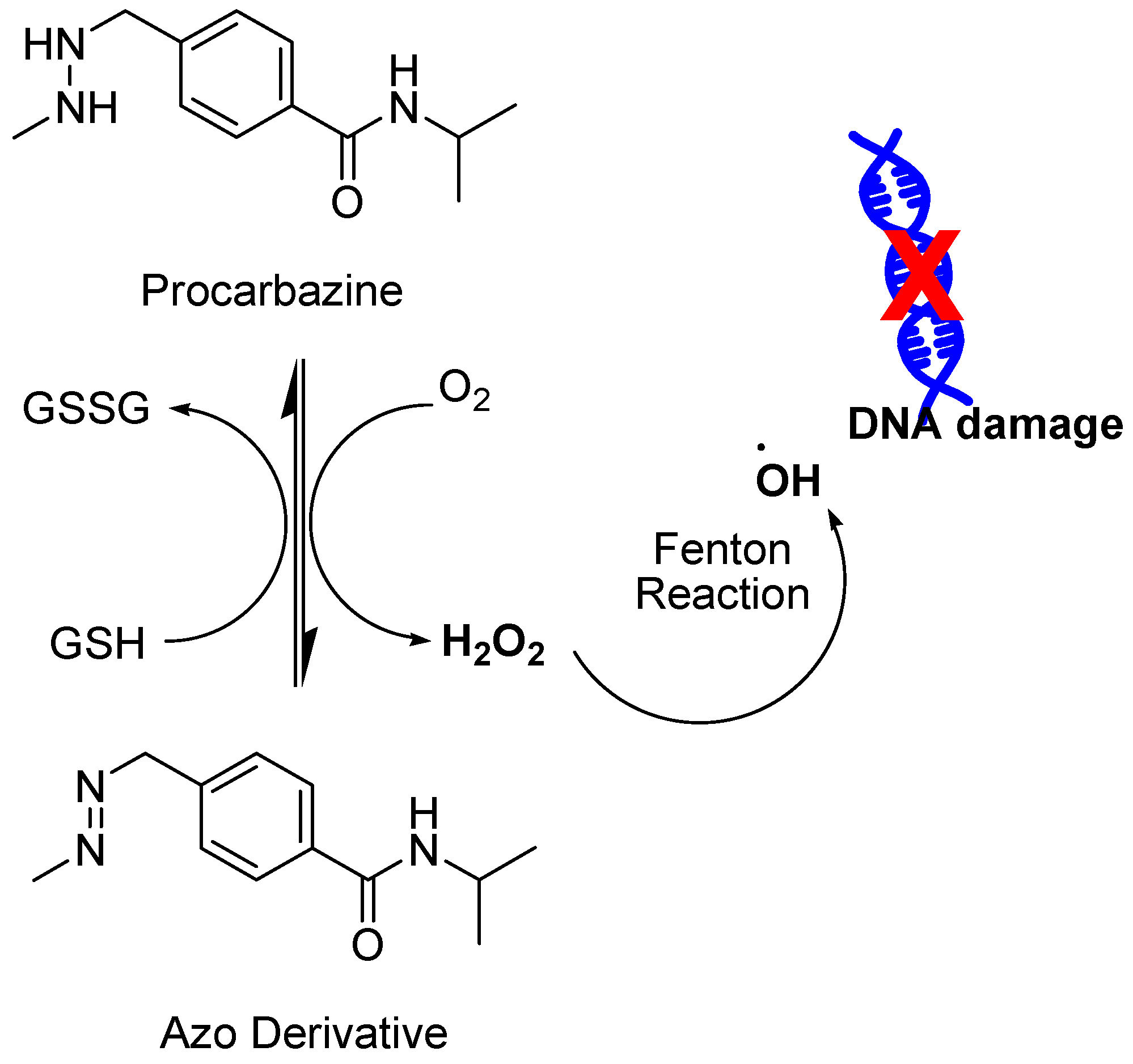
4.5. FDA-Approved Drugs
4.6. H2O2 Concentrations Induced by Representative Compounds
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Hileman, E.O.; Liu, J.; Albitar, M.; Keating, M.J.; Huang, P. Intrinsic oxidative stress in cancer cells: A biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004, 53, 209–219. [Google Scholar] [CrossRef]
- Chu, Z.Y.J.; Zheng, W.; Sun, J.; Wang, W.; Qian, H. Recent Advances on Modulation of H2O2 in Tumor Microenvironment for Enhanced Cancer Therapeutic Efficacy. Coord. Chem. Rev. 2023, 481, 215049. [Google Scholar] [CrossRef]
- Dharmaraja, A.T. Role of Reactive Oxygen Species (ROS) in Therapeutics and Drug Resistance in Cancer and Bacteria. J. Med. Chem. 2017, 60, 3221–3240. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Lopez-Lazaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies. Stresses 2022, 2, 256–274. [Google Scholar] [CrossRef]
- Nicco, C.; Laurent, A.; Chereau, C.; Weill, B.; Batteux, F. Differential modulation of normal and tumor cell proliferation by reactive oxygen species. Biomed. Pharmacother. 2005, 59, 169–174. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef]
- Chidawanyika, T.; Supattapone, S. Hydrogen Peroxide-induced Cell Death in Mammalian Cells. J. Cell. Signal 2021, 2, 206–211. [Google Scholar]
- Troyano, A.; Sancho, P.; Fernandez, C.; de Blas, E.; Bernardi, P.; Aller, P. The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell Death Differ. 2003, 10, 889–898. [Google Scholar] [CrossRef]
- Hampton, M.B.; Orrenius, S. Dual regulation of caspase activity by hydrogen peroxide: Implications for apoptosis. FEBS Lett. 1997, 414, 552–556. [Google Scholar] [CrossRef]
- Gardner, A.M.; Xu, F.H.; Fady, C.; Jacoby, F.J.; Duffey, D.C.; Tu, Y.; Lichtenstein, A. Apoptotic vs. nonapoptotic cytotoxicity induced by hydrogen peroxide. Free Radic. Biol. Med. 1997, 22, 73–83. [Google Scholar] [CrossRef]
- Nosseri, C.; Coppola, S.; Ghibelli, L. Possible involvement of poly(ADP-ribosyl) polymerase in triggering stress-induced apoptosis. Exp. Cell Res. 1994, 212, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Shacter, E. Oxidative stress inhibits apoptosis in human lymphoma cells. J. Biol. Chem. 1999, 274, 19792–19798. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H. Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1-phase arrest. Oncol. Rep. 2018, 40, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ. Res. 2007, 100, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Mehmet, H. Caspases find a new place to hide. Nature 2000, 403, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Wurstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell Res. 2012, 318, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Chen, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Lennon, S.V.; Martin, S.J.; Cotter, T.G. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif. 1991, 24, 203–214. [Google Scholar] [CrossRef]
- Kong, Q.; Lillehei, K.O. Antioxidant inhibitors for cancer therapy. Med. Hypotheses 1998, 51, 405–409. [Google Scholar] [CrossRef]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta 2007, 1776, 86–107. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.J.; Dong, C.; Shi, S. Glucose Oxidase-Related Cancer Therapies. Adv.Ther. 2020, 3, 2000110. [Google Scholar] [CrossRef]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef]
- Foglietta, F.; Serpe, L.; Canaparo, R. ROS-generating nanoplatforms as selective and tunable therapeutic weapons against cancer. Discov. Nano 2023, 18, 151. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Tian, X.; Ai, H.W. Molecular Tools to Generate Reactive Oxygen Species in Biological Systems. Bioconjug. Chem. 2019, 30, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Peng, L.; Zhou, L.; Huang, Z.; Zhou, C.; Huang, C. Oxidative Stress in Cancer Immunotherapy: Molecular Mechanisms and Potential Applications. Antioxidants 2022, 11, 853. [Google Scholar] [CrossRef]
- Giansanti, M.; Karimi, T.; Faraoni, I.; Graziani, G. High-Dose Vitamin C: Preclinical Evidence for Tailoring Treatment in Cancer Patients. Cancers 2021, 13, 1428. [Google Scholar] [CrossRef]
- Akagawa, M.; Shigemitsu, T.; Suyama, K. Production of hydrogen peroxide by polyphenols and polyphenol-rich beverages under quasi-physiological conditions. Biosci. Biotechnol. Biochem. 2003, 67, 2632–2640. [Google Scholar] [CrossRef]
- Mochizuki, M.; Yamazaki, S.; Kano, K.; Ikeda, T. Kinetic analysis and mechanistic aspects of autoxidation of catechins. Biochim. Biophys. Acta 2002, 1569, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shi, J.; Nice, E.C.; Huang, C.; Shi, Z. The Multifaceted Role of Flavonoids in Cancer Therapy: Leveraging Autophagy with a Double-Edged Sword. Antioxidants 2021, 10, 1138. [Google Scholar] [CrossRef]
- Oikawa, S.; Furukawaa, A.; Asada, H.; Hirakawa, K.; Kawanishi, S. Catechins induce oxidative damage to cellular and isolated DNA through the generation of reactive oxygen species. Free Radic. Res. 2003, 37, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Oikawa, S.; Murata, M.; Hiraku, Y.; Kawanishi, S. (-)-Epigallocatechin gallate causes oxidative damage to isolated and cellular DNA. Biochem. Pharmacol. 2003, 66, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Hasumi, K.; Woo, J.T.; Nagai, K.; Wachi, M. Generation of hydrogen peroxide primarily contributes to the induction of Fe(II)-dependent apoptosis in Jurkat cells by (-)-epigallocatechin gallate. Carcinogenesis 2004, 25, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Arakawa, H.; Maeda, M.; Satoh, K.; Kadofuku, T.; Fukuchi, K.; Gomi, K. Production of hydrogen peroxide and methionine sulfoxide by epigallocatechin gallate and antioxidants. Anticancer Res. 2001, 21, 2633–2641. [Google Scholar] [PubMed]
- Yen, G.C.; Duh, P.D.; Tsai, H.L.; Huang, S.L. Pro-oxidative properties of flavonoids in human lymphocytes. Biosci. Biotechnol. Biochem. 2003, 67, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Canada, A.T.; Giannella, E.; Nguyen, T.D.; Mason, R.P. The production of reactive oxygen species by dietary flavonols. Free Radic. Biol. Med. 1990, 9, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.H.; Tomita, I.; Watanabe, T.; Hirayama, T.; Fukui, S. Active oxygens generation by flavonoids. Biol. Pharm. Bull. 1998, 21, 93–96. [Google Scholar] [CrossRef]
- Baumann, S.; Fas, S.C.; Giaisi, M.; Muller, W.W.; Merling, A.; Gulow, K.; Edler, L.; Krammer, P.H.; Li-Weber, M. Wogonin preferentially kills malignant lymphocytes and suppresses T-cell tumor growth by inducing PLCgamma1- and Ca2+-dependent apoptosis. Blood 2008, 111, 2354–2363. [Google Scholar] [CrossRef]
- Wei, L.; Lu, N.; Dai, Q.; Rong, J.; Chen, Y.; Li, Z.; You, Q.; Guo, Q. Different apoptotic effects of wogonin via induction of H2O2 generation and Ca2+ overload in malignant hepatoma and normal hepatic cells. J. Cell. Biochem. 2010, 111, 1629–1641. [Google Scholar] [CrossRef]
- Tsai, C.F.; Yeh, W.L.; Huang, S.M.; Tan, T.W.; Lu, D.Y. Wogonin induces reactive oxygen species production and cell apoptosis in human glioma cancer cells. Int. J. Mol. Sci. 2012, 13, 9877–9892. [Google Scholar] [CrossRef] [PubMed]
- Dzah, C.S.Z.H.; Gobe, V.; Asante-Donyinah, D.A.; Duan, Y. Anti- and pro-oxidant properties of polyphenols and their role in modulating glutathione synthesis, activity and cellular redox potential: Potential synergies for disease management. Adv. Red. Res. 2024, 11, 100099. [Google Scholar] [CrossRef]
- Braicu, C.; Ladomery, M.R.; Chedea, V.S.; Irimie, A.; Berindan-Neagoe, I. The relationship between the structure and biological actions of green tea catechins. Food Chem. 2013, 141, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, K.; Liu, Z.; Huang, J. Prooxidant Effects of Epigallocatechin-3-Gallate in Health Benefits and Potential Adverse Effect. Oxid. Med. Cell. Longev. 2020, 2020, 9723686. [Google Scholar] [CrossRef] [PubMed]
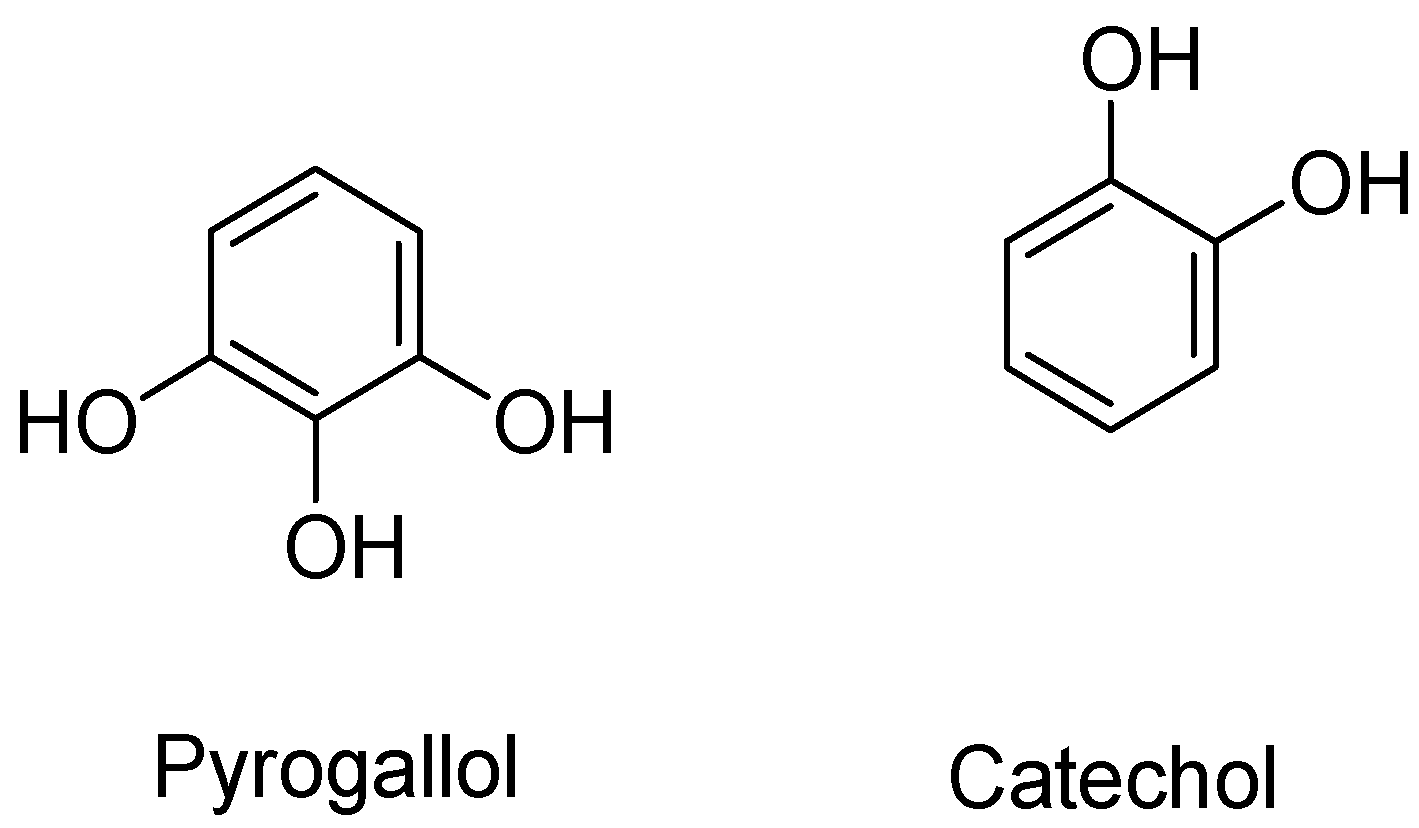
- Saeki, K.; Hayakawa, S.; Isemura, M.; Miyase, T. Importance of a pyrogallol-type structure in catechin compounds for apoptosis-inducing activity. Phytochemistry 2000, 53, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Lu, H.; Meng, X.; Ryu, J.H.; Hara, Y.; Yang, C.S. Stability, cellular uptake, biotransformation, and efflux of tea polyphenol (-)-epigallocatechin-3-gallate in HT-29 human colon adenocarcinoma cells. Cancer Res. 2002, 62, 7241–7246. [Google Scholar] [PubMed]
- Park, W.H.; Han, Y.W.; Kim, S.H.; Kim, S.Z. A superoxide anion generator, pyrogallol induces apoptosis in As4.1 cells through the depletion of intracellular GSH content. Mutat. Res. 2007, 619, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Pyrogallol as a glutathione depletor induces apoptosis in HeLa cells. Int. J. Mol. Med. 2008, 21, 721–730. [Google Scholar]
- Liu, T.P.P.; Shi, H.; Feng, J.; Zhang, X. Assembled polyphenol-based systems as advanced therapeutics. J. Poly. Sci. 2023, 62, 297–323. [Google Scholar] [CrossRef]
- Awad, H.M.; Boersma, M.G.; Boeren, S.; van Bladeren, P.J.; Vervoort, J.; Rietjens, I.M. The regioselectivity of glutathione adduct formation with flavonoid quinone/quinone methides is pH-dependent. Chem. Res. Toxicol. 2002, 15, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Galati, G.; Sabzevari, O.; Wilson, J.X.; O′Brien, P.J. Prooxidant activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicology 2002, 177, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.; Galati, G.; O′Brien, P.J. Oxygen activation during peroxidase catalysed metabolism of flavones or flavanones. Chem. Biol. Interact. 1999, 122, 15–25. [Google Scholar] [CrossRef]
- Hider, R.C.; Liu, Z.D.; Khodr, H.H. Metal chelation of polyphenols. Methods Enzymol. 2001, 335, 190–203. [Google Scholar] [PubMed]
- Pan, Y.Q.R.; Hou, M.; Xue, J.; Zhou, M.; Xu, L.; Zhang, Y. The interactions of polyphenols with Fe and their application in Fenton/Fenton-like reactions. Sep. Purif. Technol. 2022, 300, 121831. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Spencer, J.P.; Rossi, R.; Aeschbach, R.; Khan, A.; Mahmood, N.; Munoz, A.; Murcia, A.; Butler, J.; Halliwell, B. An evaluation of the antioxidant and antiviral action of extracts of rosemary and Provencal herbs. Food Chem. Toxicol. 1996, 34, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Al-Sereiti, M.R.; Abu-Amer, K.M.; Sen, P. Pharmacology of rosemary (Rosmarinus officinalis Linn.) and its therapeutic potentials. Indian J. Exp. Biol. 1999, 37, 124–130. [Google Scholar]
- Zheng, L.F.; Dai, F.; Zhou, B.; Yang, L.; Liu, Z.L. Prooxidant activity of hydroxycinnamic acids on DNA damage in the presence of Cu(II) ions: Mechanism and structure-activity relationship. Food Chem. Toxicol. 2008, 46, 149–156. [Google Scholar] [CrossRef]
- Damasceno, S.S.; Dantas, B.B.; Ribeiro-Filho, J.; Antonio, M.A.D.; Galberto, M.d.C.J. Chemical Properties of Caffeic and Ferulic Acids in Biological System: Implications in Cancer Therapy. A Review. Curr. Pharm. Des. 2017, 23, 3015–3023. [Google Scholar] [CrossRef]
- Li, Y.; Trush, M.A. Reactive oxygen-dependent DNA damage resulting from the oxidation of phenolic compounds by a copper-redox cycle mechanism. Cancer Res. 1994, 54 (Suppl. 7), 1895s–1898s. [Google Scholar]
- Pirker, K.F.; Kay, C.W.; Stolze, K.; Tunega, D.; Reichenauer, T.G.; Goodman, B.A. Free radical generation in rosmarinic acid investigated by electron paramagnetic resonance spectroscopy. Free Radic. Res. 2009, 43, 47–57. [Google Scholar] [CrossRef]
- Murakami, K.; Haneda, M.; Qiao, S.; Naruse, M.; Yoshino, M. Prooxidant action of rosmarinic acid: Transition metal-dependent generation of reactive oxygen species. Toxicol. In Vitro 2007, 21, 613–617. [Google Scholar] [CrossRef]
- Muñoz-Muñoz, J.; Garcia-Molina, F.; Ros, E.; Tudela, J.; García-Canovas, F.; Rodriguez-Lopez, J. Prooxidant and Antioxidant Activities of Rosmarinic Acid. J. Food Biochem. 2013, 37, 396–408. [Google Scholar] [CrossRef]
- Fabiani, R.; De Bartolomeo, A.; Rosignoli, P.; Servili, M.; Montedoro, G.F.; Morozzi, G. Cancer chemoprevention by hydroxytyrosol isolated from virgin olive oil through G1 cell cycle arrest and apoptosis. Eur. J. Cancer Prev. 2002, 11, 351–358. [Google Scholar] [CrossRef]
- Sun, L.; Luo, C.; Liu, J. Hydroxytyrosol induces apoptosis in human colon cancer cells through ROS generation. Food Funct. 2014, 5, 1909–1914. [Google Scholar] [CrossRef]
- Long, L.H.; Hoi, A.; Halliwell, B. Instability of, and generation of hydrogen peroxide by, phenolic compounds in cell culture media. Arch. Biochem. Biophys. 2010, 501, 162–169. [Google Scholar] [CrossRef]
- Sakihama, Y.; Cohen, M.F.; Grace, S.C.; Yamasaki, H. Plant phenolic antioxidant and prooxidant activities: Phenolics-induced oxidative damage mediated by metals in plants. Toxicology 2002, 177, 67–80. [Google Scholar] [CrossRef]
- Guichard, C.; Pedruzzi, E.; Fay, M.; Marie, J.C.; Braut-Boucher, F.; Daniel, F.; Grodet, A.; Gougerot-Pocidalo, M.A.; Chastre, E.; Kotelevets, L.; et al. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis 2006, 27, 1812–1827. [Google Scholar] [CrossRef]
- Luo, C.; Li, Y.; Wang, H.; Cui, Y.; Feng, Z.; Li, H.; Li, Y.; Wang, Y.; Wurtz, K.; Weber, P.; et al. Hydroxytyrosol promotes superoxide production and defects in autophagy leading to anti-proliferation and apoptosis on human prostate cancer cells. Curr. Cancer Drug Targets 2013, 13, 625–639. [Google Scholar] [CrossRef]
- Fabiani, R.; Fuccelli, R.; Pieravanti, F.; De Bartolomeo, A.; Morozzi, G. Production of hydrogen peroxide is responsible for the induction of apoptosis by hydroxytyrosol on HL60 cells. Mol. Nutr. Food Res. 2009, 53, 887–896. [Google Scholar] [CrossRef]
- Fabiani, R.; Sepporta, M.V.; Rosignoli, P.; De Bartolomeo, A.; Crescimanno, M.; Morozzi, G. Anti-proliferative and pro-apoptotic activities of hydroxytyrosol on different tumour cells: The role of extracellular production of hydrogen peroxide. Eur. J. Nutr. 2012, 51, 455–464. [Google Scholar] [CrossRef]
- Shingai, Y.; Fujimoto, A.; Nakamura, M.; Masuda, T. Structure and function of the oxidation products of polyphenols and identification of potent lipoxygenase inhibitors from Fe-catalyzed oxidation of resveratrol. J. Agric. Food Chem. 2011, 59, 8180–8186. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, W.C.; Kuo, C.N.; Lu, F.J. Role of redox signaling regulation in propyl gallate-induced apoptosis of human leukemia cells. Food Chem. Toxicol. 2011, 49, 494–501. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. The enhancement of propyl gallate-induced apoptosis in HeLa cells by a proteasome inhibitor MG132. Oncol. Rep. 2011, 25, 871–877. [Google Scholar]
- Han, Y.H.; Park, W.H. Propyl gallate inhibits the growth of HeLa cells via regulating intracellular GSH level. Food Chem. Toxicol. 2009, 47, 2531–2538. [Google Scholar] [CrossRef]
- Jacobi, H.; Eicke, B.; Witte, I. DNA strand break induction and enhanced cytotoxicity of propyl gallate in the presence of copper(II). Free Radic. Biol. Med. 1998, 24, 972–978. [Google Scholar] [CrossRef]
- Grzesik, M.; Bartosz, G.; Stefaniuk, I.; Pichla, M.; Namiesnik, J.; Sadowska-Bartosz, I. Dietary antioxidants as a source of hydrogen peroxide. Food Chem. 2019, 278, 692–699. [Google Scholar] [CrossRef]
- Criddle, D.N.; Gillies, S.; Baumgartner-Wilson, H.K.; Jaffar, M.; Chinje, E.C.; Passmore, S.; Chvanov, M.; Barrow, S.; Gerasimenko, O.V.; Tepikin, A.V.; et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 2006, 281, 40485–40492. [Google Scholar] [CrossRef]
- Mishra, P.K.; Park, I.; Sharma, N.; Yoo, C.M.; Lee, H.Y.; Rhee, H.W. Enzymatic Recording of Local Hydrogen Peroxide Generation Using Genetically Encodable Enzyme. Anal. Chem. 2022, 94, 14869–14877. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoek, T.L.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef]
- Lasalvia-Prisco, E.; Cucchi, S.; Vazquez, J.; Lasalvia-Galante, E.; Golomar, W.; Gordon, W. Serum markers variation consistent with autoschizis induced by ascorbic acid-menadione in patients with prostate cancer. Med. Oncol. 2003, 20, 45–52. [Google Scholar] [CrossRef]
- Checker, R.; Patwardhan, R.S.; Sharma, D.; Menon, J.; Thoh, M.; Sandur, S.K.; Sainis, K.B.; Poduval, T.B. Plumbagin, a vitamin K3 analogue, abrogates lipopolysaccharide-induced oxidative stress, inflammation and endotoxic shock via NF-kappaB suppression. Inflammation 2014, 37, 542–554. [Google Scholar] [CrossRef]
- Sand, J.M.; Bin Hafeez, B.; Jamal, M.S.; Witkowsky, O.; Siebers, E.M.; Fischer, J.; Verma, A.K. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), isolated from Plumbago zeylanica, inhibits ultraviolet radiation-induced development of squamous cell carcinomas. Carcinogenesis 2012, 33, 184–190. [Google Scholar] [CrossRef]
- Klaus, V.; Hartmann, T.; Gambini, J.; Graf, P.; Stahl, W.; Hartwig, A.; Klotz, L.O. 1,4-Naphthoquinones as inducers of oxidative damage and stress signaling in HaCaT human keratinocytes. Arch. Biochem. Biophys. 2010, 496, 93–100. [Google Scholar] [CrossRef]
- Kapur, A.; Beres, T.; Rathi, K.; Nayak, A.P.; Czarnecki, A.; Felder, M.; Gillette, A.; Ericksen, S.S.; Sampene, E.; Skala, M.C.; et al. Oxidative stress via inhibition of the mitochondrial electron transport and Nrf-2-mediated anti-oxidative response regulate the cytotoxic activity of plumbagin. Sci. Rep. 2018, 8, 1073. [Google Scholar] [CrossRef]
- Inbaraj, J.J.; Chignell, C.F. Cytotoxic action of juglone and plumbagin: A mechanistic study using HaCaT keratinocytes. Chem. Res. Toxicol. 2004, 17, 55–62. [Google Scholar] [CrossRef]
- Kappus, H. Overview of enzyme systems involved in bio-reduction of drugs and in redox cycling. Biochem. Pharmacol. 1986, 35, 1–6. [Google Scholar] [CrossRef]
- Ollinger, K.; Brunmark, A. Effect of hydroxy substituent position on 1,4-naphthoquinone toxicity to rat hepatocytes. J. Biol. Chem. 1991, 266, 21496–21503. [Google Scholar] [CrossRef]
- Gong, Q.; Hu, J.; Wang, P.; Li, X.; Zhang, X. A comprehensive review on beta-lapachone: Mechanisms, structural modifications, and therapeutic potentials. Eur. J. Med. Chem. 2021, 210, 112962. [Google Scholar] [CrossRef]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef]
- Chau, Y.P.; Shiah, S.G.; Don, M.J.; Kuo, M.L. Involvement of hydrogen peroxide in topoisomerase inhibitor beta-lapachone-induced apoptosis and differentiation in human leukemia cells. Free Radic. Biol. Med. 1998, 24, 660–670. [Google Scholar] [CrossRef]
- Gerber, D.E.; Beg, M.S.; Fattah, F.; Frankel, A.E.; Fatunde, O.; Arriaga, Y.; Dowell, J.E.; Bisen, A.; Leff, R.D.; Meek, C.C.; et al. Phase 1 study of ARQ 761, a beta-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br. J. Cancer 2018, 119, 928–936. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S. Pharmacokinetics and tolerability of MB12066, a beta-lapachone derivative targeting NAD(P)H: Quinone oxidoreductase 1: Two independent, double-blind, placebo-controlled, combined single and multiple ascending dose first-in-human clinical trials. Drug Des. Dev. Ther. 2017, 11, 3187–3195. [Google Scholar] [CrossRef]
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Sinha, B.K.; Mimnaugh, E.G.; Rajagopalan, S.; Myers, C.E. Adriamycin activation and oxygen free radical formation in human breast tumor cells: Protective role of glutathione peroxidase in adriamycin resistance. Cancer Res. 1989, 49, 3844–3848. [Google Scholar]
- Ubezio, P.; Civoli, F. Flow cytometric detection of hydrogen peroxide production induced by doxorubicin in cancer cells. Free Radic. Biol. Med. 1994, 16, 509–516. [Google Scholar] [CrossRef]
- Wagner, B.A.; Evig, C.B.; Reszka, K.J.; Buettner, G.R.; Burns, C.P. Doxorubicin increases intracellular hydrogen peroxide in PC3 prostate cancer cells. Arch. Biochem. Biophys. 2005, 440, 181–190. [Google Scholar] [CrossRef]
- Thayer, W.S. Adriamycin stimulated superoxide formation in submitochondrial particles. Chem. Biol. Interact. 1977, 19, 265–278. [Google Scholar] [CrossRef]
- Binaschi, M.; Farinosi, R.; Borgnetto, M.E.; Capranico, G. In vivo site specificity and human isoenzyme selectivity of two topoisomerase II-poisoning anthracyclines. Cancer Res. 2000, 60, 3770–3776. [Google Scholar]
- Skibo, E.B.; Xing, C. Chemistry and DNA alkylation reactions of aziridinyl quinones: Development of an efficient alkylating agent of the phosphate backbone. Biochemistry 1998, 37, 15199–15213. [Google Scholar] [CrossRef]
- Rashid, M.H.; Babu, D.; Siraki, A.G. Interactions of the antioxidant enzymes NAD(P)H: Quinone oxidoreductase 1 (NQO1) and NRH: Quinone oxidoreductase 2 (NQO2) with pharmacological agents, endogenous biochemicals and environmental contaminants. Chem. Biol. Interact. 2021, 345, 109574. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. (Eds.) Chapter 11—Drug Targeting in Anticancer Chemotherapy. In Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008; pp. 351–385. [Google Scholar]
- Webb, J.S.C.D.B.; Mowat, J.H.; Patrick, J.B.; Broschard, R.W.; Meyer, W.E.; Williams, R.P.; Wolf, C.F.; Fulmor, W.; Pidacks, C.; Lancaster, J.E. The Structures of Mitomycins A, B and C and Porfiromycin—Part I. J. Am. Chem. Soc. 1962, 84, 3185–3187. [Google Scholar] [CrossRef]
- Tomasz, M. H2O2 generation during the redox cycle of mitomycin C and dna-bound mitomycin C. Chem. Biol. Interact. 1976, 13, 89–97. [Google Scholar] [CrossRef]
- Andrez, J.C. Mitomycins syntheses: A recent update. Beilstein J. Org. Chem. 2009, 5, 33. [Google Scholar] [CrossRef]
- Remers, W.A.; Schepman, C.S. Structure-activity relationships of the mitomycins and certain synthetic analogs. J. Med. Chem. 1974, 17, 729–732. [Google Scholar] [CrossRef]
- Hornemann, U.H.M.J. Stereochemical Relationship between Mitomycins A, B, and C. J. Org. Chem. 1985, 50, 1301–1302. [Google Scholar] [CrossRef]
- Mizuno, N.S. Effects of streptonigrin on nucleic acid metabolism of tissue culture cells. Biochim. Biophys. Acta 1965, 108, 394–403. [Google Scholar] [CrossRef]
- Cone, R.; Hasan, S.K.; Lown, J.W.; Morgan, A.R. The mechanism of the degradation of DNA by streptonigrin. Can. J. Biochem. 1976, 54, 219–223. [Google Scholar] [CrossRef]
- Bolzan, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Testoni, M.I.; Bolzan, A.D.; Bianchi, M.S.; Bianchi, N.O. Effects of antioxidants on streptonigrin-induced DNA damage and clastogenesis in CHO cells. Mutat. Res. 1997, 373, 201–206. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, N.; Creagan, E.; Witzig, T.; Levine, M. Ascorbic Acid in Cancer Treatment: Let the Phoenix Fly. Cancer Cell 2018, 34, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.; Knudson, C.M.; Cullen, J.J. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra18. [Google Scholar] [CrossRef] [PubMed]
- Nauman, G.; Gray, J.C.; Parkinson, R.; Levine, M.; Paller, C.J. Systematic Review of Intravenous Ascorbate in Cancer Clinical Trials. Antioxidants 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef]
- Sawant, R.R.; Vaze, O.; D′Souza, G.G.; Rockwell, K.; Torchilin, V.P. Palmitoyl ascorbate-loaded polymeric micelles: Cancer cell targeting and cytotoxicity. Pharm. Res. 2011, 28, 301–308. [Google Scholar] [CrossRef]
- Sawant, R.R.; Vaze, O.S.; Wang, T.; D′Souza, G.G.; Rockwell, K.; Gada, K.; Khaw, B.A.; Torchilin, V.P. Palmitoyl ascorbate liposomes and free ascorbic acid: Comparison of anticancer therapeutic effects upon parenteral administration. Pharm. Res. 2012, 29, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Strafella, E.; Staffolani, S.; Santarelli, L.; Neuzil, J.; Guerrieri, R. alpha-Tocopheryl succinate promotes selective cell death induced by vitamin K3 in combination with ascorbate. Br. J. Cancer 2010, 102, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, X.; Li, C.; Huang, Y.; Xu, P.; Lloyd-Davies, L.H.; Delplancke, T.; Peng, C.; Gao, R.; Qi, H.; et al. Triethylenetetramine Synergizes with Pharmacologic Ascorbic Acid in Hydrogen Peroxide Mediated Selective Toxicity to Breast Cancer Cell. Oxid. Med. Cell. Longev. 2017, 2017, 3481710. [Google Scholar] [CrossRef]
- Li, J.; Ke, W.; Wang, L.; Huang, M.; Yin, W.; Zhang, P.; Chen, Q.; Ge, Z. Self-sufficing H2O2-responsive nanocarriers through tumor-specific H2O2 production for synergistic oxidation-chemotherapy. J. Control. Release 2016, 225, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Orvig, C.; Abrams, M.J. Medicinal inorganic chemistry: Introduction. Chem. Rev. 1999, 99, 2201–2204. [Google Scholar] [CrossRef] [PubMed]
- Yaman, M.; Kaya, G.; Yekeler, H. Distribution of trace metal concentrations in paired cancerous and non-cancerous human stomach tissues. World J. Gastroenterol. 2007, 13, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Son, Y.O.; Pratheeshkumar, P.; Shi, X. Oxidative stress and metal carcinogenesis. Free Radic. Biol. Med. 2012, 53, 742–757. [Google Scholar] [CrossRef]
- Frezza, M.; Hindo, S.; Chen, D.; Davenport, A.; Schmitt, S.; Tomco, D.; Dou, Q.P. Novel metals and metal complexes as platforms for cancer therapy. Curr. Pharm. Des. 2010, 16, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fu, L.H.; Qi, C.; Lin, J.; Huang, P. Metal peroxides for cancer treatment. Bioact. Mater. 2021, 6, 2698–2710. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Lv, G.; Bu, W. Redox dyshomeostasis strategy for tumor therapy based on nanomaterials chemistry. Chem. Sci. 2022, 13, 2202–2217. [Google Scholar] [CrossRef]
- Dugo, E.B.; Yedjou, C.G.; Stevens, J.J.; Tchounwou, P.B. Therapeutic Potential of Arsenic Trioxide (ATO) in Treatment of Hepatocellular Carcinoma: Role of Oxidative Stress in ATO-Induced Apoptosis. Ann. Clin. Pathol. 2017, 5, 1101. [Google Scholar] [PubMed]
- Chou, W.C.; Jie, C.; Kenedy, A.A.; Jones, R.J.; Trush, M.A.; Dang, C.V. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4578–4583. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Dai, J.; Chalmers-Redman, R.M.; Tatton, W.G.; Waxman, S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood 1999, 94, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Ziental, D.; Czarczynska-Goslinska, B.; Mlynarczyk, D.T.; Glowacka-Sobotta, A.; Stanisz, B.; Goslinski, T.; Sobotta, L. Titanium Dioxide Nanoparticles: Prospects and Applications in Medicine. Nanomaterials 2020, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Kormann, C.; Bahnemann, D.W.; Hoffmann, M.R. Photocatalytic production of hydrogen peroxides and organic peroxides in aqueous suspensions of titanium dioxide, zinc oxide, and desert sand. Environ. Sci. Technol. 1988, 22, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Murali, M.; Kalegowda, N.; Gowtham, H.G.; Ansari, M.A.; Alomary, M.N.; Alghamdi, S.; Shilpa, N.; Singh, S.B.; Thriveni, M.C.; Aiyaz, M.; et al. Plant-Mediated Zinc Oxide Nanoparticles: Advances in the New Millennium towards Understanding Their Therapeutic Role in Biomedical Applications. Pharmaceutics 2021, 13, 1662. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.A.; Das, J.; Sil, P.C. Zinc oxide nanoparticles: A comprehensive review on its synthesis, anticancer and drug delivery applications as well as health risks. Adv. Colloid. Interface Sci. 2020, 286, 102317. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.S.; Wang, J.F.; Song, J.; Liu, Y.; Zhu, G.; Dai, Y.; Shen, Z.; Tian, R.; Song, J.; Wang, Z.; et al. Cooperation of endogenous and exogenous reactive oxygen species induced by zinc peroxide nanoparticles to enhance oxidative stress-based cancer therapy. Theranostics 2019, 9, 7200–7209. [Google Scholar] [CrossRef]
- Mbugua, S.N. Targeting Tumor Microenvironment by Metal Peroxide Nanoparticles in Cancer Therapy. Bioinorg. Chem. Appl. 2022, 2022, 5041399. [Google Scholar] [CrossRef]
- Gao, S.; Jin, Y.; Ge, K.; Li, Z.; Liu, H.; Dai, X.; Zhang, Y.; Chen, S.; Liang, X.; Zhang, J. Self-Supply of O2 and H2O2 by a Nanocatalytic Medicine to Enhance Combined Chemo/Chemodynamic Therapy. Adv. Sci. 2019, 6, 1902137. [Google Scholar] [CrossRef]
- Lin, L.-S.; Huang, T.; Song, J.; Ou, X.-Y.; Wang, Z.; Deng, H.; Tian, R.; Liu, Y.; Wang, J.-F.; Liu, Y.; et al. Synthesis of Copper Peroxide Nanodots for H2O2 Self-Supplying Chemodynamic Therapy. J. Am. Chem. Soc. 2019, 141, 9937–9945. [Google Scholar] [CrossRef] [PubMed]
- Mathe, G.; Schweisguth, O.; Schneider, M.; Amiel, J.L.; Berumen, L.; Brule, G.; Cattan, A.; Schwarzenberg, L. Methyl-Hydrazine in Treatment of Hodgkin′s Disease and Various Forms of Haematosarcoma and Leukaemia. Lancet 1963, 2, 1077–1080. [Google Scholar] [CrossRef]
- Berneis, K.; Kofler, M.; Bollag, W.; Kaiser, A.; Langemann, A. The degradation of deoxyribonucleic acid by new tumour inhibiting compounds: The intermediate formation of hydrogen peroxide. Experientia 1963, 19, 132–133. [Google Scholar] [CrossRef] [PubMed]
- Berneis, K.; Kofler, M.; Bollag, W. The influence of chelating agents on the prooxidative effect of a hydrogen peroxide producing methylhydrazine compound. Experientia 1964, 20, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Devita, V.T., Jr.; Serpick, A.A.; Carbone, P.P. Combination chemotherapy in the treatment of advanced Hodgkin’s disease. Ann. Intern. Med. 1970, 73, 881–895. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Simon, R.M.; Hubbard, S.M.; Young, R.C.; Berard, C.W.; Moxley, J.H., 3rd; Frei, E., 3rd; Carbone, P.P.; Canellos, G.P. Curability of advanced Hodgkin′s disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann. Intern. Med. 1980, 92, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Gao, Y.; Singh, L.D.; Kunapuli, S.P.; Ravindra, R. Role of microtubules in glucose uptake by C6 glioma cells. Pharmacol. Toxicol. 1998, 83, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chereau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int. J. Cancer 2006, 119, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Diebold, B.A.; Fowler, B.; Lu, J.; Dinauer, M.C.; Bokoch, G.M. Antagonistic cross-talk between Rac and Cdc42 GTPases regulates generation of reactive oxygen species. J. Biol. Chem. 2004, 279, 28136–28142. [Google Scholar] [CrossRef]
- Zang, M.; Waelde, C.A.; Xiang, X.; Rana, A.; Wen, R.; Luo, Z. Microtubule integrity regulates Pak leading to Ras-independent activation of Raf-1. insights into mechanisms of Raf-1 activation. J. Biol. Chem. 2001, 276, 25157–25165. [Google Scholar] [CrossRef]
- Hadzic, T.; Aykin-Burns, N.; Zhu, Y.; Coleman, M.C.; Leick, K.; Jacobson, G.M.; Spitz, D.R. Paclitaxel combined with inhibitors of glucose and hydroperoxide metabolism enhances breast cancer cell killing via H2O2-mediated oxidative stress. Free Radic. Biol. Med. 2010, 48, 1024–1033. [Google Scholar] [CrossRef]
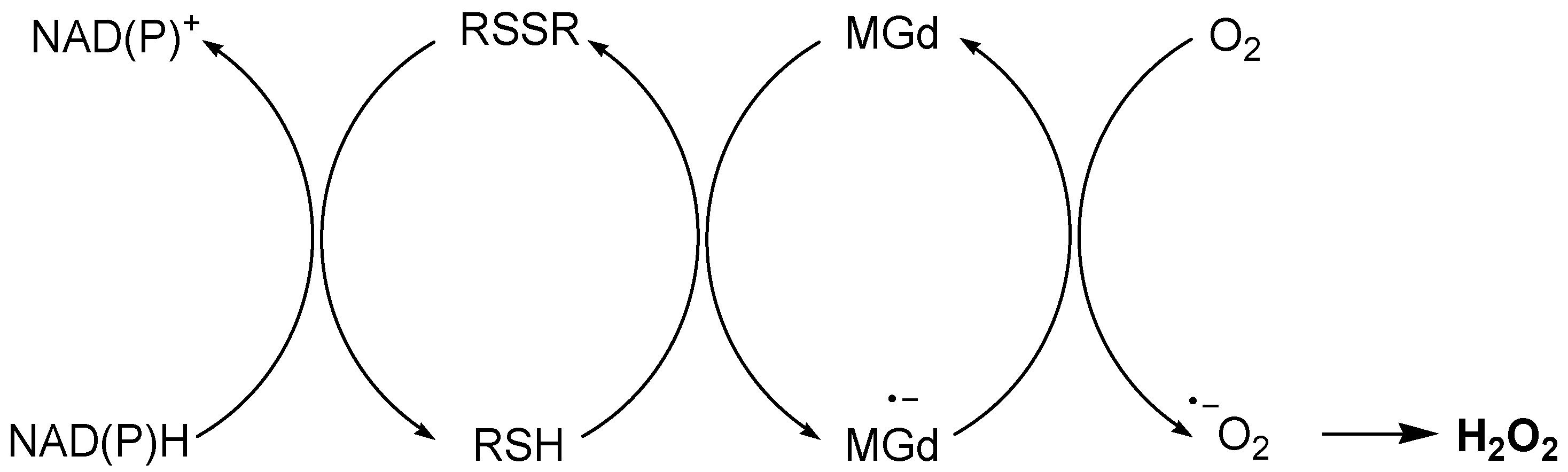
- Magda, D.; Lepp, C.; Gerasimchuk, N.; Lee, I.; Sessler, J.L.; Lin, A.; Biaglow, J.E.; Miller, R.A. Redox cycling by motexafin gadolinium enhances cellular response to ionizing radiation by forming reactive oxygen species. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 1025–1036. [Google Scholar] [CrossRef]
- Xu, S.; Zakian, K.; Thaler, H.; Matei, C.; Alfieri, A.; Chen, Y.; Koutcher, J.A. Effects of Motexafin gadolinium on tumor metabolism and radiation sensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.A.; Dashwood, R.H. Suppression of Met activation in human colon cancer cells treated with (-)-epigallocatechin-3-gallate: Minor role of hydrogen peroxide. Biochem. Biophys. Res. Commun. 2009, 389, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Huang, Y.W.; Tian, Y.; Wang, X.J.; Sheng, J. Mechanism of action of (-)-epigallocatechin-3-gallate: Auto-oxidation-dependent activation of extracellular signal-regulated kinase 1/2 in Jurkat cells. Chin. J. Nat. Med. 2014, 12, 654–662. [Google Scholar] [CrossRef]
- Skrbek, S.R.C.E.; Marko, D.; Esselen, M. Quercetin and its microbial degradation product 3,4-dihydroxyphenylacetic acid generate hydrogen peroxide modulating their stability under in vitro conditions. J. Food Nutr. Res. 2009, 48, 129–140. [Google Scholar]
- Bonilla-Porras, A.R.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Vitamin K3 and vitamin C alone or in combination induced apoptosis in leukemia cells by a similar oxidative stress signalling mechanism. Cancer Cell Int. 2011, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.; Sabarwal, A.; Narayan Mishra, J.P.; Singh, R.P. Plumbagin induces ROS-mediated apoptosis and cell cycle arrest and inhibits EMT in human cervical carcinoma cells. RSC Adv. 2018, 8, 32022–32037. [Google Scholar] [CrossRef]
- Clement, M.V.; Ramalingam, J.; Long, L.H.; Halliwell, B. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxid. Redox Signal. 2001, 3, 157–163. [Google Scholar] [CrossRef]
- El-Said, K.S.A.E.; Kanehira, K.; Taniguchi, A. Comparison of oxidative stresses mediated by different crystalline forms and surface modification of titanium dioxide nanoparticles. J. Nanomater. 2015, 2015, 703632. [Google Scholar] [CrossRef]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Ali, T. Combination Therapy for Cancer Using ROS-Activated Prodrugs and ROS-Amplifying Therapeutics. Patent Application Atty. File No. 020871-0011-WO01; US Provisional Patent No. 63/480,347. Unpublished work. 2024. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prooxidant | Dose | Cell Line | H2O2 Produced | Method | Ref. |
|---|---|---|---|---|---|
| Phenols and polyphenol analogues | |||||
| EGCG | 50 µM | Jurkat | ~1 µM | Ferrous ion oxidation--xylenol orange method | [45] |
| 10 µM | HCT116 and HT29 | 1.5 µM | Amplex Red Assay Kit | [166] | |
| 100 µM | Jurkat | 20 µM | Hydrogen Peroxide Quantitative Assay Kit from Sangon Biotech | [167] | |
| Quercetin | 70 µM | HT29 | 2 µM | Amplex Red assay kit | [168] |
| Compounds with quinone moieties | |||||
| Menadione | 10 µM | Jurkat | 20 µM | 2′,7′-dichlorofluorescein diacetate (DCFH2- DA) | [169] |
| Plumbagin | 4 µM | SiHA and HeLa | 1 mM | DCFH2-DA | [170] |
| β-Lap | 1 µM | HL-60 | 70 µM | DCFH2- DA | [101] |
| Anthracyclines | |||||
| Doxorubicin | 1 µM | PC3 | 38 pM | Amplex Red Assay Kit | [108] |
| Vitamin C | |||||
| Ascorbic Acid | 1 mM | HL-60 | 161 µM | Oxygen electrode | [171] |
| 0.2–2.0 mM | Burkitt’s lymphoma | 20–120 µM | Oxygen electrode | [123] | |
| i.v. 0.5 mg/g | rats | 0–20 µM | Boronate fluorophore peroxyxanthone (PX1) | [124] | |
| i.p. 4.0g/kg | Mice tumor | >125 µM | PX1 | [125] | |
| Metal, metal oxides, and metal peroxides | |||||
| TiO2 | 10 µg/mL | HepG2 | 150 nmol/mL | DCFH-DA assay | [172] |
| FDA-approved drugs | |||||
| Paclitaxel | 100 nM | MCF7 | 600 nM | Amplex Red Assay Kit | [173] |
| 100 nM | HL-60 | 1100 nM | Amplex Red Assay Kit | [173] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, T.; Li, D.; Ponnamperumage, T.N.F.; Peterson, A.K.; Pandey, J.; Fatima, K.; Brzezinski, J.; Jakusz, J.A.R.; Gao, H.; Koelsch, G.E.; et al. Generation of Hydrogen Peroxide in Cancer Cells: Advancing Therapeutic Approaches for Cancer Treatment. Cancers 2024, 16, 2171. https://doi.org/10.3390/cancers16122171
Ali T, Li D, Ponnamperumage TNF, Peterson AK, Pandey J, Fatima K, Brzezinski J, Jakusz JAR, Gao H, Koelsch GE, et al. Generation of Hydrogen Peroxide in Cancer Cells: Advancing Therapeutic Approaches for Cancer Treatment. Cancers. 2024; 16(12):2171. https://doi.org/10.3390/cancers16122171
Chicago/Turabian StyleAli, Taufeeque, Daniel Li, Thilini Nimasha Fernando Ponnamperumage, Alexis Kimberly Peterson, Jatin Pandey, Kulsum Fatima, John Brzezinski, Julia Anna Rose Jakusz, Hanlun Gao, Gilbert Edward Koelsch, and et al. 2024. "Generation of Hydrogen Peroxide in Cancer Cells: Advancing Therapeutic Approaches for Cancer Treatment" Cancers 16, no. 12: 2171. https://doi.org/10.3390/cancers16122171
APA StyleAli, T., Li, D., Ponnamperumage, T. N. F., Peterson, A. K., Pandey, J., Fatima, K., Brzezinski, J., Jakusz, J. A. R., Gao, H., Koelsch, G. E., Murugan, D. S., & Peng, X. (2024). Generation of Hydrogen Peroxide in Cancer Cells: Advancing Therapeutic Approaches for Cancer Treatment. Cancers, 16(12), 2171. https://doi.org/10.3390/cancers16122171