


Cholesterol Dietary Intake and Tumor Cell Homeostasis Drive Early Epithelial Tumorigenesis: A Potential Modelization of Early Prostate Tumorigenesis
 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fly Stocks and Experimental Crosses
2.2. Conditional Expression Induction
2.3. High-Cholesterol Diet
2.4. Immunohistochemistry and Imaging
2.5. Clones, Cells, and Nuclei Size
2.6. Invasive Tumor Frequency
2.7. RNA-seq Data
2.8. Statistical Analyses
3. Results
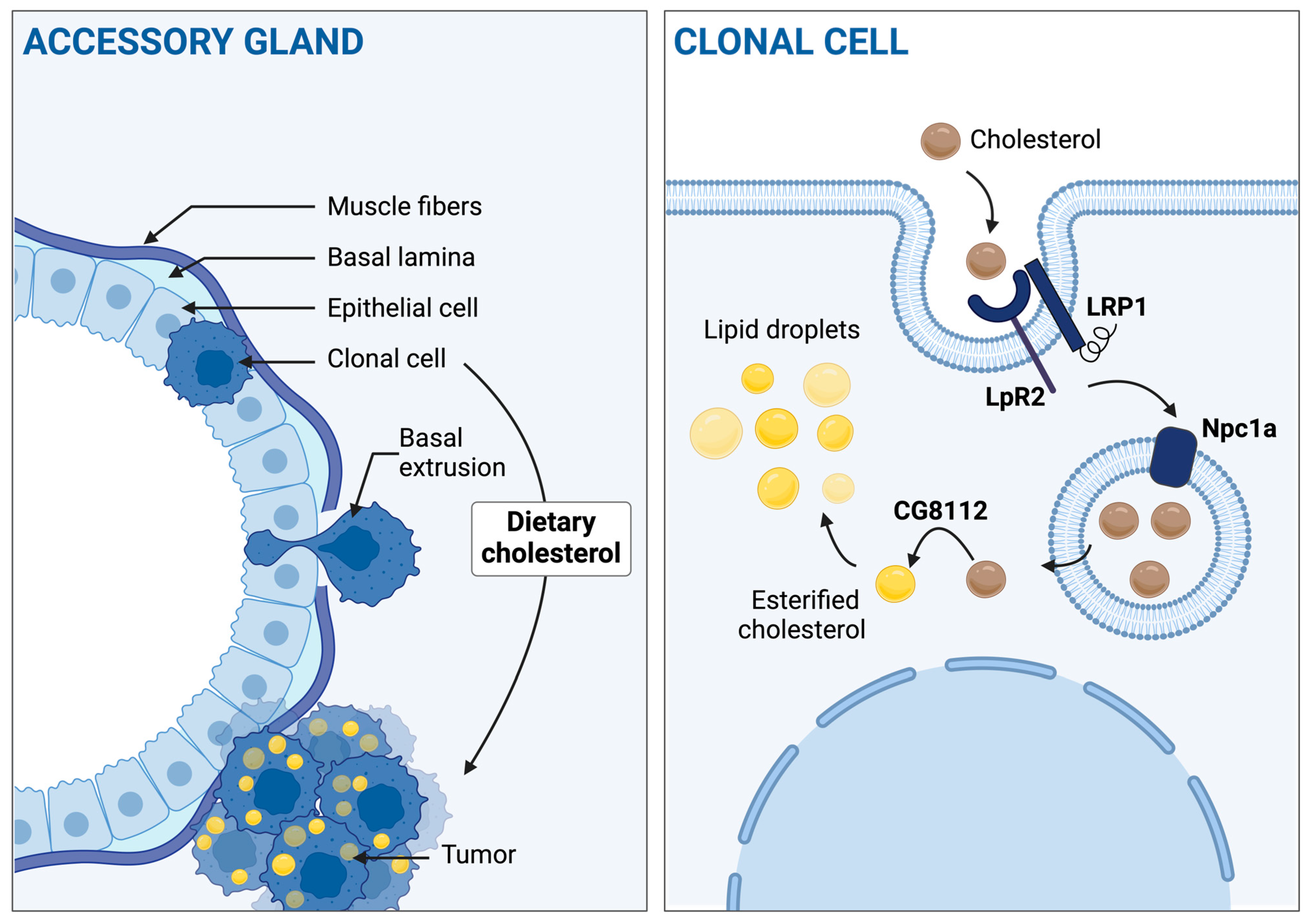
3.1. Accessory Gland Tumors Accumulate Cholesterol into Lipid Droplets
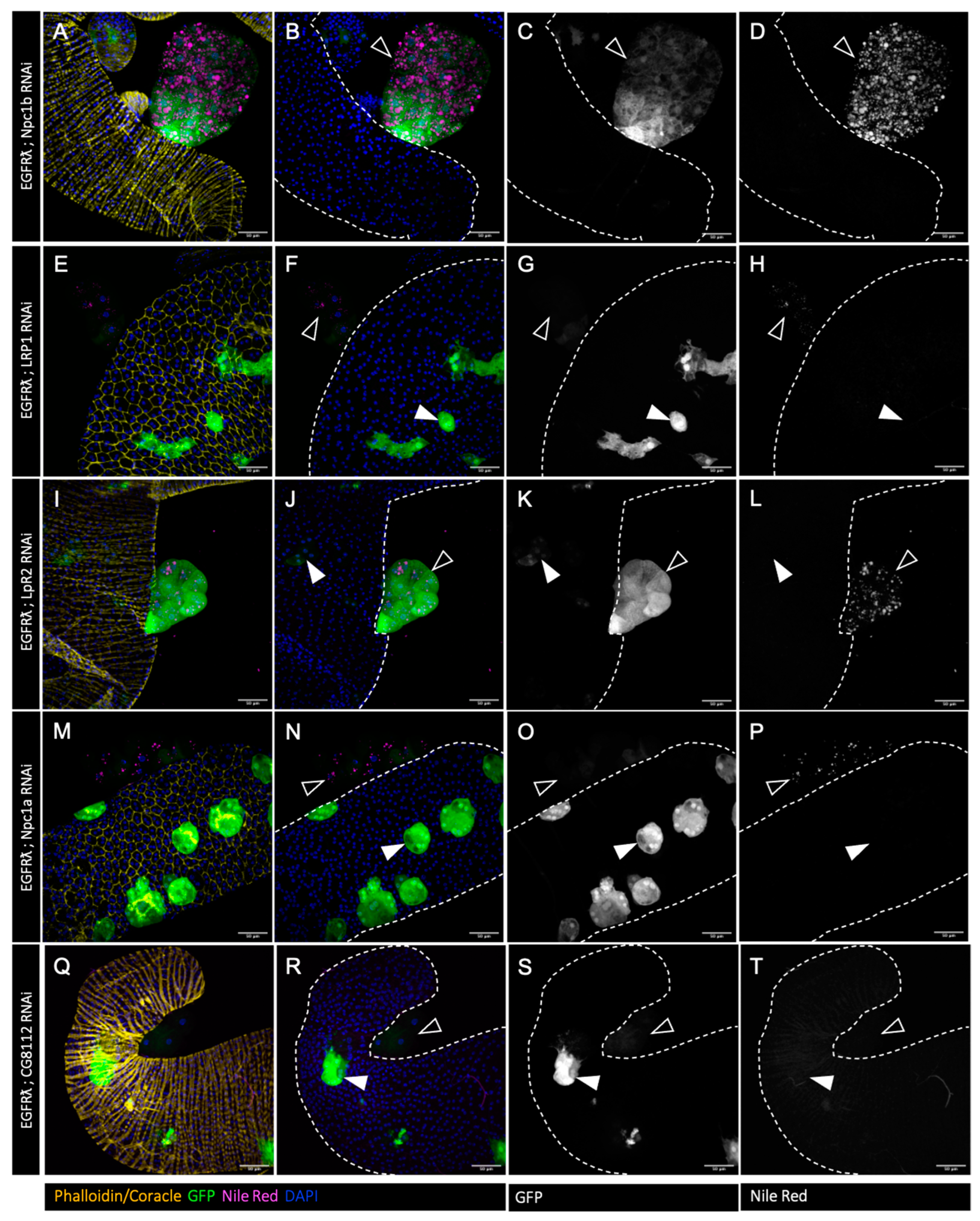
3.2. Cholesterol Homeostasis Downregulation Impairs EGFRλ-Induced Cholesterol Storage
3.3. Hyperactivation of Cell Autonomous Cholesterol Metabolism Specifically Drives Basal Extrusion
3.4. Further Cholesterol Accumulation Has No Effect on Early Tumorigenesis
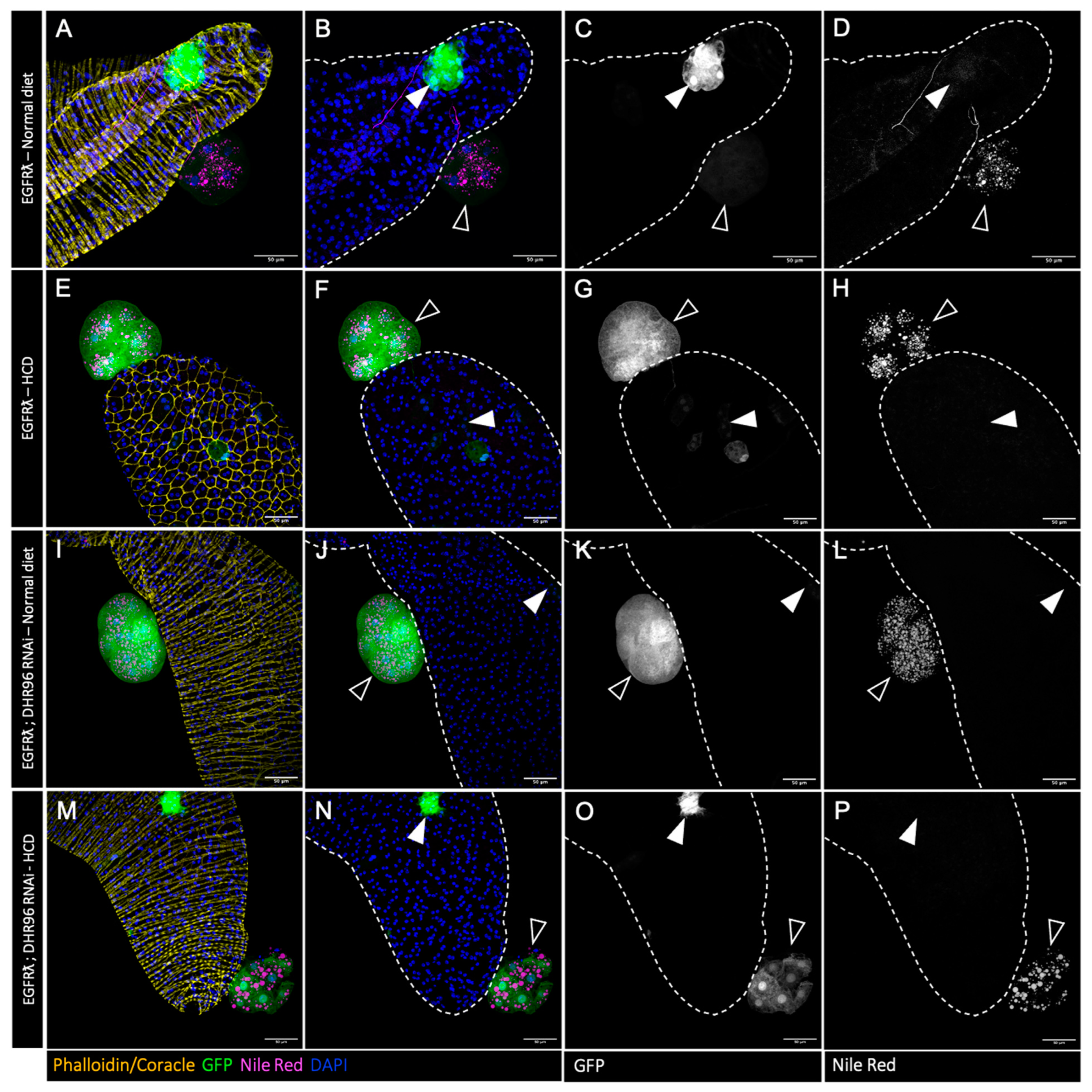
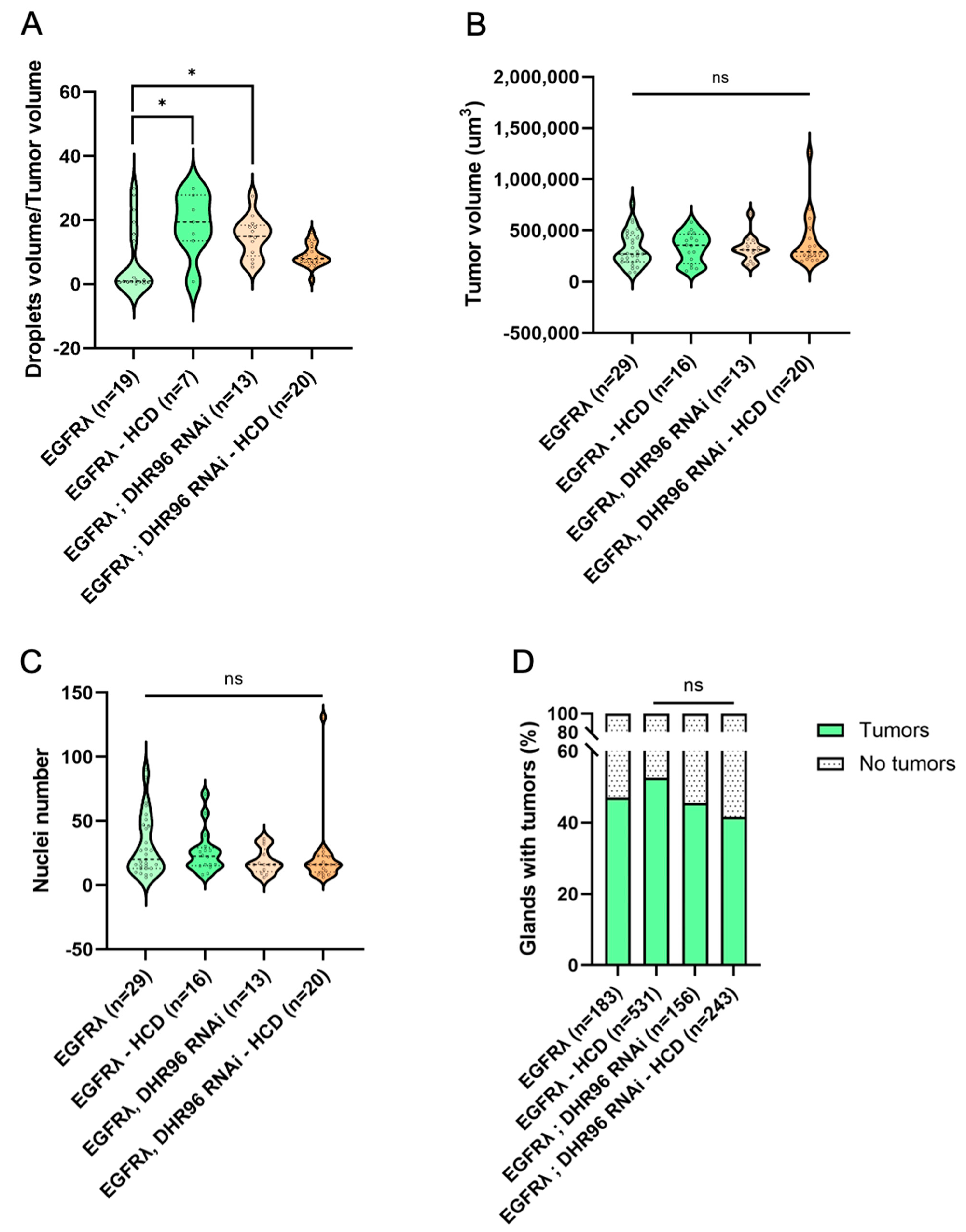
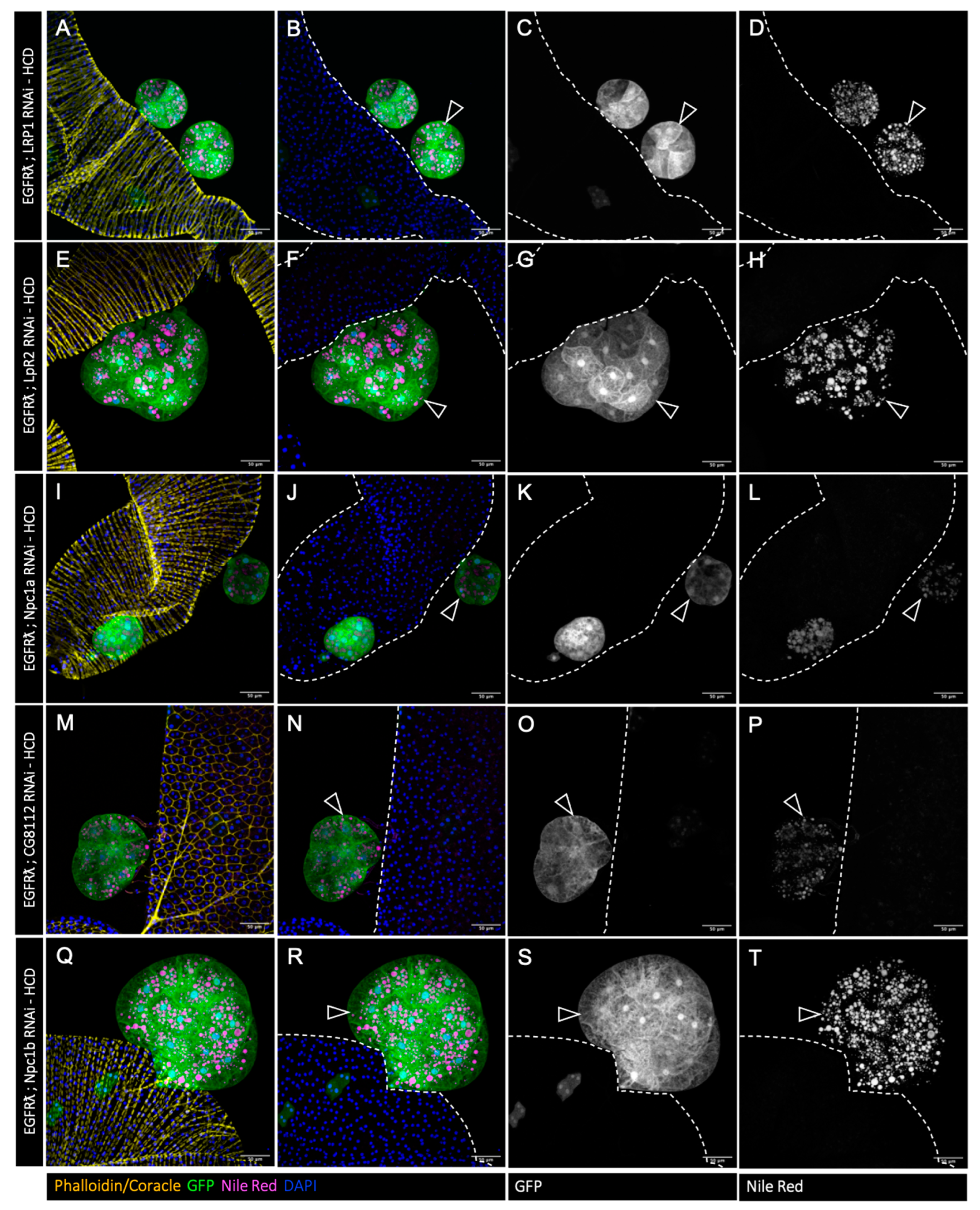
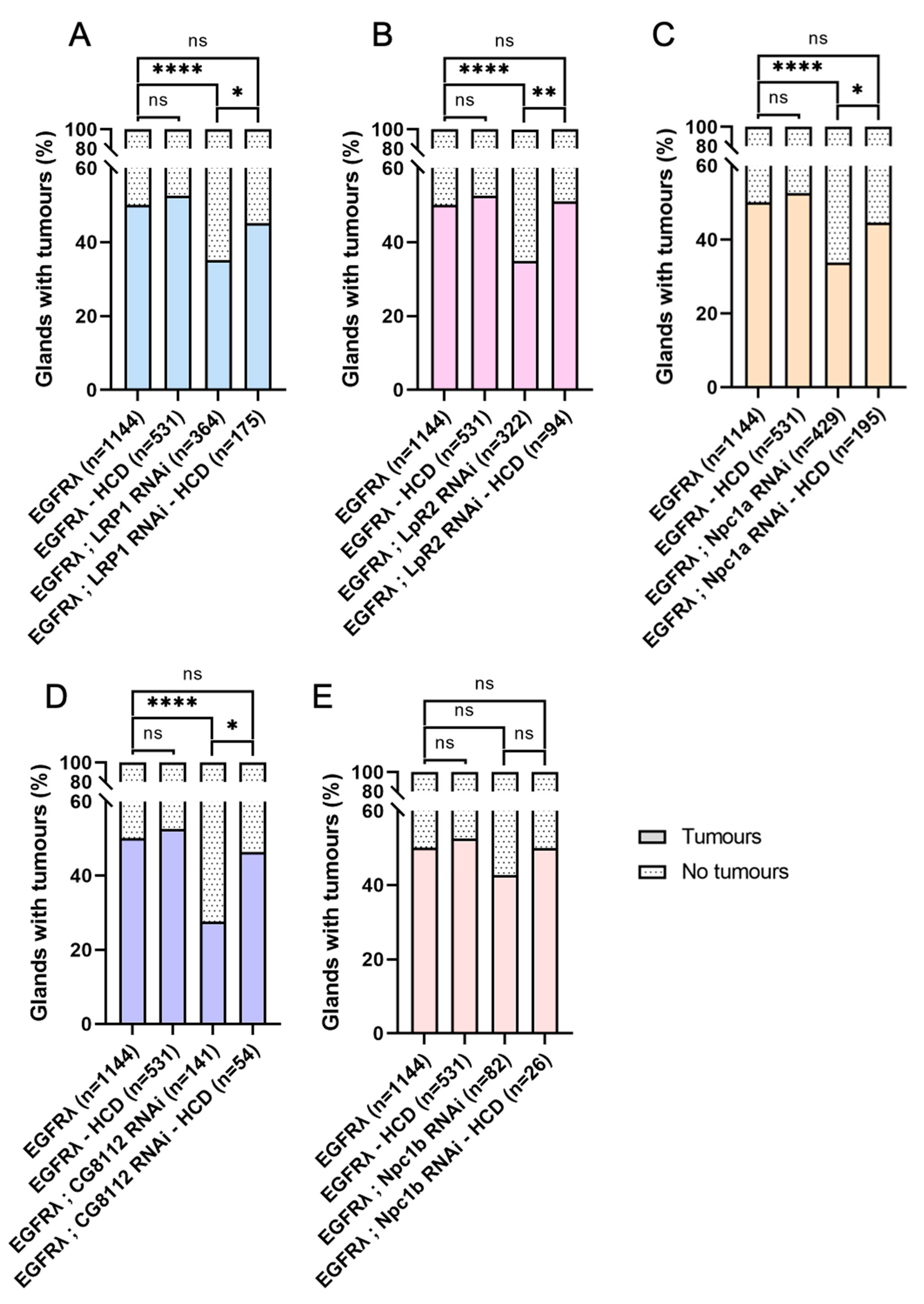
3.5. High-Cholesterol Diet Counteracts Effect of Cholesterol Metabolism Downregulation in EGFRλ-Induced Tumorigenesis
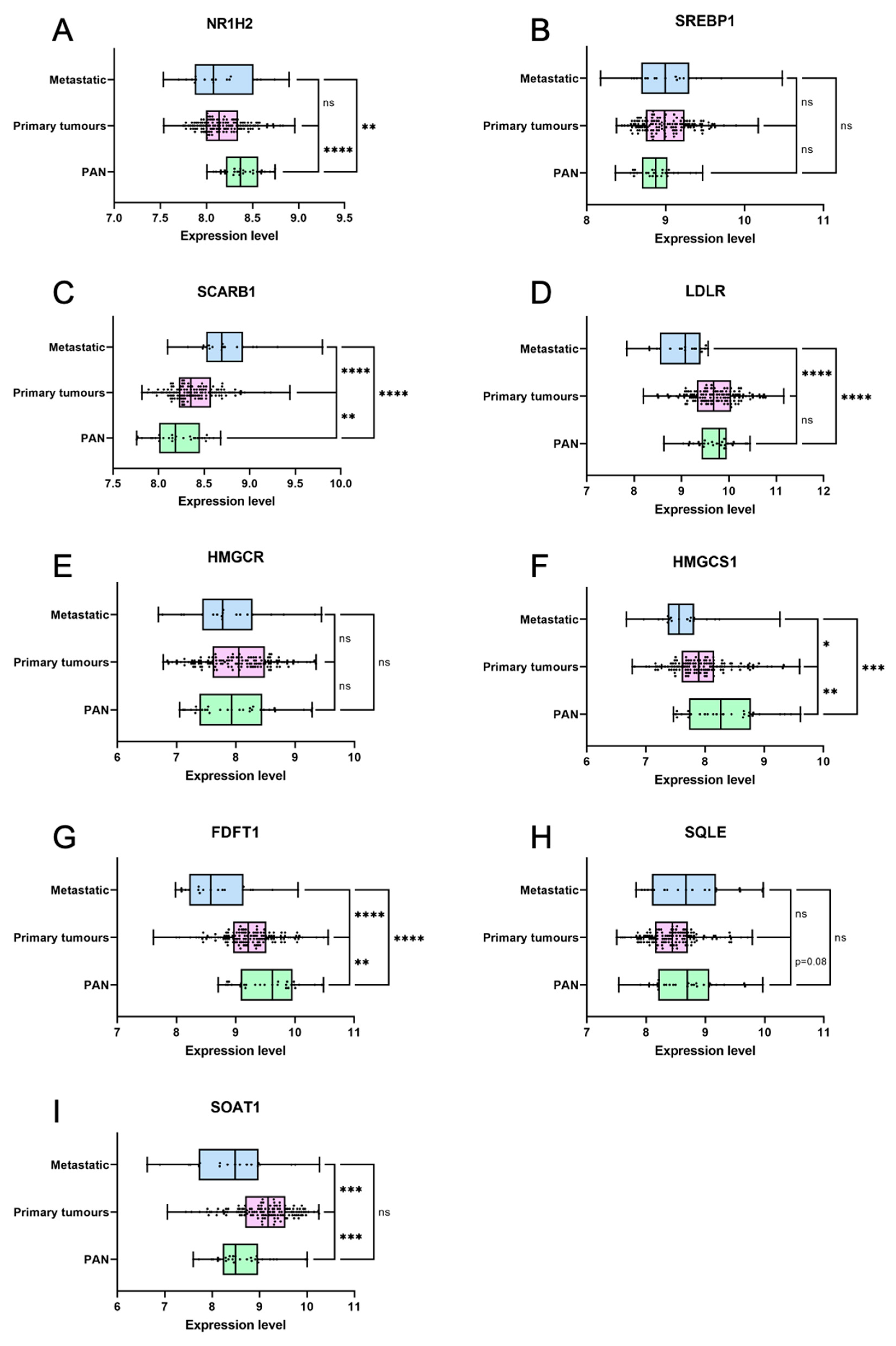
3.6. Genes Coding for Cholesterol Homeostasis and Metabolism Are Deregulated in Primary Prostate Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shimizu, H.; Ross, R.K.; Bernstein, L.; Yatani, R.; Henderson, B.E.; Mack, T.M. Cancers of the Prostate and Breast among Japanese and White Immigrants in Los Angeles County. Br. J. Cancer 1991, 63, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.S.; Goldoft, M.; Schwartz, S.M.; Weiss, N.S. Incidence of Adenocarcinoma of the Prostate in Asian Immigrants to the United States and Their Descendants. J. Urol. 1999, 161, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Clinton, S.K.; Giovannucci, E. Association between Plasma Cholesterol and Prostate Cancer in the PSA Era. Int. J. Cancer 2008, 123, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Till, C.; Goodman, P.J.; Parnes, H.L.; Figg, W.D.; Albanes, D.; Neuhouser, M.L.; Klein, E.A.; Thompson, I.M.J.; Kristal, A.R. Men with Low Serum Cholesterol Have a Lower Risk of High-Grade Prostate Cancer in the Placebo Arm of the Prostate Cancer Prevention Trial. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2807–2813. [Google Scholar] [CrossRef]
- Mondul, A.M.; Clipp, S.L.; Helzlsouer, K.J.; Platz, E.A. Association between Plasma Total Cholesterol Concentration and Incident Prostate Cancer in the CLUE II Cohort. Cancer Causes Control 2010, 21, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, C.M.; Berrington de González, A.; Freedman, N.D.; Huxley, R.; Mok, Y.; Jee, S.H.; Samet, J.M. Total Cholesterol and Cancer Risk in a Large Prospective Study in Korea. J. Clin. Oncol. 2011, 29, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Farwell, W.R.; D’Avolio, L.W.; Scranton, R.E.; Lawler, E.V.; Gaziano, J.M. Statins and Prostate Cancer Diagnosis and Grade in a Veterans Population. J. Natl. Cancer Inst. 2011, 103, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Ukomadu, C.; Dutta, A. Inhibition of Cdk2 Activating Phosphorylation by Mevastatin. J. Biol. Chem. 2003, 278, 4840–4846. [Google Scholar] [CrossRef] [PubMed]
- Platz, E.A.; Leitzmann, M.F.; Visvanathan, K.; Rimm, E.B.; Stampfer, M.J.; Willett, W.C.; Giovannucci, E. Statin Drugs and Risk of Advanced Prostate Cancer. J. Natl. Cancer Inst. 2006, 98, 1819–1825. [Google Scholar] [CrossRef]
- Flick, E.D.; Habel, L.A.; Chan, K.A.; Van Den Eeden, S.K.; Quinn, V.P.; Haque, R.; Orav, E.J.; Seeger, J.D.; Sadler, M.C.; Quesenberry, C.P.J.; et al. Statin Use and Risk of Prostate Cancer in the California Men’s Health Study Cohort. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2218–2225. [Google Scholar] [CrossRef]
- Shannon, J.; Tewoderos, S.; Garzotto, M.; Beer, T.M.; Derenick, R.; Palma, A.; Farris, P.E. Statins and Prostate Cancer Risk: A Case-Control Study. Am. J. Epidemiol. 2005, 162, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Shafique, K.; McLoone, P.; Qureshi, K.; Leung, H.; Hart, C.; Morrison, D.S. Cholesterol and the Risk of Grade-Specific Prostate Cancer Incidence: Evidence from Two Large Prospective Cohort Studies with up to 37 Years’ Follow Up. BMC Cancer 2012, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- YuPeng, L.; YuXue, Z.; PengFei, L.; Cheng, C.; YaShuang, Z.; DaPeng, L.; Chen, D. Cholesterol Levels in Blood and the Risk of Prostate Cancer: A Meta-Analysis of 14 Prospective Studies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1086–1093. [Google Scholar] [CrossRef]
- Liu, H.; Shui, I.M.; Keum, N.; Shen, X.; Wu, K.; Clinton, S.K.; Cao, Y.; Song, M.; Zhang, X.; Platz, E.A.; et al. Plasma Total Cholesterol Concentration and Risk of Higher-Grade Prostate Cancer: A Nested Case-Control Study and a Dose-Response Meta-Analysis. Int. J. Cancer 2023, 153, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Meijer, D.; van Moorselaar, R.J.A.; Vis, A.N.; Bijnsdorp, I. V Prostate Cancer Development Is Not Affected by Statin Use in Patients with Elevated PSA Levels. Cancers 2019, 11, 953. [Google Scholar] [CrossRef] [PubMed]
- Caro-Maldonado, A.; Camacho, L.; Zabala-Letona, A.; Torrano, V.; Fernández-Ruiz, S.; Zamacola-Bascaran, K.; Arreal, L.; Valcárcel-Jiménez, L.; Martín-Martín, N.; Flores, J.M.; et al. Low-Dose Statin Treatment Increases Prostate Cancer Aggressiveness. Oncotarget 2018, 9, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Celhay, O.; Bousset, L.; Guy, L.; Kemeny, J.-L.; Leoni, V.; Caccia, C.; Trousson, A.; Damon-Soubeyrant, C.; De Haze, A.; Sabourin, L.; et al. Individual Comparison of Cholesterol Metabolism in Normal and TumourTumor Areas in Radical Prostatectomy Specimens from Patients with Prostate Cancer: Results of the CHOMECAP Study. Eur. Urol. Oncol. 2019, 2, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Lin, J.; Lu, M.L.; Solomon, K.R.; Freeman, M.R. Cholesterol-Rich Lipid Rafts Mediate Akt-Regulated Survival in Prostate Cancer Cells. Cancer Res. 2002, 62, 2227–2231. [Google Scholar] [PubMed]
- Patra, S.K. Dissecting Lipid Raft Facilitated Cell Signaling Pathways in Cancer. Biochim. Biophys. Acta 2008, 1785, 182–206. [Google Scholar] [CrossRef]
- Llaverias, G.; Danilo, C.; Wang, Y.; Witkiewicz, A.K.; Daumer, K.; Lisanti, M.P.; Frank, P.G. A Western-Type Diet Accelerates Tumor Progression in an Autochthonous Mouse Model of Prostate Cancer. Am. J. Pathol. 2010, 177, 3180–3191. [Google Scholar] [CrossRef]
- Oh, H.Y.; Lee, E.J.; Yoon, S.; Chung, B.H.; Cho, K.S.; Hong, S.J. Cholesterol Level of Lipid Raft Microdomains Regulates Apoptotic Cell Death in Prostate Cancer Cells through EGFR-Mediated Akt and ERK Signal Transduction. Prostate 2007, 67, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.J.C.; Alves, G.; Viennois, E.; Bernard, S.; Communal, Y.; Sion, B.; Marceau, G.; Damon, C.; Mouzat, K.; Caira, F.; et al. Liver X Receptor Activation Downregulates AKT Survival Signaling in Lipid Rafts and Induces Apoptosis of Prostate Cancer Cells. Oncogene 2010, 29, 2712–2723. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.J.C.; Dufour, J.; Alves, G.; Viennois, E.; De Boussac, H.; Trousson, A.; Volle, D.H.; Caira, F.; Val, P.; Arnaud, P.; et al. Liver x Receptors Protect from Development of Prostatic Intra-Epithelial Neoplasia in Mice. PLoS Genet. 2013, 9, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Rambur, A.; Lours-Calet, C.; Beaudoin, C.; Buñay, J.; Vialat, M.; Mirouse, V.; Trousson, A.; Renaud, Y.; Lobaccaro, J.-M.A.; Baron, S.; et al. Sequential Ras/MAPK and PI3K/AKT/MTOR Pathways Recruitment Drives Basal Extrusion in the Prostate-like Gland of Drosophila. Nat. Commun. 2020, 11, 2300. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.D.; Cagan, R.L. Drosophila Lung Cancer Models Identify Trametinib plus Statin as Candidate Therapeutic. Cell Rep. 2016, 14, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Bangi, E.; Murgia, C.; Teague, A.G.S.; Sansom, O.J.; Cagan, R.L. Functional Exploration of Colorectal Cancer Genomes Using Drosophila. Nat. Commun. 2016, 7, 13615. [Google Scholar] [CrossRef] [PubMed]
- Kalb, J.M.; DiBenedetto, A.J.; Wolfner, M.F. Probing the Function of Drosophila Melanogaster Accessory Glands by Directed Cell Ablation. Proc. Natl. Acad. Sci. USA 1993, 90, 8093–8097. [Google Scholar] [CrossRef] [PubMed]
- Wolfner, M.F. Tokens of Love: Functions and Regulation of Drosophila Male Accessory Gland Products. Insect Biochem. Mol. Biol. 1997, 27, 179–192. [Google Scholar] [CrossRef]
- Rylett, C.M.; Walker, M.J.; Howell, G.J.; Shirras, A.D.; Isaac, R.E. Male Accessory Glands of Drosophila Melanogaster Make a Secreted Angiotensin I-Converting Enzyme (ANCE), Suggesting a Role for the Peptide-Processing Enzyme in Seminal Fluid. J. Exp. Biol. 2007, 210, 3601–3606. [Google Scholar] [CrossRef] [PubMed]
- Rewitz, K.F.; O’Connor, M.B.; Gilbert, L.I. Molecular Evolution of the Insect Halloween Family of Cytochrome P450s: Phylogeny, Gene Organization and Functional Conservation. Insect Biochem. Mol. Biol. 2007, 37, 741–753. [Google Scholar] [CrossRef]
- Sharma, V.; Pandey, A.K.; Kumar, A.; Misra, S.; Gupta, H.P.K.; Gupta, S.; Singh, A.; Buehner, N.A.; Ravi Ram, K. Functional Male Accessory Glands and Fertility in Drosophila Require Novel Ecdysone Receptor. PLoS Genet. 2017, 13, e1006788. [Google Scholar] [CrossRef] [PubMed]
- Molano-Fernández, M.; Hickson, I.D.; Herranz, H. Cyclin E Overexpression in the Drosophila Accessory Gland Induces Tissue Dysplasia. Front. Cell Dev. Biol. 2022, 10, 992253. [Google Scholar] [CrossRef] [PubMed]
- Rambur, A.; Vialat, M.; Beaudoin, C.; Lours-Calet, C.; Lobaccaro, J.-M.; Baron, S.; Morel, L.; de Joussineau, C. Drosophila Accessory Gland: A Complementary In Vivo Model to Bring New Insight to Prostate Cancer. Cells 2021, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Leiblich, A.; Goberdhan, D.C.I.; Hamdy, F. The Drosophila Accessory Gland as a Model for Prostate Cancer and Other Pathologies. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 121, pp. 339–375. [Google Scholar]
- Ito, S.; Ueda, T.; Ueno, A.; Nakagawa, H.; Taniguchi, H.; Kayukawa, N.; Miki, T. A Genetic Screen in Drosophila for Regulators of Human Prostate Cancer Progression. Biochem. Biophys. Res. Commun. 2014, 451, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Leiblich, A.; Wainwright, S.M.; Mendes, C.C.; Sarma, D.; Hellberg, J.E.E.U.; Gandy, C.; Goberdhan, D.C.I.; Hamdy, F.C.; Wilson, C. Rbf/E2F1 Control Growth and Endoreplication via Steroid-Independent Ecdysone Receptor Signalling in Drosophila Prostate-like Secondary Cells. PLoS Genet. 2023, 19, e1010815. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, E.T.; Moeller, M.E.; Yamanaka, N.; Ou, Q.; Laursen, J.M.; Soenderholm, C.; Zhuo, R.; Phelps, B.; Tang, K.; Zeng, J.; et al. A Drosophila Genome-Wide Screen Identifies Regulators of Steroid Hormone Production and Developmental Timing. Dev. Cell 2016, 37, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.; Schwudke, D.; Sampaio, J.L.; Palm, W.; Riezman, I.; Dey, G.; Gupta, G.D.; Mayor, S.; Riezman, H.; Shevchenko, A.; et al. Survival Strategies of a Sterol Auxotroph. Development 2010, 137, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Niwa, Y.S. The Fruit Fly Drosophila Melanogaster as a Model System to Study Cholesterol Metabolism and Homeostasis. Cholesterol 2011, 2011, 176802. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K. The Biological Synthesis of Cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef]
- Clark, A.J.; Block, K. The Absence of Sterol Synthesis in Insects. J. Biol. Chem. 1959, 234, 2578–2582. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Haenszel, W.; Kurihara, M. Studies of Japanese Migrants. I. Mortality from Cancer and Other Diseases among Japanese in the United States. J. Natl. Cancer Inst. 1968, 40, 43–68. [Google Scholar]
- Mostaghel, E.A.; Solomon, K.R.; Pelton, K.; Freeman, M.R.; Montgomery, R.B. Impact of Circulating Cholesterol Levels on Growth and Intratumoral Androgen Concentration of Prostate Tumors. PLoS ONE 2012, 7, e30062. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ruelcke, J.E.; Choi, E.; Sharpe, L.J.; Nassar, Z.D.; Bielefeldt-Ohmann, H.; Parat, M.-O.; Shah, A.; Francois, M.; Inder, K.L.; et al. Diet-Induced Hypercholesterolemia Promotes Androgen-Independent Prostate Cancer Metastasis via IQGAP1 and Caveolin-1. Oncotarget 2015, 6, 7438–7453. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, L.; Dai, T.; Pei, X.; Xia, L.; Zeng, G.; Ye, M.; Liu, K.; Zeng, F.; Han, W.; et al. SQLE Mediates Metabolic Reprogramming to Promote LN Metastasis in Castration-Resistant Prostate Cancer. Onco. Targets Ther. 2021, 14, 4285–4295. [Google Scholar] [CrossRef] [PubMed]
- Slattum, G.M.; Rosenblatt, J. TumourTumor Cell Invasion: An Emerging Role for Basal Epithelial Cell Extrusion. Nat. Rev. Cancer 2014, 14, 495–501. [Google Scholar] [CrossRef]
- Fadul, J.; Rosenblatt, J. The Forces and Fates of Extruding Cells. Curr. Opin. Cell Biol. 2018, 54, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Slattum, G.; Gu, Y.; Sabbadini, R.; Rosenblatt, J. Autophagy in Oncogenic K-Ras Promotes Basal Extrusion of Epithelial Cells by Degrading S1P. Curr. Biol. 2014, 24, 19–28. [Google Scholar] [CrossRef]
- Fadul, J.; Zulueta-Coarasa, T.; Slattum, G.M.; Redd, N.M.; Jin, M.F.; Redd, M.J.; Daetwyler, S.; Hedeen, D.; Huisken, J.; Rosenblatt, J. KRas-Transformed Epithelia Cells Invade and Partially Dedifferentiate by Basal Cell Extrusion. Nat. Commun. 2021, 12, 7180. [Google Scholar] [CrossRef]
- Shirai, T.; Sekai, M.; Kozawa, K.; Sato, N.; Tanimura, N.; Kon, S.; Matsumoto, T.; Murakami, T.; Ito, S.; Tilston-Lunel, A.; et al. Basal Extrusion of Single-Oncogenic Mutant Cells Induces Dome-like Structures with Altered Microenvironments. Cancer Sci. 2022, 113, 3710–3721. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Lu, J.; Sui, L.; Wang, D.; Yin, M.; Hoffmann, I.; Legler, A.; Pflugfelder, G.O. The Orthologous Tbx Transcription Factors Omb and TBX2 Induce Epithelial Cell Migration and Extrusion in Vivo without Involvement of Matrix Metalloproteinases. Oncotarget 2014, 5, 11998–12015. [Google Scholar] [CrossRef] [PubMed]
- Hendley, A.M.; Wang, Y.J.; Polireddy, K.; Alsina, J.; Ahmed, I.; Lafaro, K.J.; Zhang, H.; Roy, N.; Savidge, S.G.; Cao, Y.; et al. P120 Catenin Suppresses Basal Epithelial Cell Extrusion in Invasive Pancreatic Neoplasia. Cancer Res. 2016, 76, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Kim, J.; Adam, R.M.; Solomon, K.R.; Freeman, M.R. Cholesterol Targeting Alters Lipid Raft Composition and Cell Survival in Prostate Cancer Cells and Xenografts. J. Clin. Investig. 2005, 115, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C. V Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Graziani, S.R.; Igreja, F.A.F.; Hegg, R.; Meneghetti, C.; Brandizzi, L.I.; Barboza, R.; Amâncio, R.F.; Pinotti, J.A.; Maranhão, R.C. Uptake of a Cholesterol-Rich Emulsion by Breast Cancer. Gynecol. Oncol. 2002, 85, 493–497. [Google Scholar] [CrossRef]
- Bangi, E.; Ang, C.; Smibert, P.; Uzilov, A.V.; Teague, A.G.; Antipin, Y.; Chen, R.; Hecht, C.; Gruszczynski, N.; Yon, W.J.; et al. A Personalized Platform Identifies Trametinib plus Zoledronate for a Patient with KRAS-Mutant Metastatic Colorectal Cancer. Sci. Adv. 2019, 5, eaav6528. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vialat, M.; Baabdaty, E.; Trousson, A.; Kocer, A.; Lobaccaro, J.-M.A.; Baron, S.; Morel, L.; de Joussineau, C. Cholesterol Dietary Intake and Tumor Cell Homeostasis Drive Early Epithelial Tumorigenesis: A Potential Modelization of Early Prostate Tumorigenesis. Cancers 2024, 16, 2153. https://doi.org/10.3390/cancers16112153
Vialat M, Baabdaty E, Trousson A, Kocer A, Lobaccaro J-MA, Baron S, Morel L, de Joussineau C. Cholesterol Dietary Intake and Tumor Cell Homeostasis Drive Early Epithelial Tumorigenesis: A Potential Modelization of Early Prostate Tumorigenesis. Cancers. 2024; 16(11):2153. https://doi.org/10.3390/cancers16112153
Chicago/Turabian StyleVialat, Marine, Elissa Baabdaty, Amalia Trousson, Ayhan Kocer, Jean-Marc A. Lobaccaro, Silvère Baron, Laurent Morel, and Cyrille de Joussineau. 2024. "Cholesterol Dietary Intake and Tumor Cell Homeostasis Drive Early Epithelial Tumorigenesis: A Potential Modelization of Early Prostate Tumorigenesis" Cancers 16, no. 11: 2153. https://doi.org/10.3390/cancers16112153
APA StyleVialat, M., Baabdaty, E., Trousson, A., Kocer, A., Lobaccaro, J.-M. A., Baron, S., Morel, L., & de Joussineau, C. (2024). Cholesterol Dietary Intake and Tumor Cell Homeostasis Drive Early Epithelial Tumorigenesis: A Potential Modelization of Early Prostate Tumorigenesis. Cancers, 16(11), 2153. https://doi.org/10.3390/cancers16112153