The Role of Claudin-1 in Enhancing Pancreatic Cancer Aggressiveness and Drug Resistance via Metabolic Pathway Modulation

 ,
,  , , and
, , and
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Immunohistochemical Analysis of Surgical Specimens and Immunocytochemistry of Cell Blocks
2.3. Cell Culture and Treatment
2.4. Western Blotting
2.5. Cell Proliferation and Plate Colony Formation Assay
2.6. Wound-Healing, Migration, and Invasion Assays
2.7. Proteome Analysis
2.8. Immunocytochemistry
2.9. Cellular Fractionation and Co-Immunoprecipitation Assays
2.10. RNA Interference
2.11. Apoptosis Assay
2.12. Statistical Analysis
2.13. Ethics Statement and Patient Consent
3. Results
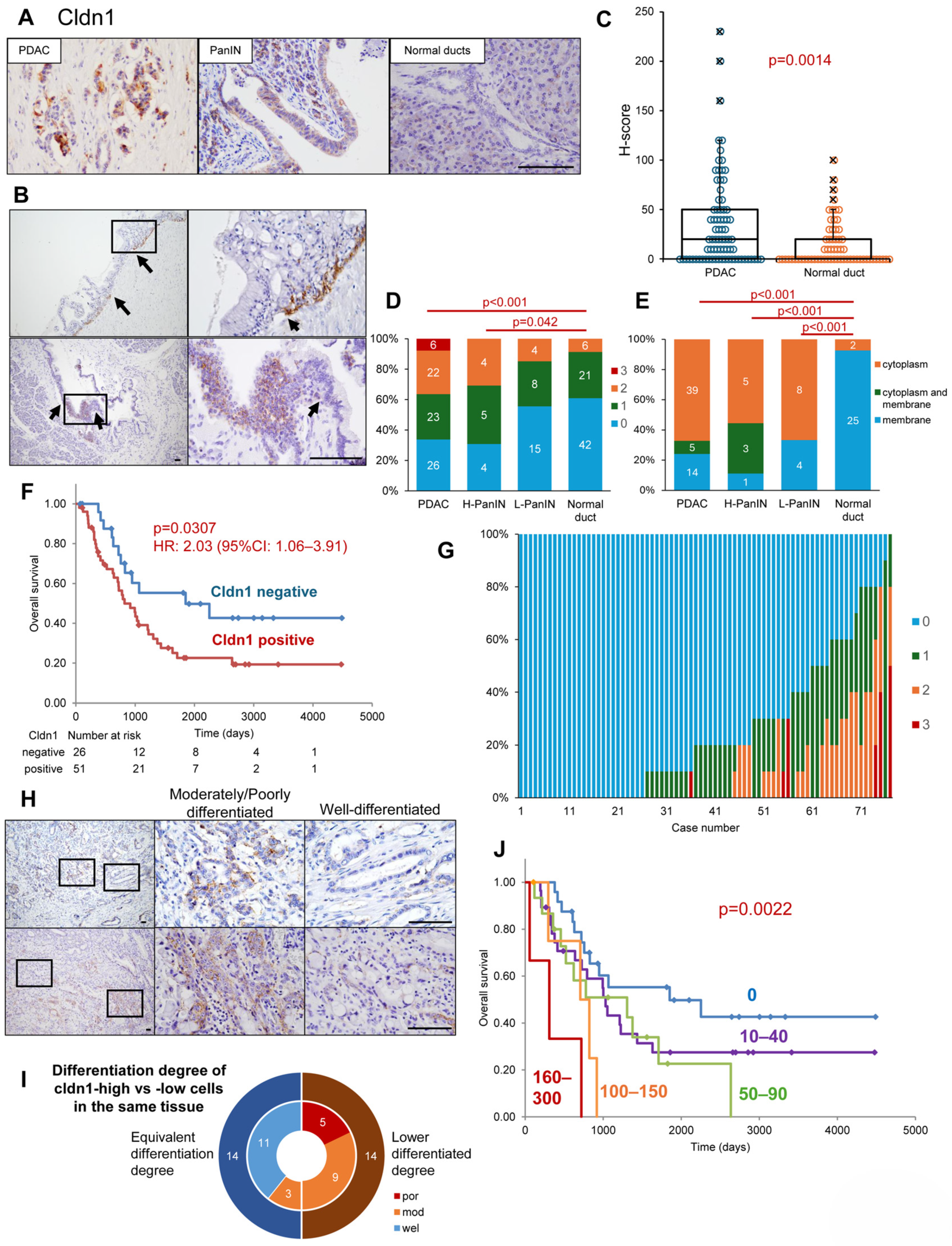
3.1. Expression Profiles of Cldn1 in Surgical Specimens of PDAC, PanIN, and Normal Ducts
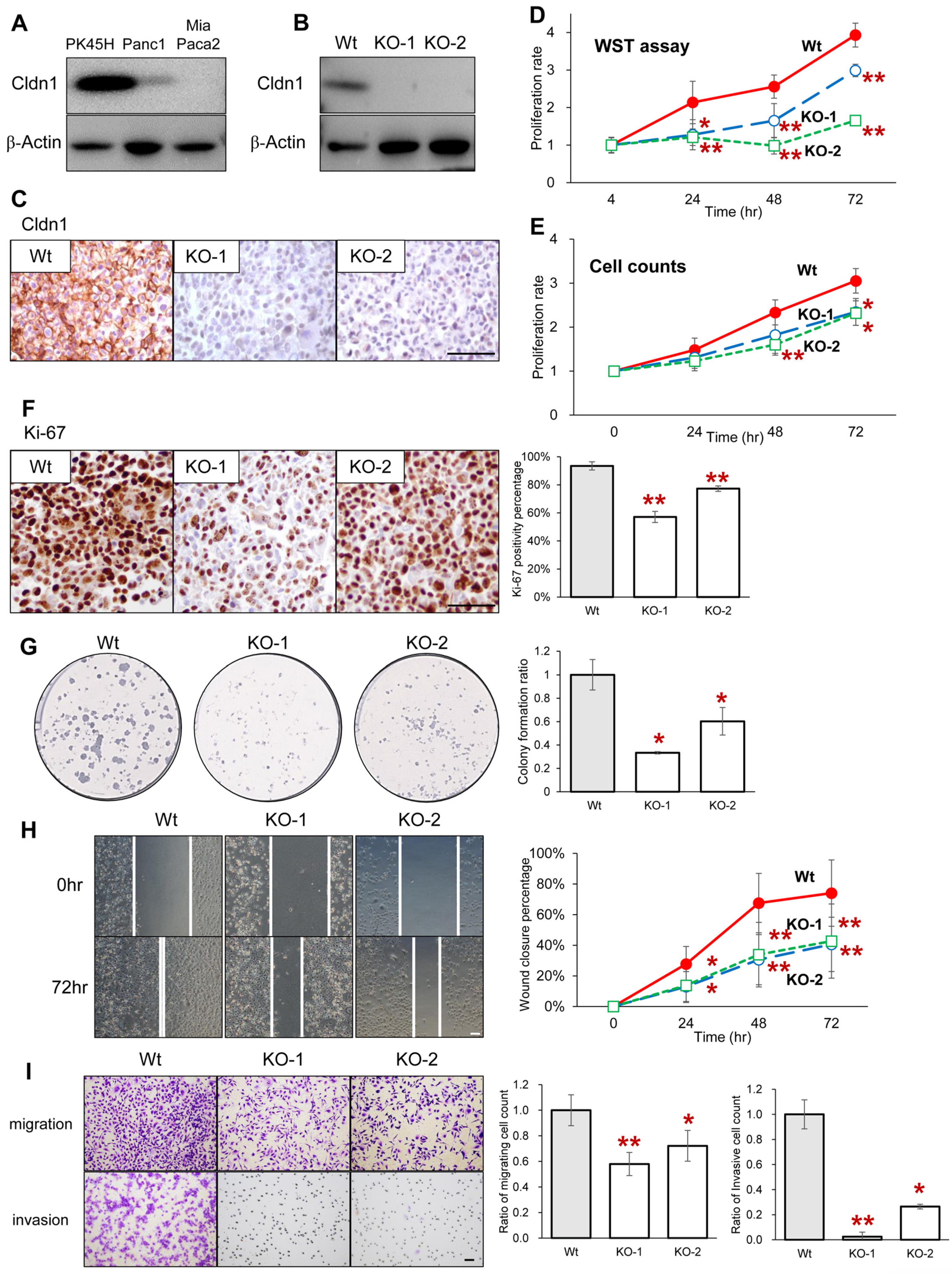
3.2. Cldn1 Contributes to the Proliferation, Migration, and Invasion of Pancreatic Cancer Cells
3.3. Comprehensive Proteome Analysis Showed AKRs as Candidates That Interact with Cldn1 in Pancreatic Cancer Cells
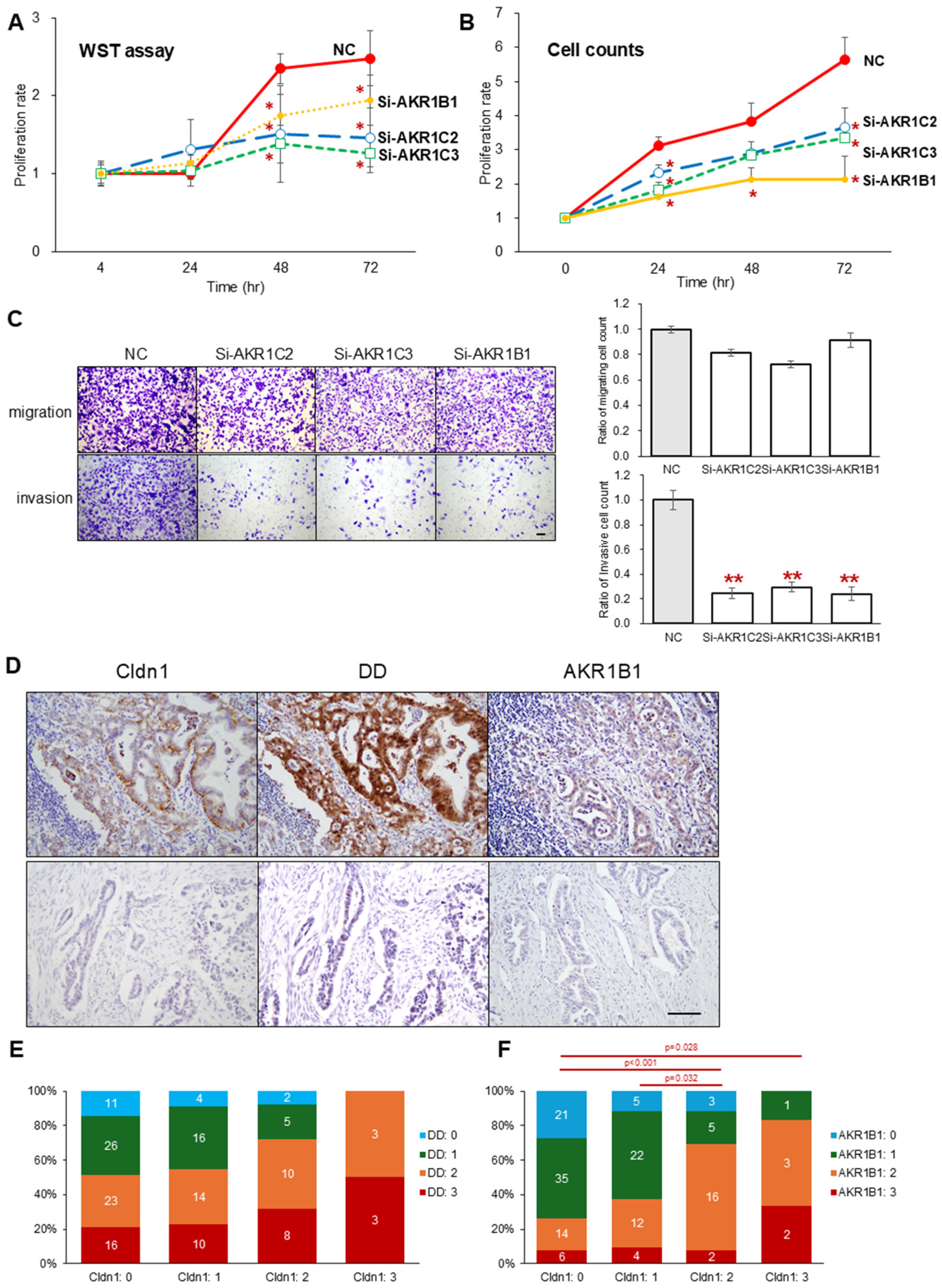
3.4. Cldn1 KO and AKR Superfamily Knockdown Attenuate Drug Resistance
3.5. Knockdown of the AKR Superfamily Also Attenuates the Proliferation and Invasion Abilities of PK45H Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | pancreatic ductal adenocarcinoma |
| Cldn1 | claudin-1 |
| AKR | aldo-keto reductase |
| EMT | epithelial-mesenchymal transition |
| DD | dihydrodiol dehydrogenase |
| H-score | histological score |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DAPI | 4,6-diamidino-2-phenylindole |
References
- Bengtsson, A.; Andersson, R.; Ansari, D. The Actual 5-Year Survivors of Pancreatic Ductal Adenocarcinoma Based on Real-World Data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Cancer Information Service, National Cancer Center, Japan. Annual Report of Hospital-Based Cancer Registries. Available online: https://hbcr-survival.ganjoho.jp/ (accessed on 2 July 2024).
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic Developments in Pancreatic Cancer: Current and Future Perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the Outcomes of Pancreatic Cancer Surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Blackford, A.L.; Canto, M.I.; Klein, A.P.; Hruban, R.H.; Goggins, M. Recent Trends in the Incidence and Survival of Stage 1A Pancreatic Cancer: A Surveillance, Epidemiology, and End Results Analysis. J. Natl. Cancer Inst. 2020, 112, 1162–1169. [Google Scholar] [CrossRef]
- Ducreux, M.; Seufferlein, T.; Van Laethem, J.-L.; Laurent-Puig, P.; Smolenschi, C.; Malka, D.; Boige, V.; Hollebecque, A.; Conroy, T. Systemic Treatment of Pancreatic Cancer Revisited. Semin. Oncol. 2019, 46, 28–38. [Google Scholar] [CrossRef]
- Furuse, M.; Tsukita, S. Claudins in Occluding Junctions of Humans and Flies. Trends Cell Biol. 2006, 16, 181–188. [Google Scholar] [CrossRef]
- Magara, K.; Takasawa, A.; Takasawa, K.; Aoyama, T.; Ota, M.; Kyuno, D.; Ono, Y.; Murakami, T.; Yamamoto, S.; Nakamori, Y.; et al. Multilayered Proteomics Reveals That JAM-A Promotes Breast Cancer Progression via Regulation of Amino Acid Transporter LAT1. Cancer Sci. 2024, 115, 3153–3168. [Google Scholar] [CrossRef]
- Huang, Y.-T.; Hsu, Y.-T.; Wu, P.-Y.; Yeh, Y.-M.; Lin, P.-C.; Hsu, K.-F.; Shen, M.-R. Tight Junction Protein Cingulin Variant Is Associated with Cancer Susceptibility by Overexpressed IQGAP1 and Rac1-Dependent Epithelial-Mesenchymal Transition. J. Exp. Clin. Cancer Res. 2024, 43, 65. [Google Scholar] [CrossRef]
- Tabariès, S.; Robert, A.; Marcil, A.; Ling, B.; Acchione, M.; Lippens, J.; Pagé, M.; Fortin, A.; Meury, L.; Coutu, M.; et al. Anti-Claudin-2 Antibody-Drug Conjugates for the Treatment of Colorectal Cancer Liver Metastasis. Mol. Cancer Ther. 2024, 23, 1459–1470. [Google Scholar] [CrossRef]
- Tabariès, S.; Dupuy, F.; Dong, Z.; Monast, A.; Annis, M.G.; Spicer, J.; Ferri, L.E.; Omeroglu, A.; Basik, M.; Amir, E.; et al. Claudin-2 Promotes Breast Cancer Liver Metastasis by Facilitating Tumor Cell Interactions with Hepatocytes. Mol. Cell. Biol. 2012, 32, 2979–2991. [Google Scholar] [CrossRef]
- Medrano-Gonzálezl, P.A.; Cruz-Villegas, F.; Alarcón Del Carmen, A.; Montaño, L.F.; Rendón-Huerta, E.P. Claudin-6 Increases SNAI1, NANOG and SOX2 Gene Expression in Human Gastric Adenocarcinoma AGS Cells. Mol. Biol. Rep. 2022, 49, 11663–11674. [Google Scholar] [CrossRef]
- Ito, Y.; Takasawa, A.; Takasawa, K.; Murakami, T.; Akimoto, T.; Kyuno, D.; Kawata, Y.; Shano, K.; Kirisawa, K.; Ota, M.; et al. Aberrant Expression of Claudin-6 Contributes to Malignant Potentials and Drug Resistance of Cervical Adenocarcinoma. Cancer Sci. 2022, 113, 1519–1530. [Google Scholar] [CrossRef]
- Takasawa, A.; Murata, M.; Takasawa, K.; Ono, Y.; Osanai, M.; Tanaka, S.; Nojima, M.; Kono, T.; Hirata, K.; Kojima, T.; et al. Nuclear Localization of Tricellulin Promotes the Oncogenic Property of Pancreatic Cancer. Sci. Rep. 2016, 6, 33582. [Google Scholar] [CrossRef]
- Takasawa, A.; Takasawa, K.; Murata, M.; Osanai, M.; Sawada, N. Emerging Roles of Transmembrane-Type Tight Junction Proteins in Cancers. Pathol. Int. 2023, 73, 331–340. [Google Scholar] [CrossRef]
- Zhou, B.; Moodie, A.; Blanchard, A.A.A.; Leygue, E.; Myal, Y. Claudin 1 in Breast Cancer: New Insights. J. Clin. Med. Res. 2015, 4, 1960–1976. [Google Scholar] [CrossRef]
- Takasawa, K.; Takasawa, A.; Akimoto, T.; Magara, K.; Aoyama, T.; Kitajima, H.; Murakami, T.; Ono, Y.; Kyuno, D.; Suzuki, H.; et al. Regulatory Roles of Claudin-1 in Cell Adhesion and Microvilli Formation. Biochem. Biophys. Res. Commun. 2021, 565, 36–42. [Google Scholar] [CrossRef]
- Bhat, A.A.; Syed, N.; Therachiyil, L.; Nisar, S.; Hashem, S.; Macha, M.A.; Yadav, S.K.; Krishnankutty, R.; Muralitharan, S.; Al-Naemi, H.; et al. Claudin-1, A Double-Edged Sword in Cancer. Int. J. Mol. Sci. 2020, 21, 569. [Google Scholar] [CrossRef]
- Wang, C.; Wu, N.; Pei, B.; Ma, X.; Yang, W. Claudin and Pancreatic Cancer. Front. Oncol. 2023, 13, 1136227. [Google Scholar] [CrossRef]
- Łukaszewicz-Zając, M.; Mroczko, B. Claudins-Promising Biomarkers for Selected Gastrointestinal (GI) Malignancies? Cancers 2023, 16, 152. [Google Scholar] [CrossRef]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours; John Wiley & Sons: Hoboken, NJ, USA, 2017; ISBN 9781119263579. [Google Scholar]
- Japan Pancreas Society (Ed.) General Rules for the Study of Pancreatic Cancer, 8th ed.; Kanehara & Co, Ltd.: Tokyo, Japan, 2023. [Google Scholar]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for Designing CRISPR/Cas Guide RNA with Reduced off-Target Sites. Bioinformatics 2015, 31, 1120–1123. [Google Scholar] [CrossRef]
- Arlt, A.; Vorndamm, J.; Breitenbroich, M.; Fölsch, U.R.; Kalthoff, H.; Schmidt, W.E.; Schäfer, H. Inhibition of NF-KappaB Sensitizes Human Pancreatic Carcinoma Cells to Apoptosis Induced by Etoposide (VP16) or Doxorubicin. Oncogene 2001, 20, 859–868. [Google Scholar] [CrossRef]
- Zhang, S.-H.; Huang, Q. Etoposide Induces Apoptosis via the Mitochondrial- and Caspase-Dependent Pathways and in Non-Cancer Stem Cells in Panc-1 Pancreatic Cancer Cells. Oncol. Rep. 2013, 30, 2765–2770. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]
- Ohgane, K.; Yoshioka, H. Quantification of Gel Bands by an Image J Macro, Band/Peak Quantification Tool. protocols.io 2019. [Google Scholar] [CrossRef]
- Brosseau, N.; Andreev, E.; Ramotar, D. Uptake Assays to Monitor Anthracyclines Entry into Mammalian Cells. Bio Protoc. 2017, 7, e2555. [Google Scholar] [CrossRef]
- Montagnani Marelli, M.; Manea, M.; Moretti, R.M.; Marzagalli, M.; Limonta, P. Oxime Bond-Linked Daunorubicin-GnRH-III Bioconjugates Exert Antitumor Activity in Castration-Resistant Prostate Cancer Cells via the Type I GnRH Receptor. Int. J. Oncol. 2015, 46, 243–253. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the Freely Available Easy-to-Use Software “EZR” for Medical Statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Posta, M.; Győrffy, B. Analysis of a Large Cohort of Pancreatic Cancer Transcriptomic Profiles to Reveal the Strongest Prognostic Factors. Clin. Transl. Sci. 2023, 16, 1479–1491. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef]
- Penning, T.M.; Jonnalagadda, S.; Trippier, P.C.; Rizner, T.L. Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol. Rev. 2021, 73, 1150–1171. [Google Scholar] [CrossRef]
- Wang, D.-W.; Zhang, W.-H.; Danil, G.; Yang, K.; Hu, J.-K. The Role and Mechanism of Claudins in Cancer. Front. Oncol. 2022, 12, 1051497. [Google Scholar] [CrossRef]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of Tight Junctions in the Epithelial-to-Mesenchymal Transition of Cancer Cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Primeaux, M.; Liu, X.; Gowrikumar, S.; Fatima, I.; Fisher, K.W.; Bastola, D.; Vecchio, A.J.; Singh, A.B.; Dhawan, P. Claudin-1 Interacts with EPHA2 to Promote Cancer Stemness and Chemoresistance in Colorectal Cancer. Cancer Lett. 2023, 579, 216479. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Zhou, S.; Liu, X.; Li, G.; Song, B.; Xu, W. Claudin-1/4 as Directly Target Gene of HIF-1α Can Feedback Regulating HIF-1α by PI3K-AKT-MTOR and Impact the Proliferation of Esophageal Squamous Cell Though Rho GTPase and p-JNK Pathway. Cancer Gene Ther. 2022, 29, 665–682. [Google Scholar] [CrossRef]
- Yamamoto, D.; Kayamori, K.; Sakamoto, K.; Tsuchiya, M.; Ikeda, T.; Harada, H.; Yoda, T.; Watabe, T.; Hara-Yokoyama, M. Intracellular Claudin-1 at the Invasive Front of Tongue Squamous Cell Carcinoma Is Associated with Lymph Node Metastasis. Cancer Sci. 2020, 111, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Blanchard, A.; Wang, N.; Ma, X.; Han, J.; Schroedter, I.; Leygue, E.; Myal, Y. Claudin 1 Promotes Migration and Increases Sensitivity to Tamoxifen and Anticancer Drugs in Luminal-like Human Breast Cancer Cells MCF7. Cancer Investig. 2015, 33, 429–439. [Google Scholar] [CrossRef]
- Suh, Y.; Yoon, C.-H.; Kim, R.-K.; Lim, E.-J.; Oh, Y.S.; Hwang, S.-G.; An, S.; Yoon, G.; Gye, M.C.; Yi, J.-M.; et al. Claudin-1 Induces Epithelial-Mesenchymal Transition through Activation of the c-Abl-ERK Signaling Pathway in Human Liver Cells. Oncogene 2013, 32, 4873–4882. [Google Scholar] [CrossRef]
- French, A.D.; Fiori, J.L.; Camilli, T.C.; Leotlela, P.D.; O’Connell, M.P.; Frank, B.P.; Subaran, S.; Indig, F.E.; Taub, D.D.; Weeraratna, A.T. PKC and PKA Phosphorylation Affect the Subcellular Localization of Claudin-1 in Melanoma Cells. Int. J. Med. Sci. 2009, 6, 93–101. [Google Scholar] [CrossRef]
- Chang, J.W.; Seo, S.T.; Im, M.A.; Won, H.-R.; Liu, L.; Oh, C.; Jin, Y.L.; Piao, Y.; Kim, H.J.; Kim, J.T.; et al. Claudin-1 Mediates Progression by Regulating EMT through AMPK/TGF-β Signaling in Head and Neck Squamous Cell Carcinoma. Transl. Res. 2022, 247, 58–78. [Google Scholar] [CrossRef] [PubMed]
- Fatima, I.; Uppada, J.P.; Chhonker, Y.S.; Gowrikumar, S.; Barman, S.; Roy, S.; Tolentino, K.T.; Palermo, N.; Natarajan, A.; Beauchamp, D.R.; et al. Identification and Characterization of a First-Generation Inhibitor of Claudin-1 in Colon Cancer Progression and Metastasis. Biomed. Pharmacother. 2023, 159, 114255. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Hashimoto, S.; Eguchi, H.; Morikawa, Y.; Suenami, K.; Yoshino, Y.; Matsunaga, T.; Endo, S.; Ikari, A. Enhancement of Chemoresistance by Claudin-1-Mediated Formation of Amino Acid Barriers in Human Lung Adenocarcinoma A549 Cells. Arch. Biochem. Biophys. 2024, 759, 110106. [Google Scholar] [CrossRef]
- Nagaoka, Y.; Oshiro, K.; Yoshino, Y.; Matsunaga, T.; Endo, S.; Ikari, A. Activation of the TGF-Β1/EMT Signaling Pathway by Claudin-1 Overexpression Reduces Doxorubicin Sensitivity in Small Cell Lung Cancer SBC-3 Cells. Arch. Biochem. Biophys. 2024, 751, 109824. [Google Scholar] [CrossRef]
- Kondo, J.; Sato, F.; Kusumi, T.; Liu, Y.; Motonari, O.; Sato, T.; Kijima, H. Claudin-1 Expression Is Induced by Tumor Necrosis Factor-Alpha in Human Pancreatic Cancer Cells. Int. J. Mol. Med. 2008, 22, 645–649. [Google Scholar]
- Zhu, L.; Tang, N.; Hang, H.; Zhou, Y.; Dong, J.; Yang, Y.; Mao, L.; Qiu, Y.; Fu, X.; Cao, W. Loss of Claudin-1 Incurred by DNMT Aberration Promotes Pancreatic Cancer Progression. Cancer Lett. 2024, 586, 216611. [Google Scholar] [CrossRef]
- Myal, Y.; Leygue, E.; Blanchard, A.A. Claudin 1 in Breast Tumorigenesis: Revelation of a Possible Novel “Claudin High” Subset of Breast Cancers. J. Biomed. Biotechnol. 2010, 2010, 956897. [Google Scholar] [CrossRef] [PubMed]
- Nagini, S.; Kallamadi, P.R.; Tanagala, K.K.K.; Reddy, G.B. Aldo-Keto Reductases: Role in Cancer Development and Theranostics. Oncol. Res. 2024, 32, 1287–1308. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, Y.; Song, J.; Sun, D.; Zhang, Y.-Y. Function, Drug Resistance and Prognostic Effect of AKR1C2 in Human Cancer. Neoplasma 2023, 70, 319–332. [Google Scholar] [CrossRef]
- Chang, T.S.; Lin, H.K.; Rogers, K.A.; Brame, L.S.; Yeh, M.M.; Yang, Q.; Fung, K.M. Expression of Aldo-Keto Reductase Family 1 Member C3 (AKR1C3) in Neuroendocrine Tumors & Adenocarcinomas of Pancreas, Gastrointestinal Tract, and Lung. Int. J. Clin. Exp. Pathol. 2013, 6, 2419–2429. [Google Scholar] [PubMed]
- Khayami, R.; Hashemi, S.R.; Kerachian, M.A. Role of Aldo-Keto Reductase Family 1 Member B1 (AKR1B1) in the Cancer Process and Its Therapeutic Potential. J. Cell. Mol. Med. 2020, 24, 8890–8902. [Google Scholar] [CrossRef]
- Shirato, A.; Kikugawa, T.; Miura, N.; Tanji, N.; Takemori, N.; Higashiyama, S.; Yokoyama, M. Cisplatin Resistance by Induction of Aldo-Keto Reductase Family 1 Member C2 in Human Bladder Cancer Cells. Oncol. Lett. 2014, 7, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Kuang, P.; Zhou, C.; Li, X.; Ren, S.; Li, B.; Wang, Y.; Li, J.; Tang, L.; Zhang, J.; Zhao, Y. Proteomics-Based Identification of Secreted Protein Dihydrodiol Dehydrogenase 2 as a Potential Biomarker for Predicting Cisplatin Efficacy in Advanced NSCLC Patients. Lung Cancer 2012, 77, 427–432. [Google Scholar] [CrossRef]
- Deng, H.B.; Adikari, M.; Parekh, H.K.; Simpkins, H. Ubiquitous Induction of Resistance to Platinum Drugs in Human Ovarian, Cervical, Germ-Cell and Lung Carcinoma Tumor Cells Overexpressing Isoforms 1 and 2 of Dihydrodiol Dehydrogenase. Cancer Chemother. Pharmacol. 2004, 54, 301–307. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Huang, T.-J.; Zhang, X.-K.; Xie, Y.-J.; Lin, S.-T.; Luo, F.-F.; Meng, D.-F.; Hu, H.; Wang, J.; Peng, L.-X.; et al. AKR1C2 Acts as a Targetable Oncogene in Esophageal Squamous Cell Carcinoma via Activating PI3K/AKT Signaling Pathway. J. Cell. Mol. Med. 2020, 24, 9999–10012. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.; Zhang, W.; Li, J.; Liu, H.; Hao, M.; Wang, J.; Zhang, H.; Yang, G.; Hao, M.; et al. AEG-1 Promotes Metastasis through Downstream AKR1C2 and NF1 in Liver Cancer. Oncol. Res. 2014, 22, 203–211. [Google Scholar] [CrossRef]
- Pippione, A.C.; Kovachka, S.; Vigato, C.; Bertarini, L.; Mannella, I.; Sainas, S.; Rolando, B.; Denasio, E.; Piercy-Mycock, H.; Romalho, L.; et al. Structure-Guided Optimization of 3-Hydroxybenzoisoxazole Derivatives as Inhibitors of Aldo-Keto Reductase 1C3 (AKR1C3) to Target Prostate Cancer. Eur. J. Med. Chem. 2024, 268, 116193. [Google Scholar] [CrossRef]
- Yamashita, N.; Kanno, Y.; Saito, N.; Terai, K.; Sanada, N.; Kizu, R.; Hiruta, N.; Park, Y.; Bujo, H.; Nemoto, K. Aryl Hydrocarbon Receptor Counteracts Pharmacological Efficacy of Doxorubicin via Enhanced AKR1C3 Expression in Triple Negative Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2019, 516, 693–698. [Google Scholar] [CrossRef]
- Maddeboina, K.; Jonnalagadda, S.K.; Morsy, A.; Duan, L.; Chhonker, Y.S.; Murry, D.J.; Penning, T.M.; Trippier, P.C. Aldo-Keto Reductase 1C3 Inhibitor Prodrug Improves Pharmacokinetic Profile and Demonstrates in Vivo Efficacy in a Prostate Cancer Xenograft Model. J. Med. Chem. 2023, 66, 9894–9915. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, X.; Fu, Q.; Cao, Q.; Chen, X.; Wang, M.; Yu, J.; Long, J.; Yao, J.; Liu, H.; et al. AKR1B1 Promotes Basal-like Breast Cancer Progression by a Positive Feedback Loop That Activates the EMT Program. J. Exp. Med. 2017, 214, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Xu, M.-X.; Qian, T.-Y.; Zhu, S.-Z.; Jiang, F.; Liu, Z.-X.; Xu, W.-S.; Zhou, J.; Xiao, M.-B. The AKR1B1 Inhibitor Epalrestat Suppresses the Progression of Cervical Cancer. Mol. Biol. Rep. 2020, 47, 6091–6103. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, L.; Cheng, Z.; Sun, Y.; Zhou, Y.; Cao, J. Aberrant Expression of AKR1B1 Indicates Poor Prognosis and Promotes Gastric Cancer Progression by Regulating the AKT-MTOR Pathway. Aging 2023, 15, 9661–9675. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.B.; Jin, D.D.; Jiao, Y.J.; Ni, W.K.; Liu, J.X.; Qu, L.S.; Lu, C.H.; Ni, R.Z.; Jiang, F.; Chen, W.C. Beta2-AR Regulates the Expression of AKR1B1 in Human Pancreatic Cancer Cells and Promotes Their Proliferation via the ERK1/2 Pathway. Mol. Biol. Rep. 2018, 45, 1863–1871. [Google Scholar] [CrossRef]
- Daunys, S.; Matulis, D.; Petrikaitė, V. Synergistic Activity of Hsp90 Inhibitors and Anticancer Agents in Pancreatic Cancer Cell Cultures. Sci. Rep. 2019, 9, 16177. [Google Scholar] [CrossRef]
- Hofman, J.; Malcekova, B.; Skarka, A.; Novotna, E.; Wsol, V. Anthracycline Resistance Mediated by Reductive Metabolism in Cancer Cells: The Role of Aldo-Keto Reductase 1C3. Toxicol. Appl. Pharmacol. 2014, 278, 238–248. [Google Scholar] [CrossRef]
- Yadav, U.C.S.; Naura, A.S.; Aguilera-Aguirre, L.; Boldogh, I.; Boulares, H.A.; Calhoun, W.J.; Ramana, K.V.; Srivastava, S.K. Aldose Reductase Inhibition Prevents Allergic Airway Remodeling through PI3K/AKT/GSK3β Pathway in Mice. PLoS ONE 2013, 8, e57442. [Google Scholar] [CrossRef]
- Pannequin, J.; Delaunay, N.; Darido, C.; Maurice, T.; Crespy, P.; Frohman, M.A.; Balda, M.S.; Matter, K.; Joubert, D.; Bourgaux, J.-F.; et al. Phosphatidylethanol Accumulation Promotes Intestinal Hyperplasia by Inducing ZONAB-Mediated Cell Density Increase in Response to Chronic Ethanol Exposure. Mol. Cancer Res. 2007, 5, 1147–1157. [Google Scholar] [CrossRef]
- Zeng, C.M.; Chang, L.L.; Ying, M.D.; Cao, J.; He, Q.J.; Zhu, H.; Yang, B. Aldo-Keto Reductase AKR1C1-AKR1C4: Functions, Regulation, and Intervention for Anti-Cancer Therapy. Front. Pharmacol. 2017, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.D.; Zhang, Y. Regulation of Aldo-Keto Reductases in Human Diseases. Front. Pharmacol. 2012, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Zhao, Y.; He, Y.; Deng, Y.; Gan, X.; Li, P.; Chen, X.; Ding, Z. Exosomal MiR-184 Facilitates Bladder Cancer Progression by Targeting AKR1C3 and Inducing Immune Escape via IRF2-CXCL10 Axis. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167627. [Google Scholar] [CrossRef]
- Fukumoto, I.; Kinoshita, T.; Hanazawa, T.; Kikkawa, N.; Chiyomaru, T.; Enokida, H.; Yamamoto, N.; Goto, Y.; Nishikawa, R.; Nakagawa, M.; et al. Identification of Tumour Suppressive MicroRNA-451a in Hypopharyngeal Squamous Cell Carcinoma Based on MicroRNA Expression Signature. Br. J. Cancer 2014, 111, 386–394. [Google Scholar] [CrossRef]
- Majer, A.; Blanchard, A.A.; Medina, S.; Booth, S.A.; Myal, Y. Claudin 1 Expression Levels Affect MiRNA Dynamics in Human Basal-like Breast Cancer Cells. DNA Cell Biol. 2016, 35, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Huang, C.; Cui Zhou, D.; Hu, Y.; Lih, T.M.; Savage, S.R.; Krug, K.; Clark, D.J.; Schnaubelt, M.; Chen, L.; et al. Proteogenomic Characterization of Pancreatic Ductal Adenocarcinoma. Cell 2021, 184, 5031–5052.e26. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-Exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Univariate Analysis | Multivariate Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variables | Hazard Ratio | 95%CI (Lower) | 95%CI (Upper) | p-Value | Hazard Ratio | 95%CI (Lower) | 95%CI (Upper) | p-Value | ||
| Cldn1 positive | 2.03 | 1.06 | 3.91 | 0.033 | * | 2.21 | 1.13 | 4.32 | 0.021 | * |
| Male | 0.74 | 0.42 | 1.29 | 0.284 | ||||||
| Age > 65 | 1.04 | 0.58 | 1.88 | 0.890 | ||||||
| ASA: 3 | 1.21 | 0.48 | 3.06 | 0.684 | ||||||
| BMI > 25 | 1.09 | 0.54 | 2.18 | 0.816 | ||||||
| Tumor location: Head | 1.17 | 0.64 | 2.12 | 0.616 | ||||||
| Resectability: BR/UR | 1.44 | 0.82 | 2.55 | 0.208 | ||||||
| Neoadjuvant treatment | 1.14 | 0.63 | 2.06 | 0.655 | ||||||
| Poorly differentiated carcinoma | 1.21 | 0.51 | 2.84 | 0.669 | ||||||
| Lymphatic invasion | 1.38 | 0.77 | 2.48 | 0.275 | ||||||
| Vascular invasion | 1.18 | 0.66 | 2.11 | 0.585 | ||||||
| Neural invasion | 0.93 | 0.46 | 1.87 | 0.843 | ||||||
| Pleural invasion | 0.98 | 0.50 | 1.92 | 0.955 | ||||||
| Portal vein invasion | 0.90 | 0.45 | 1.80 | 0.761 | ||||||
| T-stage: pT3-4 | 1.74 | 0.89 | 3.40 | 0.108 | ||||||
| N-stage: pN1 | 1.98 | 1.08 | 3.63 | 0.027 | * | |||||
| TNM stage: pStage III/IV | 2.89 | 1.59 | 5.27 | <0.001 | ** | 2.85 | 1.50 | 5.42 | 0.001 | ** |
| High pre-operative CA19-9 (>37 U/mL) | 1.35 | 0.76 | 2.43 | 0.308 | ||||||
| High post-operative CA19-9 (>37 U/mL) | 3.66 | 1.96 | 6.85 | <0.001 | ** | 3.32 | 1.73 | 6.36 | <0.001 | ** |
| Accession | Protein Names | Gene Names | Ratio (KO-1/wt) | p Value (KO-1vs wt) | Ratio (KO-2/wt) | p Value (KO-2vs wt) |
|---|---|---|---|---|---|---|
| P47985 | Cytochrome b-c1 complex subunit Rieske, mitochondrial | UQCRFS1 | 0.001 | 3.8 × 10−5 | 0.001 | 3.8 × 10−5 |
| Q14697-2 | Isoform 2 of Neutral alpha-glucosidase AB | GANAB | 0.003 | 8.5 × 10−6 | 0.44 | 7.1 × 10−3 |
| P52895 | Aldo-keto reductase family 1 member C2 | AKR1C2 | 0.06 | 2.4 × 10−6 | 0.01 | 8.7 × 10−7 |
| P40261 | Nicotinamide N-methyltransferase | NNMT | 0.09 | 9.2 × 10−11 | 0.15 | 7.0 × 10−11 |
| O00469-1 | procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 | PLOD2 | 0.21 | 3.1 × 10−3 | 0.31 | 6.0 × 10−3 |
| P42330 | Aldo-keto reductase family 1 member C3 | AKR1C3 | 0.37 | 1.4 × 10−7 | 0.19 | 9.2 × 10−10 |
| P45954 | Short/branched chain specific acyl-CoA dehydrogenase, mitochondrial | ACADSB | 0.37 | 2.0 × 10−2 | 0.40 | 4.7 × 10−2 |
| O75828 | Carbonyl reductase [NADPH] 3 | CBR3 | 0.40 | 7.4 × 10−7 | 0.39 | 1.5 × 10−8 |
| Q02127 | Dihydroorotate dehydrogenase (Quinone), mitochondrial | DHODH | 0.46 | 3.0 × 10−2 | 0.31 | 2.6 × 10−3 |
| P48163 | NADP-dependent malic enzyme | ME1 | 0.49 | 2.6 × 10−5 | 0.34 | 4.1 × 10−5 |
| P08243-1 | Asparagine synthetase [glutamine-hydrolyzing] | ASNS | 0.50 | 2.5 × 10−8 | 0.54 | 1.6 × 10−7 |
| O14521 | Succinate dehydrogenase [ubiquinone] cytochrome b small subunit, mitochondrial | SDHD | 0.51 | 2.3 × 10−4 | 0.47 | 8.2 × 10−5 |
| Q15125 | 3-beta-hydroxysteroid-Delta(8), Delta(7)-isomerase | EBP | 0.54 | 9.6 × 10−5 | 0.60 | 5.7 × 10−4 |
| Q01581 | Hydroxymethylglutaryl-CoA synthase, cytoplasmic | HMGCS1 | 0.55 | 5.7 × 10−6 | 0.45 | 1.7 × 10−3 |
| P15121 | Aldose reductase | AKR1B1 | 0.56 | 1.2 × 10−8 | 0.31 | 3.0 × 10−7 |
| Q9BWD1 | Acetyl-CoA acetyltransferase, cytosolic | ACAT2 | 0.58 | 6.1 × 10−4 | 0.66 | 8.2 × 10−4 |
| P52788-1 | Spermine synthase | SMS | 0.58 | 1.3 × 10−4 | 0.43 | 5.3 × 10−7 |
| P30566 | Adenylosuccinate lyase | ADSL | 0.59 | 6.8 × 10−6 | 0.31 | 5.3 × 10−8 |
| P09601 | Heme oxygenase 1 | HMOX1 | 0.59 | 6.5 × 10−3 | 0.13 | 1.7 × 10−4 |
| P04181-1 | Ornithine aminotransferase, mitochondrial | OAT | 0.60 | 3.0 × 10−4 | 0.64 | 4.4 × 10−4 |
| P15531 | Nucleoside diphosphate kinase A | NME1 | 0.60 | 5.7 × 10−4 | 0.50 | 6.4 × 10−4 |
| O00330-1 | Pyruvate dehydrogenase protein X component, mitochondrial | PDHX | 0.60 | 2.1 × 10−6 | 0.66 | 1.4 × 10−3 |
| Q14693-1 | Phosphatidate phosphatase LPIN1 | LPIN1 | 0.61 | 2.8 × 10−3 | 0.39 | 2.7 × 10−4 |
| Q96EM0 | Trans-3-Hydroxy-L-proline dehydratase | L3HYPDH | 0.64 | 1.9 × 10−4 | 0.31 | 1.1 × 10−4 |
| P13674-1 | Prolyl 4-hydroxylase subunit alpha-1 | P4HA1 | 0.64 | 3.2 × 10−5 | 0.62 | 1.2 × 10−5 |
| Q969N2 | GPI transamidase component PIG-T | PIGT | 0.65 | 3.3 × 10−3 | 0.44 | 3.5 × 10−4 |
| P53701 | Cytochrome c-type heme lyase | HCCS | 0.65 | 8.1 × 10−4 | 0.38 | 4.2 × 10−5 |
| Q7L5N7 | Lysophosphatidylcholine acyltransferase 2 | LPCAT2 | 0.65 | 2.3 × 10−5 | 0.58 | 7.8 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyuno, D.; Asano, H.; Okumura, R.; Takasawa, K.; Takasawa, A.; Konno, T.; Nakamori, Y.; Magara, K.; Ono, Y.; Imamura, M.; et al. The Role of Claudin-1 in Enhancing Pancreatic Cancer Aggressiveness and Drug Resistance via Metabolic Pathway Modulation. Cancers 2025, 17, 1469. https://doi.org/10.3390/cancers17091469
Kyuno D, Asano H, Okumura R, Takasawa K, Takasawa A, Konno T, Nakamori Y, Magara K, Ono Y, Imamura M, et al. The Role of Claudin-1 in Enhancing Pancreatic Cancer Aggressiveness and Drug Resistance via Metabolic Pathway Modulation. Cancers. 2025; 17(9):1469. https://doi.org/10.3390/cancers17091469
Chicago/Turabian StyleKyuno, Daisuke, Hinae Asano, Reona Okumura, Kumi Takasawa, Akira Takasawa, Takumi Konno, Yuna Nakamori, Kazufumi Magara, Yusuke Ono, Masafumi Imamura, and et al. 2025. "The Role of Claudin-1 in Enhancing Pancreatic Cancer Aggressiveness and Drug Resistance via Metabolic Pathway Modulation" Cancers 17, no. 9: 1469. https://doi.org/10.3390/cancers17091469
APA StyleKyuno, D., Asano, H., Okumura, R., Takasawa, K., Takasawa, A., Konno, T., Nakamori, Y., Magara, K., Ono, Y., Imamura, M., Kimura, Y., Kojima, T., & Osanai, M. (2025). The Role of Claudin-1 in Enhancing Pancreatic Cancer Aggressiveness and Drug Resistance via Metabolic Pathway Modulation. Cancers, 17(9), 1469. https://doi.org/10.3390/cancers17091469