Understanding the Significance of Hypoxia-Inducible Factors (HIFs) in Glioblastoma: A Systematic Review

 ,
,  ,
,  , ,
, ,  , , , and
, , , and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Registration
2.2. Search Strategy
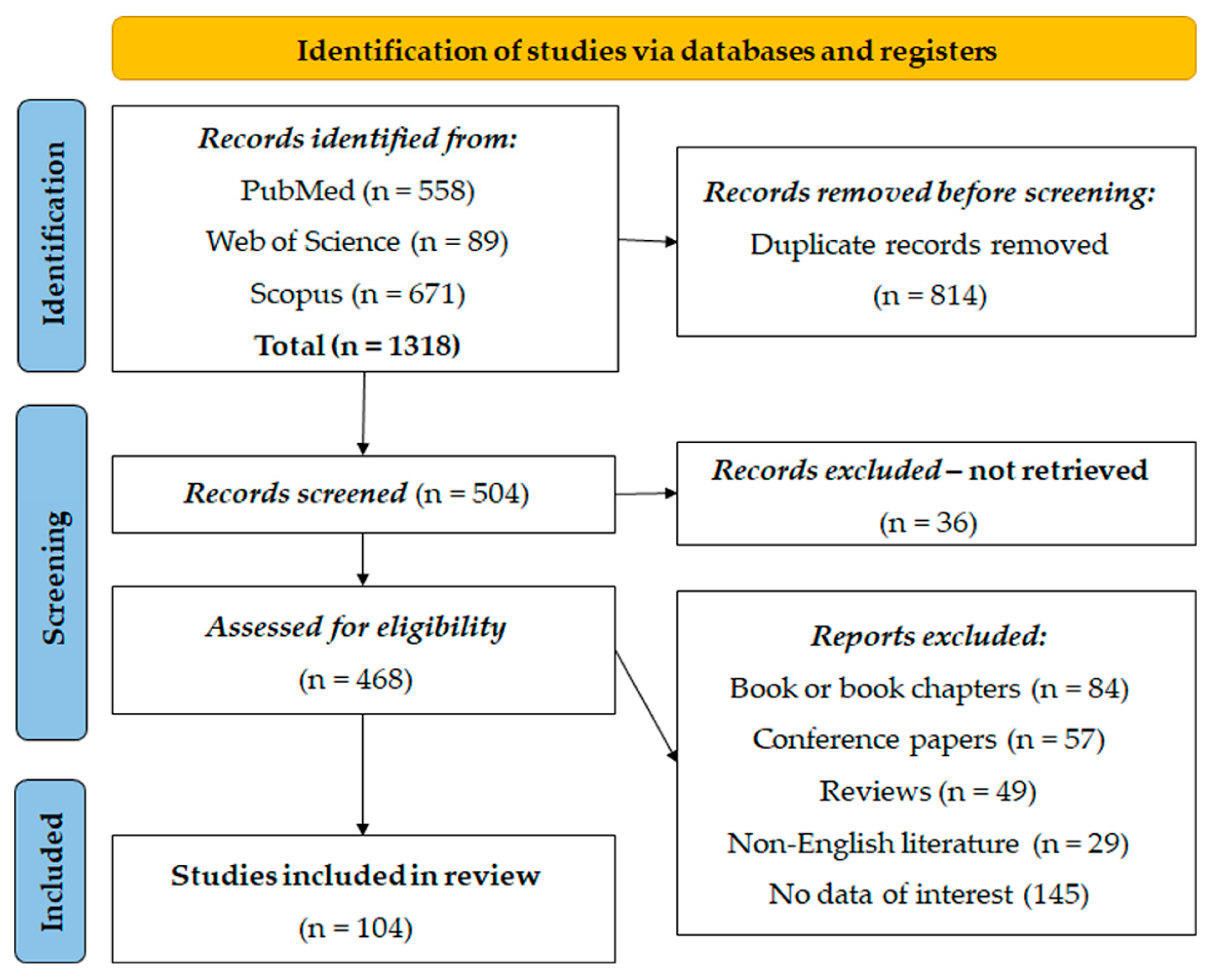
2.3. Study Selection
2.3.1. Inclusion and Exclusion Criteria
2.3.2. Included Studies
2.4. Data Extraction
2.5. Statistical Analysis
3. Results
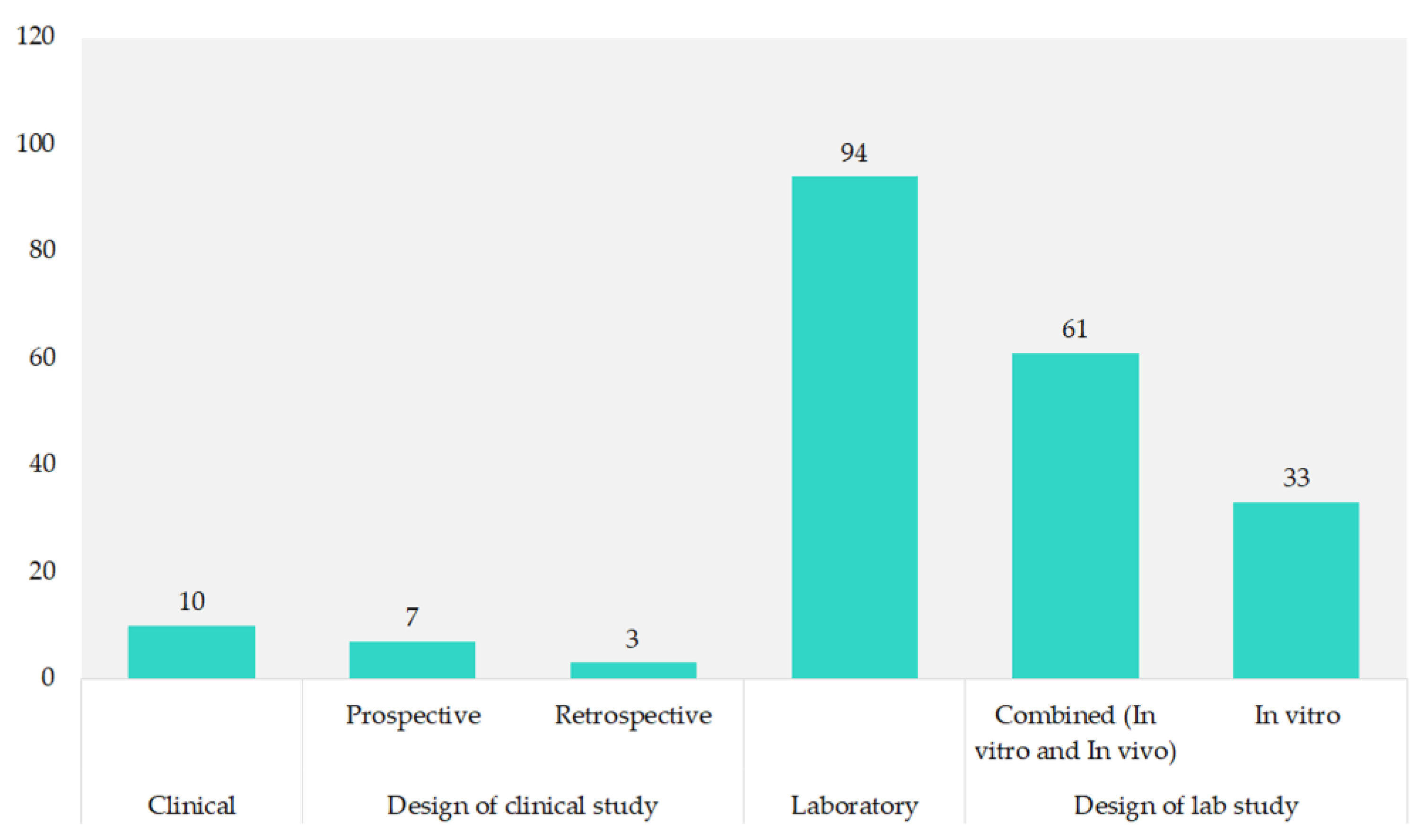
3.1. Included Studies’ Characteristics
3.2. Role of HIF-Related Gene Modification in the Treatment of Glioblastoma
3.2.1. HIF’s Mechanisms Explored in Genetic Studies
3.2.2. Effect of Gene Modifications Related to HIFs
3.3. Role of HIF-Related Targeted and Systematic Therapy of Glioblastoma
3.4. Role of Combined Gene and Targeted or Systematic Therapy of Glioblastoma
3.5. Role of HIFs in Clinal Studies of Glioblastoma
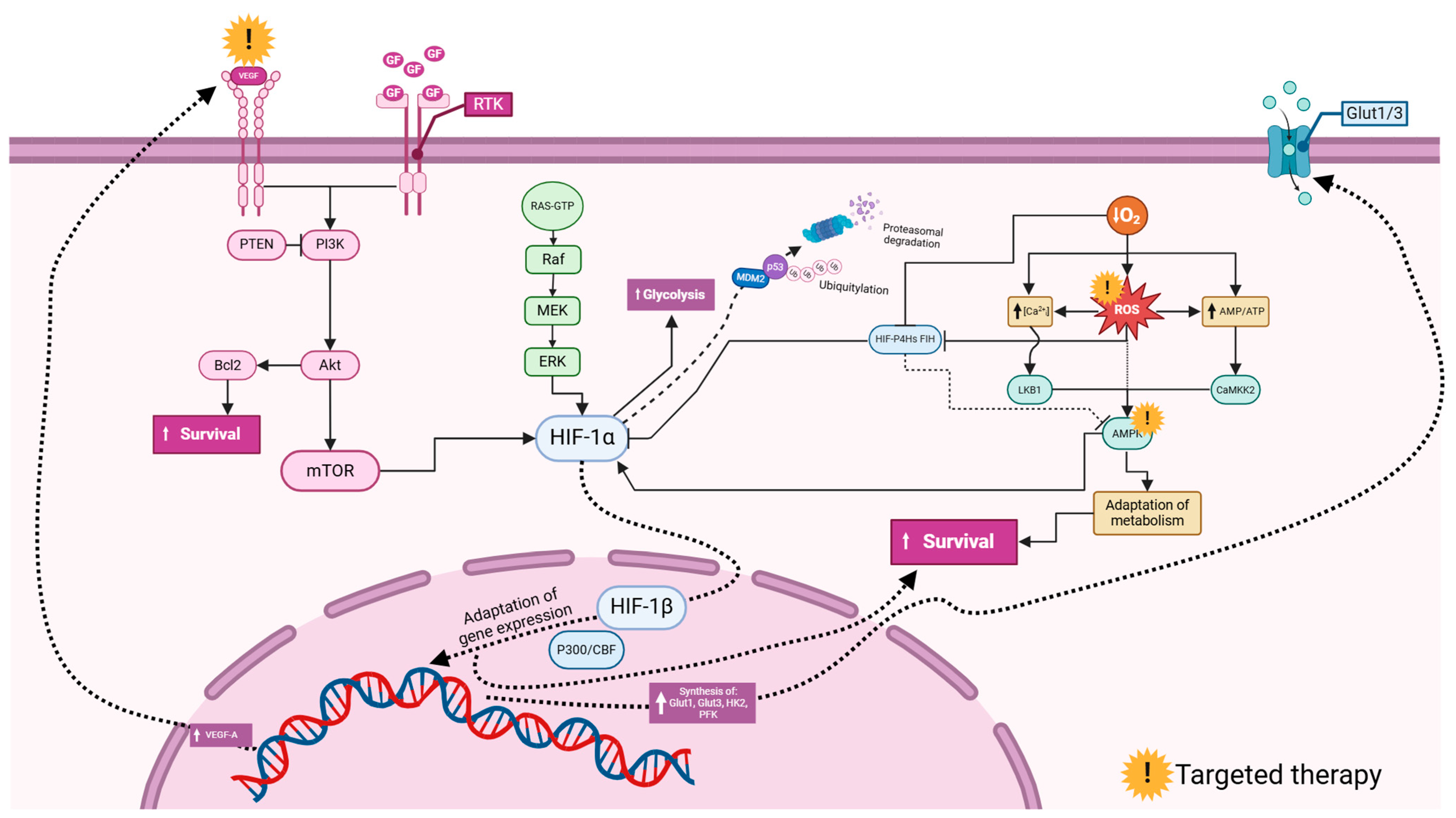
3.6. Common HIF-Related Pathways in Glioblastoma
4. Discussion
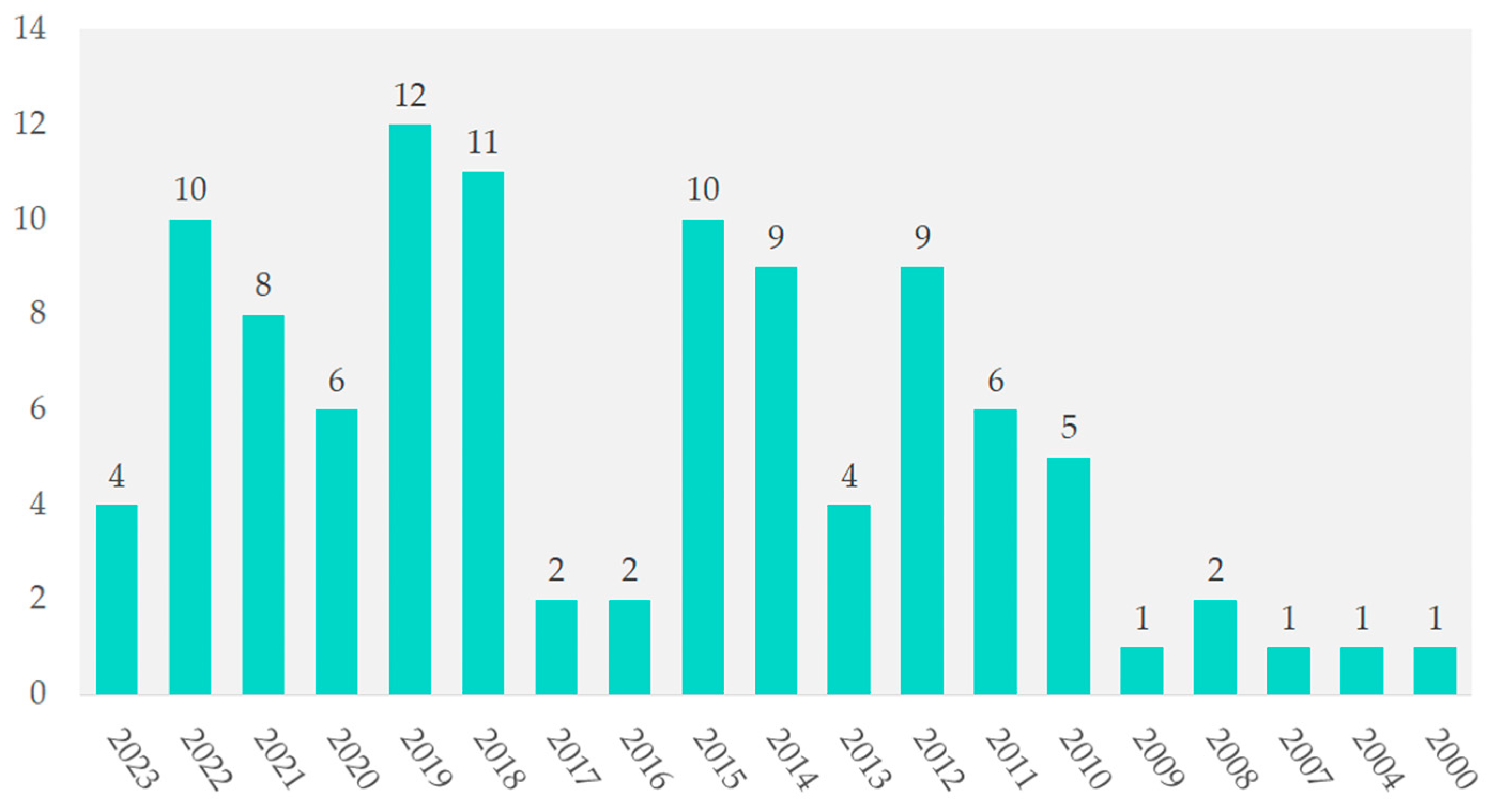
4.1. Research Trends
4.2. The Impact of HIF-Related Gene Modification on Glioblastoma Therapeutics
4.3. Exploring HIF-Related Targeted and Systemic Therapies for Glioblastoma in Experimental Settings
4.4. Insights into HIF-Associated Discoveries from Clinical Investigations in GBM
4.5. Advantages, Disadvantages, and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
| Search | (Glioblastoma) AND (Hypoxia-Inducible Factors) |
| Base: MEDLINE (PubMed) | |
| Filter | None |
| Search query | (“glioblastoma”[MeSH Terms] OR “glioblastoma”[All Fields] OR “glioblastomas”[All Fields]) AND ((“hypoxia”[MeSH Terms] OR “hypoxia”[All Fields] OR “hypoxia s”[All Fields] OR “hypoxias”[All Fields]) AND (“induce”[All Fields] OR “induced”[All Fields] OR “inducer”[All Fields] OR “inducers”[All Fields] OR “induces”[All Fields] OR “inducibilities”[All Fields] OR “inducibility”[All Fields] OR “inducible”[All Fields] OR “inducing”[All Fields]) AND (“factor”[All Fields] OR “factor s”[All Fields] OR “factors”[All Fields])) |
| Results | 558 papers |
| Base: Web of Science | |
| Filter | None |
| Search query | TS = (“glioblastoma” OR “glioblastomas”) AND TS = (“hypoxia inducible factors” OR “hypoxia-inducible factors” OR “HIFs”) |
| Results | 89 papers |
| Base: Scopus | |
| Filter | None |
| Search query | TITLE-ABS-KEY(“glioblastoma”) AND TITLE-ABS-KEY(“hypoxia inducible factors” OR “HIFs”) |
| Results | 671 papers |
Appendix B
| Section/Topic | # | Checklist Item | Reported on Page # |
| Title | |||
| Title | 1 | Identify the report as a systematic review, meta-analysis, or both. | 1 |
| Abstract | |||
| Structured summary | 2 | Provide a structured summary including, as applicable: background; objectives; data sources; study eligibility criteria, participants, and interventions; study appraisal and synthesis methods; results; limitations; conclusions and implications of key findings; and systematic review registration number. | 1 |
| Introduction | |||
| Rationale | 3 | Describe the rationale for the review in the context of what is already known. | 2 |
| Objectives | 4 | Provide an explicit statement of the questions being addressed with reference to the participants, interventions, comparisons, outcomes, and study design (PICOS). | 2 |
| Methods | |||
| Protocol and registration | 5 | Indicate if a review protocol exists, if and where it can be accessed (e.g., Web address), and, if available, provide registration information including registration number. | 2 |
| Eligibility criteria | 6 | Specify study characteristics (e.g., PICOS and length of follow-up) and report characteristics (e.g., years considered, language and publication status) used as criteria for eligibility, giving rationale. | 3 |
| Information sources | 7 | Describe all information sources (e.g., databases with dates of coverage and contact with study authors to identify additional studies) in the search and date last searched. | 3 |
| Search | 8 | Present a full electronic search strategy for at least one database, including any limits used, such that it could be repeated. | Appendix A |
| Study selection | 9 | State the process for selecting studies (i.e., screening, eligibility, included in the systematic review, and, if applicable, included in the meta-analysis). | 3 |
| Data collection process | 10 | Describe the method of data extraction from the reports (e.g., piloted forms, independently and in duplicate) and any processes for obtaining and confirming data from the investigators. | 3 |
| Data items | 11 | List and define all variables for which data were sought (e.g., PICOS and funding sources) and any assumptions and simplifications made. | 3 |
| Risk of bias in individual studies | 12 | Describe the methods used for assessing the risk of bias in individual studies (including the specification of whether this was performed at the study or outcome level), and how this information is to be used in any data synthesis. | N/A |
| Summary measures | 13 | State the principal summary measures (e.g., risk ratio and the difference in means). | N/A |
| Synthesis of results | 14 | Describe the methods of handling data and combining the results of studies, if performed, including the measures of consistency (e.g., I2) for each meta-analysis. | N/A |
| Risk of bias across studies | 15 | Specify any assessment of the risk of bias that may affect the cumulative evidence (e.g., publication bias and selective reporting within studies). | N/A |
| Additional analyses | 16 | Describe the methods of additional analyses (e.g., sensitivity or subgroup analyses and meta-regression), if performed, indicating which were pre-specified. | N/A |
| Results | |||
| Study selection | 17 | Give the number of studies screened, assessed for eligibility, and included in the review, with reasons for exclusions at each stage, ideally with a flow diagram. | Figure 1 |
| Study characteristics | 18 | For each study, present characteristics for which data were extracted (e.g., study size, PICOS, and follow-up period) and provide the citations. | 3 |
| Risk of bias within studies | 19 | Present data on the risk of bias of each study and, if available, any outcome level assessment (see item 12). | N/A |
| Results of individual studies | 20 | For all outcomes considered (benefits or harms), present, for each study: (a) simple summary data for each intervention group and (b) effect estimates and confidence intervals, ideally with a forest plot. | Table 1, Table 2 and Table 3 |
| Synthesis of results | 21 | Present the results of each meta-analysis performed, including confidence intervals and the measures of consistency. | N/A |
| Risk of bias across studies | 22 | Present the results of any assessment of the risk of bias across studies (see item 15). | N/A |
| Additional analysis | 23 | Give the results of additional analyses, if performed (e.g., sensitivity or subgroup analyses and meta-regression [see item 16]). | N/A |
| Discussion | |||
| Summary of evidence | 24 | Summarize the main findings including the strength of evidence for each main outcome; consider their relevance to key groups (e.g., healthcare providers, users, and policy makers). | 4–8 |
| Limitations | 25 | Discuss limitations at the study and outcome level (e.g., risk of bias), and at the review level (e.g., the incomplete retrieval of identified research and reporting bias). | 10 |
| Conclusions | 26 | Provide a general interpretation of the results in the context of other evidence, and implications for future research. | 11 |
| Funding | |||
| Funding | 27 | Describe the sources of funding for the systematic review and other support (e.g., supply of data); and the role of funders for the systematic review. | 11 |
| N/A—Not Available | |||
References
- Shah, S. Novel Therapies in Glioblastoma Treatment: Review of Glioblastoma; Current Treatment Options; and Novel Oncolytic Viral Therapies. Med. Sci. 2024, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Begagić, E.; Bečulić, H.; Đuzić, N.; Džidić-Krivić, A.; Pugonja, R.; Muharemović, A.; Jaganjac, B.; Salković, N.; Sefo, H.; Pojskić, M. CRISPR/Cas9-Mediated Gene Therapy for Glioblastoma: A Scoping Review. Biomedicines 2024, 12, 238. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Q.; Pierrevelcin, M.; Messe, M.; Lhermitte, B.; Blandin, A.F.; Papin, C.; Coca, A.; Dontenwill, M.; Entz-Werlé, N. Hypoxia Inducible Factors’ Signaling in Pediatric High-Grade Gliomas: Role, Modelization and Innovative Targeted Approaches. Cancers 2020, 12, 979. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Renfrow, J.J.; Soike, M.H.; Debinski, W.; Ramkissoon, S.H.; Mott, R.T.; Frenkel, M.B.; Sarkaria, J.N.; Lesser, G.J.; Strowd, R.E. Hypoxia-inducible factor 2α: A novel target in gliomas. Future Med. Chem. 2018, 10, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.L.; Mumert, M.L.; Gillespie, D.L.; Kinney, A.Y.; Schabel, M.C.; Salzman, K.L. Preoperative dynamic contrast-enhanced MRI correlates with molecular markers of hypoxia and vascularity in specific areas of intratumoral microenvironment and is predictive of patient outcome. Neuro Oncol. 2014, 16, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, E.R.; Zhang, Z.; Fink, J.R.; Muzi, M.; Hanna, L.; Greco, E.; Prah, M.; Schmainda, K.M.; Mintz, A.; Kostakoglu, L.; et al. ACRIN 6684: Assessment of Tumor Hypoxia in Newly Diagnosed Glioblastoma Using 18F-FMISO PET and MRI. Clin. Cancer Res. 2016, 22, 5079–5086. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Anobile, D.P.; Montenovo, G.; Pecoraro, C.; Franczak, M.; Ait Iddouch, W.; Peters, G.J.; Riganti, C.; Giovannetti, E. Splicing deregulation, microRNA and notch aberrations: Fighting the three-headed dog to overcome drug resistance in malignant mesothelioma. Expert Rev. Clin. Pharmacol. 2022, 15, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, K.; Xiang, J.; Zhang, M.; Zhu, M.; Xi, Q. Hypoxia induces immunosuppression, metastasis and drug resistance in pancreatic cancers. Cancer Lett. 2023, 571, 216345. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef] [PubMed]
- Matthew, J.P.; Joanne, E.M.; Patrick, M.B.; Isabelle, B.; Tammy, C.H.; Cynthia, D.M.; Larissa, S.; Jennifer, M.T.; Elie, A.A.; Sue, E.B.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, O.R.; Peters, G.-J.Y.; Bakker, C.J.; Carlsson, R.; Coles, N.A.; Corker, K.S.; Feldman, G.; Moreau, D.; Nordström, T.; Pickering, J.S.; et al. Increasing the transparency of systematic reviews: Presenting a generalized registration form. Syst. Rev. 2023, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Urushihara, Y.; Murata, Y.; Fujishima, Y.; Hosoi, Y. AMPK increases expression of ATM through transcriptional factor Sp1 and induces radioresistance under severe hypoxia in glioblastoma cell lines. Biochem. Biophys. Res. Commun. 2022, 590, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.H.; Shih, C.M.; Liu, A.J.; Chen, K.C. Hypoxia-inducible lncRNA MIR210HG interacting with OCT1 is involved in glioblastoma multiforme malignancy. Cancer Sci. 2022, 113, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ogura, Y.; Tanaka, K.; Nagashima, H.; Sasayama, T.; Endo, M.; Minami, Y. Ror1 is expressed inducibly by Notch and hypoxia signaling and regulates stem cell-like property of glioblastoma cells. Cancer Sci. 2023, 114, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Pandey, P.; Jha, P.; Dwivedi, V.; Sarkar, C.; Kulshreshtha, R. Hypoxic signature of microRNAs in glioblastoma: Insights from small RNA deep sequencing. BMC Genom. 2014, 15, 686. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.M.; Uno, M.; Oba-Shinjo, S.M.; Clara, C.A.; de Almeida Galatro, T.F.; Rosemberg, S.; Teixeira, M.J.; Nagahashi Marie, S.K. CXCR7 and CXCR4 Expressions in Infiltrative Astrocytomas and Their Interactions with HIF1α Expression and IDH1 Mutation. Pathol. Oncol. Res. 2015, 21, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Eckerich, C.; Zapf, S.; Fillbrandt, R.; Loges, S.; Westphal, M.; Lamszus, K. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int. J. Cancer 2007, 121, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Inukai, M.; Yokoi, A.; Ishizuka, Y.; Hashimura, M.; Matsumoto, T.; Oguri, Y.; Nakagawa, M.; Ishibashi, Y.; Ito, T.; Kumabe, T.; et al. A functional role of S100A4/non-muscle myosin IIA axis for pro-tumorigenic vascular functions in glioblastoma. Cell Commun. Signal 2022, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Weisz, A.; Kurashima, Y.; Hashimoto, K.; Ogura, T.; D’Acquisto, F.; Addeo, R.; Makuuchi, M.; Esumi, H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: Control of hypoxia-inducible factor-1 activity by nitric oxide. Blood 2000, 95, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Q.; Xu, S.; Wu, J.; Huang, Q.; Song, P.; Duan, F. Down-regulation of BAG3 inhibits proliferation and promotes apoptosis of glioblastoma multiforme through BAG3/HSP70/HIF-1α signaling pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 4305–4318. [Google Scholar] [PubMed]
- Méndez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.C.; Newcomb, E.W.; Zagzag, D. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1α Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e224. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.R.; Sadik, A.; Tykocinski, L.O.; Dietze, J.; Poschet, G.; Heiland, I.; Opitz, C.A. Hypoxia Inducible Factor 1α Inhibits the Expression of Immunosuppressive Tryptophan-2,3-Dioxygenase in Glioblastoma. Front. Immunol. 2019, 10, 2762. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Savino, M.; Falchetti, M.L.; Illi, B.; Bozzo, F.; Valle, C.; Helmer-Citterich, M.; Ferrè, F.; Nasi, S.; Levi, A. c-MYC inhibition impairs hypoxia response in glioblastoma multiforme. Oncotarget 2016, 7, 33257–33271. [Google Scholar] [CrossRef] [PubMed]
- Nie, W.; Luo, X.; Lu, D.; Yuan, P.; Liu, B.; Xu, H.; Ye, M. Casein kinase 1α 1 is involved in the progression of glioblastoma through HIF-1α-mediated autophagy. J. Neurophysiol. 2022, 128, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Noch, E.; Bookland, M.; Khalili, K. Astrocyte-elevated gene-1 (AEG-1) induction by hypoxia and glucose deprivation in glioblastoma. Cancer Biol. Ther. 2011, 11, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Rampazzo, E.; Abbadi, S.; Della Puppa, A.; Scienza, R.; D’Avella, D.; Denaro, L.; Te Kronnie, G.; Panchision, D.M.; Basso, G. Molecular mechanisms of HIF-1alpha modulation induced by oxygen tension and BMP2 in glioblastoma derived cells. PLoS ONE 2009, 4, e6206. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1α is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Polat, B.; Stein, S.; Guckenberger, M.; Hagemann, C.; Staab, A.; Katzer, A.; Anacker, J.; Flentje, M.; Vordermark, D. Inhibition of N-Myc down regulated gene 1 in in vitro cultured human glioblastoma cells. World J. Clin. Oncol. 2012, 3, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Sesen, J.; Cammas, A.; Scotland, S.J.; Elefterion, B.; Lemarié, A.; Millevoi, S.; Mathew, L.K.; Seva, C.; Toulas, C.; Moyal, E.C.; et al. Int6/eIF3e is essential for proliferation and survival of human glioblastoma cells. Int. J. Mol. Sci. 2014, 15, 2172–2190. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Hu, F.; Huang, R.; Mackman, N.; Horowitz, J.M.; Jensen, R.L.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res. 2006, 66, 7067–7074. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Fan, J.; Yang, H.; Zhao, C.; Niu, W.; Fang, Z.; Chen, X. Heterogeneity of subsets in glioblastoma mediated by Smad3 palmitoylation. Oncogenesis 2021, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Voss, D.M.; Sloan, A.; Spina, R.; Ames, H.M.; Bar, E.E. The Alternative Splicing Factor, MBNL1, Inhibits Glioblastoma Tumor Initiation and Progression by Reducing Hypoxia-Induced Stemness. Cancer Res. 2020, 80, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yan, Q.; Liao, B.; Zhao, L.; Xiong, S.; Wang, J.; Zou, D.; Pan, J.; Wu, L.; Deng, Y.; et al. The HIF1α/HIF2α-miR210-3p network regulates glioblastoma cell proliferation, dedifferentiation and chemoresistance through EGF under hypoxic conditions. Cell Death Dis. 2020, 11, 992. [Google Scholar] [CrossRef]
- Bae, W.Y.; Choi, J.S.; Nam, S.; Jeong, J.W. β-arrestin 2 stimulates degradation of HIF-1α and modulates tumor progression of glioblastoma. Cell Death Differ. 2021, 28, 3092–3104. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, Y.; She, X.; Sun, Y.; Fan, L.; Ren, X.; Fu, H.; Liu, C.; Li, P.; Zhao, C.; et al. Hypermethylated gene ANKDD1A is a candidate tumor suppressor that interacts with FIH1 and decreases HIF1α stability to inhibit cell autophagy in the glioblastoma multiforme hypoxia microenvironment. Oncogene 2019, 38, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Ozaki, S.; Kanemura, Y.; Suehiro, S.; Ohtsuka, Y.; Kohno, S.; Ohue, S.; et al. Hypoxia-induced phenotypic transition from highly invasive to less invasive tumors in glioma stem-like cells: Significance of CD44 and osteopontin as therapeutic targets in glioblastoma. Transl. Oncol. 2021, 14, 101137. [Google Scholar] [CrossRef] [PubMed]
- Choksi, S.; Lin, Y.; Pobezinskaya, Y.; Chen, L.; Park, C.; Morgan, M.; Li, T.; Jitkaew, S.; Cao, X.; Kim, Y.S.; et al. A HIF-1 target, ATIA, protects cells from apoptosis by modulating the mitochondrial thioredoxin, TRX2. Mol. Cell 2011, 42, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, E.; Chong, K.; Ryu, S.W.; Kim, C.; Choi, K.; Kim, J.H.; Choi, C. Atypical induction of HIF-1α expression by pericellular Notch1 signaling suffices for the malignancy of glioblastoma multiforme cells. Cell Mol. Life Sci. 2022, 79, 537. [Google Scholar] [CrossRef]
- Katakowski, M.; Charteris, N.; Chopp, M.; Khain, E. Density-Dependent Regulation of Glioma Cell Proliferation and Invasion Mediated by miR-9. Cancer Microenviron. 2016, 9, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, J.; Zhu, L.; Pan, H.; Li, W.; Zhou, Y.; Cong, Z.; Yan, F.; Chen, S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int. J. Cancer 2014, 135, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Saati, M.; Bensalah-Pigeon, H.; Ben M’Barek, K.; Gitton-Quent, O.; Bertrand, R.; Busso, D.; Mouthon, M.A.; Collura, A.; Junier, M.P.; et al. The HIF1α/JMY pathway promotes glioblastoma stem-like cell invasiveness after irradiation. Sci. Rep. 2020, 10, 18742. [Google Scholar] [CrossRef]
- Hu, Q.; Liu, F.; Yan, T.; Wu, M.; Ye, M.; Shi, G.; Lv, S.; Zhu, X. MicroRNA-576-3p inhibits the migration and proangiogenic abilities of hypoxia-treated glioma cells through hypoxia-inducible factor-1α. Int. J. Mol. Med. 2019, 43, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Paul, A.; Sen, E. Tumor necrosis factor α-induced hypoxia-inducible factor 1α-β-catenin axis regulates major histocompatibility complex class I gene activation through chromatin remodeling. Mol. Cell Biol. 2013, 33, 2718–2731. [Google Scholar] [CrossRef] [PubMed]
- Evagelou, S.L.; Bebenek, O.; Specker, E.J.; Uniacke, J. DEAD Box Protein Family Member DDX28 Is a Negative Regulator of Hypoxia-Inducible Factor 2α- and Eukaryotic Initiation Factor 4E2-Directed Hypoxic Translation. Mol. Cell Biol. 2020, 40, e00610-19. [Google Scholar] [CrossRef] [PubMed]
- Ikemori, R.Y.; Machado, C.M.; Furuzawa, K.M.; Nonogaki, S.; Osinaga, E.; Umezawa, K.; de Carvalho, M.A.; Verinaud, L.; Chammas, R. Galectin-3 up-regulation in hypoxic and nutrient deprived microenvironments promotes cell survival. PLoS ONE 2014, 9, e111592. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Yu, X.; Huang, H.; Zhou, W.; Xiang, C.; Huang, H.; Miele, L.; Liu, Z.; Bebek, G.; Bao, S.; et al. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. Cell Stem Cell 2018, 22, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Bordji, K.; Grandval, A.; Cuhna-Alves, L.; Lechapt-Zalcman, E.; Bernaudin, M. Hypoxia-inducible factor-2α (HIF-2α), but not HIF-1α, is essential for hypoxic induction of class III β-tubulin expression in human glioblastoma cells. FEBS J. 2014, 281, 5220–5236. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Heller, S.; Wanka, C.; Rieger, J.; Steinbach, J.P. Knockdown of the TP53-Induced Glycolysis and Apoptosis Regulator (TIGAR) Sensitizes Glioma Cells to Hypoxia, Irradiation and Temozolomide. Int. J. Mol. Sci. 2019, 20, 1061. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Potdar, A.A.; Gong, Y.; Eswarappa, S.M.; Donnola, S.; Lathia, J.D.; Hambardzumyan, D.; Rich, J.N.; Fox, P.L. Profilin-1 phosphorylation directs angiocrine expression and glioblastoma progression through HIF-1α accumulation. Nat. Cell Biol. 2014, 16, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhu, K.; Yang, Z.; Zhou, Y.; Xia, Z.; Ren, J.; Zhao, Y.; Wu, G.; Liu, C. Hypoxia-Induced Autophagy Is Involved in Radioresistance via HIF1A-Associated Beclin-1 in Glioblastoma Multiforme. Heliyon 2023, 9, e12820. [Google Scholar] [CrossRef] [PubMed]
- Coma, S.; Shimizu, A.; Klagsbrun, M. Hypoxia induces tumor and endothelial cell migration in a semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor neuropilin 2. Cell Adhes. Migr. 2011, 5, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Chen, Y.; Lai, H.T.; Wu, S.Y.; Wang, J.E.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.M.; et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.S.; Lim, K.J.; Price, A.C.; Orr, B.A.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2014, 33, 4433–4441. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.J.; Chiang, J.Y.; Chang, H.J.; Chen, D.C.; Wang, H.L.; Yang, H.A.; Wei, K.Y.; Huang, Y.C.; Wang, C.C.; Wei, S.T.; et al. Cellular and exosomal GPx1 are essential for controlling hydrogen peroxide balance and alleviating oxidative stress in hypoxic glioblastoma. Redox Biol. 2023, 65, 102831. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh, A.R.; Durden, D.L. MDM2 regulates hypoxic hypoxia-inducible factor 1α stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J. Biol. Chem. 2014, 289, 22785–22797. [Google Scholar] [CrossRef] [PubMed]
- Lulli, V.; Buccarelli, M.; Ilari, R.; Castellani, G.; De Dominicis, C.; Di Giamberardino, A.; QG, D.A.; Giannetti, S.; Martini, M.; Stumpo, V.; et al. Mir-370-3p Impairs Glioblastoma Stem-Like Cell Malignancy Regulating a Complex Interplay between HMGA2/HIF1A and the Oncogenic Long Non-Coding RNA (lncRNA) NEAT1. Int. J. Mol. Sci. 2020, 21, 3610. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Zhang, Y.; Celiku, O.; Zhang, W.; Song, H.; Williams, B.J.; Giles, A.J.; Rich, J.N.; Abounader, R.; Gilbert, M.R.; et al. Mitochondrial NIX Promotes Tumor Survival in the Hypoxic Niche of Glioblastoma. Cancer Res. 2019, 79, 5218–5232. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Kuang, Y.; Li, L.; Li, H.; Zhao, T.; He, Y.; Di, C.; Kang, J.; Yuan, L.; Yu, B.; et al. A positive feedback circuit comprising p21 and HIF-1α aggravates hypoxia-induced radioresistance of glioblastoma by promoting Glut1/LDHA-mediated glycolysis. FASEB J. 2022, 36, e22229. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Pantisano, V.; Puca, R.; Porru, M.; Aiello, A.; Grasselli, A.; Leonetti, C.; Safran, M.; Rechavi, G.; Givol, D.; et al. Zinc downregulates HIF-1α and inhibits its activity in tumor cells in vitro and in vivo. PLoS ONE 2010, 5, e15048. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Saccone, S.; Federico, C.; Rasà, D.M.; Caltabiano, R.; Broggi, G.; Giunta, S.; Musumeci, G.; D’Agata, V. Effect of PACAP on Hypoxia-Induced Angiogenesis and Epithelial-Mesenchymal Transition in Glioblastoma. Biomedicines 2021, 9, 965. [Google Scholar] [CrossRef]
- Ma, S.; Wang, F.; Dong, J.; Wang, N.; Tao, S.; Du, J.; Hu, S. Inhibition of hypoxia-inducible factor 1 by acriflavine renders glioblastoma sensitive for photodynamic therapy. J. Photochem. Photobiol. B 2022, 234, 112537. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Maugeri, G.; Magrì, B.; Giunta, S.; Saccone, S.; Federico, C.; Pricoco, E.; Broggi, G.; Caltabiano, R.; Musumeci, G.; et al. Modulatory activity of ADNP on the hypoxia-induced angiogenic process in glioblastoma. Int. J. Oncol. 2023, 62, 14. [Google Scholar] [CrossRef]
- D’Alessio, A.; Proietti, G.; Lama, G.; Biamonte, F.; Lauriola, L.; Moscato, U.; Vescovi, A.; Mangiola, A.; Angelucci, C.; Sica, G. Analysis of angiogenesis related factors in glioblastoma, peritumoral tissue and their derived cancer stem cells. Oncotarget 2016, 7, 78541–78556. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, I.; Limongi, C.; Piscopo, P.; Crestini, A.; Guerriero, C.; Fiore, M.; Conti, L.; Confaloni, A.; Tata, A.M. M2 Receptor Activation Counteracts the Glioblastoma Cancer Stem Cell Response to Hypoxia Condition. Int. J. Mol. Sci. 2020, 21, 1700. [Google Scholar] [CrossRef] [PubMed]
- Gagner, J.P.; Sarfraz, Y.; Ortenzi, V.; Alotaibi, F.M.; Chiriboga, L.A.; Tayyib, A.T.; Douglas, G.J.; Chevalier, E.; Romagnoli, B.; Tuffin, G.; et al. Multifaceted C-X-C Chemokine Receptor 4 (CXCR4) Inhibition Interferes with Anti-Vascular Endothelial Growth Factor Therapy-Induced Glioma Dissemination. Am. J. Pathol. 2017, 187, 2080–2094. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Luo, J.; Wang, Z.; Cai, X. Borneol promotes autophagic degradation of HIF-1α and enhances chemotherapy sensitivity in malignant glioma. PeerJ 2024, 12, e16691. [Google Scholar] [CrossRef] [PubMed]
- Douglas, C.; Lomeli, N.; Lepe, J.; Di, K.; Nandwana, N.K.; Vu, T.; Pham, J.; Kenney, M.C.; Das, B.; Bota, D.A. Discovery and Validation of Novel LonP1 and Proteasome Inhibitor in IDH1-R132H Malignant Astrocytoma Models. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Arienti, C.; Pignatta, S.; Zanoni, M.; Zamagni, A.; Cortesi, M.; Sarnelli, A.; Romeo, A.; Arpa, D.; Longobardi, P.; Bartolini, D.; et al. High-pressure oxygen rewires glucose metabolism of patient-derived glioblastoma cells and fuels inflammasome response. Cancer Lett. 2021, 506, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.W.; Liao, A.; Qutub, A.A. Simulation predicts IGFBP2-HIF1α interaction drives glioblastoma growth. PLoS Comput. Biol. 2015, 11, e1004169. [Google Scholar] [CrossRef]
- Lund, E.L.; Høg, A.; Olsen, M.W.; Hansen, L.T.; Engelholm, S.A.; Kristjansen, P.E. Differential regulation of VEGF, HIF1alpha and angiopoietin-1, -2 and -4 by hypoxia and ionizing radiation in human glioblastoma. Int. J. Cancer 2004, 108, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, C.P.; Burkhardt, J.K.; Shin, B.J.; Gürsel, D.B.; Mubita, L.; Gorrepati, R.; Brennan, C.; Holland, E.C.; Boockvar, J.A. Protein phosphatase 2A mediates dormancy of glioblastoma multiforme-derived tumor stem-like cells during hypoxia. PLoS ONE 2012, 7, e30059. [Google Scholar] [CrossRef]
- Bi, L.; Liu, Y.; Yang, Q.; Zhou, X.; Li, H.; Liu, Y.; Li, J.; Lu, Y.; Tang, H. Paris saponin H inhibits the proliferation of glioma cells through the A1 and A3 adenosine receptor-mediated pathway. Int. J. Mol. Med. 2021, 47, 30. [Google Scholar] [CrossRef] [PubMed]
- Khoei, S.; Shoja, M.; Mostaar, A.; Faeghi, F. Effects of resveratrol and methoxyamine on the radiosensitivity of iododeoxyuridine in U87MG glioblastoma cell line. Exp. Biol. Med. 2016, 241, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xu, X.; Li, C.; Li, C.; Li, Y.; Yin, S.; Yu, S.; Chen, X.Q. Mannose synergizes with chemoradiotherapy to cure cancer via metabolically targeting HIF-1 in a novel triple-negative glioblastoma mouse model. Clin. Transl. Med. 2020, 10, e226. [Google Scholar] [CrossRef] [PubMed]
- Dačević, M.; Isaković, A.; Podolski-Renić, A.; Isaković, A.M.; Stanković, T.; Milošević, Z.; Rakić, L.; Ruždijić, S.; Pešić, M. Purine nucleoside analog--sulfinosine modulates diverse mechanisms of cancer progression in multi-drug resistant cancer cell lines. PLoS ONE 2013, 8, e54044. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Kimura, T.; Sadahiro, H.; Kawano, H.; Takubo, K.; Suzuki, M.; Ikeda, E. Histological Characterization of the Tumorigenic “Peri-Necrotic Niche” Harboring Quiescent Stem-Like Tumor Cells in Glioblastoma. PLoS ONE 2016, 11, e0147366. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, D.; Zhang, Z.; Wang, Y.; Zhang, Z.; Liang, Z.; Liu, F.; Chen, L. Photodynamic therapy enhances the cytotoxicity of temozolomide against glioblastoma via reprogramming anaerobic glycolysis. Photodiagn. Photodyn. Ther. 2023, 42, 103342. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Ye, D.; Gessler, F.A.; Lee, Y.J.; Peruzzotti-Jametti, L.; Baumgarten, P.; Johnson, K.R.; Maric, D.; Yang, W.; Kögel, D.; et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci. Rep. 2017, 7, 7425. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Di Vito, M.; Frati, A.; Pellegrini, L.; De Santis, E.; Sette, G.; Eramo, A.; Sale, P.; Mari, E.; Santoro, A.; et al. Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J. Neuroinflamm. 2011, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Muh, C.R.; Joshi, S.; Singh, A.R.; Kesari, S.; Durden, D.L.; Makale, M.T. PTEN status mediates 2ME2 anti-tumor efficacy in preclinical glioblastoma models: Role of HIF1α suppression. J. Neurooncol. 2014, 116, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Maity, A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 2006, 8, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Ishibashi, H.; Nakamura, H.; Yachie, A.; Ohno-Shosaku, T. Hypoxia-induced inhibition of the endocannabinoid system in glioblastoma cells. Oncol. Rep. 2017, 38, 3702–3708. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lai, S.W.; Shen, C.K.; Chen, C.W.; Tsai, C.F.; Liu, Y.S.; Lu, D.Y.; Huang, B.R. Fenofibrate inhibits hypoxia-inducible factor-1 alpha and carbonic anhydrase expression through activation of AMP-activated protein kinase/HO-1/Sirt1 pathway in glioblastoma cells. Environ. Toxicol. 2021, 36, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Gagner, J.P.; Lechpammer, M.; Zagzag, D. Induction and Assessment of Hypoxia in Glioblastoma Cells In Vitro. Methods Mol. Biol. 2018, 1741, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ding, X.; Ye, H.; Wang, J.; Shao, J.; Huang, T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway. Neuroreport 2018, 29, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Guo, S.; Khan, F.; Dunterman, M.; Ali, H.; Liu, Y.; Huang, Y.; Chen, P. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep. Med. 2023, 4, 101238. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.W.; Wang, C.C.; Wu, C.P.; Lin, Y.J.; Lee, Y.C.; Cheng, Y.W.; Hsieh, C.H. Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function of ABCB1. Neuro Oncol. 2012, 14, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Barliya, T.; Mandel, M.; Livnat, T.; Weinberger, D.; Lavie, G. Degradation of HIF-1alpha under hypoxia combined with induction of Hsp90 polyubiquitination in cancer cells by hypericin: A unique cancer therapy. PLoS ONE 2011, 6, e22849. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shyu, W.C.; Chiang, C.Y.; Kuo, J.W.; Shen, W.C.; Liu, R.S. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS ONE 2011, 6, e23945. [Google Scholar] [CrossRef] [PubMed]
- Kannappan, V.; Liu, Y.; Wang, Z.; Azar, K.; Kurusamy, S.; Kilari, R.S.; Armesilla, A.L.; Morris, M.R.; Najlah, M.; Liu, P.; et al. PLGA-Nano-Encapsulated Disulfiram Inhibits Hypoxia-Induced NF-κB, Cancer Stem Cells, and Targets Glioblastoma In Vitro and In Vivo. Mol. Cancer Ther. 2022, 21, 1273–1284. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Caragher, S.P.; Shireman, J.M.; Huang, M.; Miska, J.; Atashi, F.; Baisiwala, S.; Hong Park, C.; Saathoff, M.R.; Warnke, L.; Xiao, T.; et al. Activation of Dopamine Receptor 2 Prompts Transcriptomic and Metabolic Plasticity in Glioblastoma. J. Neurosci. 2019, 39, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Wang, Y.; Ge, P.; Bailey, C.; Zhang, P.; Zhang, D.; Meng, Z.; Qi, C.; Chen, Q.; Chen, J.; et al. The HIF1α-PDGFD-PDGFRα axis controls glioblastoma growth at normoxia/mild-hypoxia and confers sensitivity to targeted therapy by echinomycin. J. Exp. Clin. Cancer Res. 2021, 40, 278. [Google Scholar] [CrossRef]
- Han, D.; Wei, W.; Chen, X.; Zhang, Y.; Wang, Y.; Zhang, J.; Wang, X.; Yu, T.; Hu, Q.; Liu, N.; et al. NF-κB/RelA-PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget 2015, 6, 26119–26128. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Lin, Y.J.; Wu, C.P.; Lee, H.T.; Shyu, W.C.; Wang, C.C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin. Cancer Res. 2015, 21, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Bandopadhyay, G.; Coyle, B.; Grabowska, A. A HIF-independent, CD133-mediated mechanism of cisplatin resistance in glioblastoma cells. Cell. Oncol. 2018, 41, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Cheul Oh, S.; Giles, A.J.; Jung, J.; Gilbert, M.R.; Park, D.M. Cardiac glycosides suppress the maintenance of stemness and malignancy via inhibiting HIF-1α in human glioma stem cells. Oncotarget 2017, 8, 40233–40245. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Wang, C.C.; Lin, Y.J.; Wu, C.P.; Hsieh, C.H. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J. Transl. Med. 2015, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Zhang, M.; Feng, Y.K.; Wang, X.J. IDH1-R132H Suppresses Glioblastoma Malignancy through FAT1-ROS-HIF-1α Signaling. Neurol. India 2020, 68, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Pan, M.H.; Wang, L.; Li, W.; Jiang, C.; He, J.; Abouzid, K.; Liu, L.Z.; Shi, Z.; Jiang, B.H. Hypoxia-mediated mitochondria apoptosis inhibition induces temozolomide treatment resistance through miR-26a/Bad/Bax axis. Cell Death Dis. 2018, 9, 1128. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Luo, Z.; Lin, Y.; Chen, H.; Chen, T.; Xu, L.; Orgurek, S.; Berry, K.; Dzieciatkowska, M.; Reisz, J.A.; et al. PRMT3 drives glioblastoma progression by enhancing HIF1A and glycolytic metabolism. Cell Death Dis. 2022, 13, 943. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Boso, D.; Rampazzo, E.; Zanon, C.; Bresolin, S.; Maule, F.; Porcù, E.; Cani, A.; Della Puppa, A.; Trentin, L.; Basso, G.; et al. HIF-1α/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics 2019, 9, 4860–4877. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, X.F. HIF-miR-215-KDM1B promotes glioma-initiating cell adaptation to hypoxia. Cell Cycle 2016, 15, 1939–1940. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cheng, X.; Wang, X.; Wang, J.; Wen, X.; Xie, C.; Liao, C. Clinical implications of hypoxia-inducible factor-1α and caveolin-1 overexpression in isocitrate dehydrogenase-wild type glioblastoma multiforme. Oncol. Lett. 2019, 17, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Bache, M.; Rot, S.; Keßler, J.; Güttler, A.; Wichmann, H.; Greither, T.; Wach, S.; Taubert, H.; Söling, A.; Bilkenroth, U.; et al. mRNA expression levels of hypoxia-induced and stem cell-associated genes in human glioblastoma. Oncol. Rep. 2015, 33, 3155–3161. [Google Scholar] [CrossRef] [PubMed]
- Erpolat, O.P.; Gocun, P.U.; Akmansu, M.; Ozgun, G.; Akyol, G. Hypoxia-related molecules HIF-1α, CA9, and osteopontin: Predictors of survival in patients with high-grade glioma. Strahlenther. Onkol. 2013, 189, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kaynar, M.Y.; Sanus, G.Z.; Hnimoglu, H.; Kacira, T.; Kemerdere, R.; Atukeren, P.; Gumustas, K.; Canbaz, B.; Tanriverdi, T. Expression of hypoxia inducible factor-1alpha in tumors of patients with glioblastoma multiforme and transitional meningioma. J. Clin. Neurosci. 2008, 15, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Matsuo, T.; Nagata, I. Protocol of radiotherapy for glioblastoma according to the expression of HIF-1. Brain Tumor Pathol. 2004, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, H.; Zhu, J.; Tang, Y.; Zhou, Y.; Zhu, L.; Gao, C.; Li, W.; You, W.; Yu, B.; et al. Correlation of Nrf2 and HIF-1α in glioblastoma and their relationships to clinicopathologic features and survival. Neurol. Res. 2013, 35, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Sfifou, F.; Hakkou, E.M.; Bouaiti, E.A.; Slaoui, M.; Errihani, H.; Al Bouzidi, A.; Abouqal, R.; El Ouahabi, A.; Cherradi, N. Correlation of immunohistochemical expression of HIF-1alpha and IDH1 with clinicopathological and therapeutic data of moroccan glioblastoma and survival analysis. Ann. Med. Surg. 2021, 69, 102731. [Google Scholar] [CrossRef] [PubMed]
- Potharaju, M.; Mathavan, A.; Mangaleswaran, B.; Patil, S.; John, R.; Ghosh, S.; Kalavakonda, C.; Ghosh, M.; Verma, R.S. Clinicopathological Analysis of HIF-1alpha and TERT on Survival Outcome in Glioblastoma Patients: A Prospective, Single Institution Study. J. Cancer 2019, 10, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Clara, C.A.; Marie, S.K.; de Almeida, J.R.; Wakamatsu, A.; Oba-Shinjo, S.M.; Uno, M.; Neville, M.; Rosemberg, S. Angiogenesis and expression of PDGF-C, VEGF, CD105 and HIF-1α in human glioblastoma. Neuropathology 2014, 34, 343–352. [Google Scholar] [CrossRef] [PubMed]
- El-Benhawy, S.A.; Sakr, O.A.; Fahmy, E.I.; Ali, R.A.; Hussein, M.S.; Nassar, E.M.; Salem, S.M.; Abu-Samra, N.; Elzawawy, S. Assessment of Serum Hypoxia Biomarkers Pre- and Post-radiotherapy in Patients with Brain Tumors. J. Mol. Neurosci. 2022, 72, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Begagić, E.; Pugonja, R.; Bečulić, H.; Čeliković, A.; Tandir Lihić, L.; Kadić Vukas, S.; Čejvan, L.; Skomorac, R.; Selimović, E.; Jaganjac, B.; et al. Molecular Targeted Therapies in Glioblastoma Multiforme: A Systematic Overview of Global Trends and Findings. Brain Sci. 2023, 13, 1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Yu, H.Y.; Zhao, G.X.; Jiang, X.Z.; Gao, G.; Wei, B.J. Global research trends in immunotherapy for glioma: A comprehensive visualization and bibliometric analysis. Front. Endocrinol. 2023, 14, 1273634. [Google Scholar] [CrossRef] [PubMed]
- Łaba, A.E.; Ziółkowski, P. Trends in glioblastoma treatment research: An analysis of clinical trials and literature. Neurol. Neurochir. Pol. 2021, 55, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol. 2013, 15, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C. Use of laboratory studies for the design, explanation, and validation of human micronutrient intervention studies. J. Nutr. 2012, 142, 157s–160s. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martinez, O.; García-Montero, C.; Callejón-Peláez, E.; Sáez, M.A.; Álvarez-Mon, M.A.; García-Honduvilla, N.; Monserrat, J.; Álvarez-Mon, M.; Bujan, J.; et al. A General Overview on the Hyperbaric Oxygen Therapy: Applications, Mechanisms and Translational Opportunities. Medicina 2021, 57, 864. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Tsuneyama, K.; Yen, M.-H.; Lee, J.-T.; Chen, J.-L.; Huang, S.-M. Hyperbaric oxygen suppressed tumor progression through the improvement of tumor hypoxia and induction of tumor apoptosis in A549-cell-transferred lung cancer. Sci. Rep. 2021, 11, 12033. [Google Scholar] [CrossRef]
- Herrera-Campos, A.B.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Fernández-Cortés, M.; Montuenga, L.M.; Oliver, F.J.; Garcia-Diaz, A. Implications of Hyperoxia over the Tumor Microenvironment: An Overview Highlighting the Importance of the Immune System. Cancers 2022, 14, 2740. [Google Scholar] [CrossRef]
- Trejo-Solis, C.; Silva-Adaya, D.; Serrano-García, N.; Magaña-Maldonado, R.; Jimenez-Farfan, D.; Ferreira-Guerrero, E.; Cruz-Salgado, A.; Castillo-Rodriguez, R.A. Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma. Int. J. Mol. Sci. 2023, 24, 17633. [Google Scholar] [CrossRef] [PubMed]
- Beylerli, O.; Beilerli, A.; Shumadalova, A.; Wang, X.; Yang, M.; Sun, H.; Teng, L. Therapeutic effect of natural polyphenols against glioblastoma. Front. Cell Dev. Biol. 2022, 10, 1036809. [Google Scholar] [CrossRef] [PubMed]
- Luís, Â.; Amaral, L.; Domingues, F.; Pereira, L.; Cascalheira, J.F. Action of Curcumin on Glioblastoma Growth: A Systematic Review with Meta-Analysis of Animal Model Studies. Biomedicines 2024, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, F.; Omata, D.; Munakata, L.; Kageyama, S.; Maruyama, K.; Kudo, N.; Suzuki, R. Brain Delivery of Cisplatin Using Microbubbles in Combination with Ultrasound as an Effective Therapy for Glioblastoma. Pharmaceuticals 2023, 16, 1599. [Google Scholar] [CrossRef] [PubMed]
- Chédeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Muniraj, N.; Siddharth, S.; Sharma, D. Bioactive Compounds: Multi-Targeting Silver Bullets for Preventing and Treating Breast Cancer. Cancers 2019, 11, 1563. [Google Scholar] [CrossRef] [PubMed]
- Di Nunzio, M.R.; Douhal, A. Robust Inclusion Complex of Topotecan Comprised within a Rhodamine-Labeled β-Cyclodextrin: Competing Proton and Energy Transfer Processes. Pharmaceutics 2023, 15, 1620. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T. Current Status and Future Perspective in Glioma Invasion Research. Brain Sci. 2024, 14, 309. [Google Scholar] [CrossRef] [PubMed]
- Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewska, S.; Sławski, J.; Collawn, J.F.; Bartoszewski, R. HIF-1-Induced hsa-miR-429: Understanding Its Direct Targets as the Key to Developing Cancer Diagnostics and Therapies. Cancers 2023, 15, 2903. [Google Scholar] [CrossRef] [PubMed]
- Englert-Golon, M.; Tokłowicz, M.; Żbikowska, A.; Sajdak, S.; Kotwicka, M.; Andrusiewicz, M. Differential Expression of HIF1A, EPAS1, and VEGF Genes in Benign and Malignant Ovarian Neoplasia. Cancers 2022, 14, 4899. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Ramar, V.; Guo, S.; Hudson, B.; Liu, M. Progress in Glioma Stem Cell Research. Cancers 2024, 16, 102. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Oishi, T.; Koizumi, S.; Kurozumi, K. Molecular Mechanisms and Clinical Challenges of Glioma Invasion. Brain Sci. 2022, 12, 291. [Google Scholar] [CrossRef] [PubMed]
- Qannita, R.A.; Alalami, A.I.; Harb, A.A.; Aleidi, S.M.; Taneera, J.; Abu-Gharbieh, E.; El-Huneidi, W.; Saleh, M.A.; Alzoubi, K.H.; Semreen, M.H.; et al. Targeting Hypoxia-Inducible Factor-1 (HIF-1) in Cancer: Emerging Therapeutic Strategies and Pathway Regulation. Pharmaceuticals 2024, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef]
- Pantazopoulou, V.; Jeannot, P.; Rosberg, R.; Berg, T.J.; Pietras, A. Hypoxia-Induced Reactivity of Tumor-Associated Astrocytes Affects Glioma Cell Properties. Cells 2021, 10, 613. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Moreno-Murciano, P.; Oriol-Caballo, M.; López-Blanch, R.; Pineda, B.; Gutiérrez-Arroyo, J.L.; Loras, A.; Gonzalez-Bonet, L.G.; Martinez-Cadenas, C.; Estrela, J.M.; et al. Glioblastoma Therapy: Past, Present and Future. Int. J. Mol. Sci. 2024, 25, 2529. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | Search Strategy |
|---|---|
| P (population or problem) | Glioblastoma |
| I (intervention) | Hypoxia-inducible factors |
| C (comparison) | None |
| O (outcome) | None |
| S (study design) | Original investigations |
| Reference | Country (Year) | Study Design | Species | Cell Line(s) | Targeted HIF | Related Factor | Role of HIF and Related Factors | Gene Modification | Effect of Gene Modification |
|---|---|---|---|---|---|---|---|---|---|
| Hashimoto et al. [16] | Japan (2022) | Lab (IV) | CL | T98G and A172 | HIF1α | AMPK and ATM | AMPK boosts ATM expression via Sp1 transcription factor, eliciting radioresistance in severe hypoxia. | KD | AMPKα KD under severe hypoxia decreases Sp1 and ATM expression, whereas Sp1 KD suppresses ATM, Src, EGFR, and Akt expression, ultimately diminishing radioresistance. |
| Ho et al. [17] | Taiwan (2021) | Lab (C) | Mice and CL | U-87, U-118, and PDM-123 | HIF1α | MIR210HG, OCT1, IGFBP2, and FGFR1 | MIR210HG participates in hypoxia-mediated glioma invasion, cancer stemness, and TMZ resistance. It also promotes the transcription activity of OCT1, regulating the expressions of the oncogenes IGFBP2 and FGFR1. | KD | The overexpression of MIR210HG in normoxia boosts the activities of IGFBP2 and FGFR1 promoters, an effect that is inhibited by the suppression of OCT1. In hypoxia, the promoter activities of IGFBP2 and FGFR1 are reduced when MIR210HG or OCT1 is knocked down. |
| Ishikawa et al. [18] | Japan (2022) | Lab (IV) | CL | T98G, A172, and U87 | HIF1α | Ror1 (Wnt5a-Ror1 axis) | HIF1α activates Ror1 transcription by binding to its promoter regions in glioblastoma, influencing cancer progression via cell proliferation and migration regulation. | KD | KD of HIF1α inhibited the expression of Ror1, in particular under hypoxic conditions. |
| Agrawal et al. [19] | India (2014) | Lab (IV) | CL | U251, U87, and A172 | HIF1α | miR-210-3p | miR-210-3p promotes the survival, aggressiveness, and therapy resistance of glioblastoma cells. The regulation of miR-210-3p is HIF1α dependent and, on the other hand, miR-210-3p promotes HIF transcriptional activity. | OE | Increase in the expression of the HIF target genes VEGF and CA9 in response to miR-210-3p overexpression and their downregulation in response to miR-210-3p inhibition. |
| Bianco et al. [20] | Brazil (2015) | Lab (IV) | CL | U87 | HIF1α | CXCR7, CXCR4, and IDH1 | CXCR7 expression in astrocytoma varies with malignancy; HIF1α boosts CXCR7 and CXCR4, whereas IDH1mut lowers them, suggesting CXCR7 involvement in astrocytoma tumorigenesis. | OE | HIF1α overexpression was linked to higher CXCR7 and CXCR4 expressions, while IDH1 mutation was associated with lower levels; CXCR7 overexpressed in astrocytoma and correlated with CXCR4/IDH1 in AGII and with CXCR4/IDH1/HIF1α in glioblastoma, with no survival correlation. |
| Eckerich et al. [21] | Germany (2007) | Lab (IV) | CL | U87 and U251 | HIF1α | C-Met and SF/HGF | SF/HGF, a multifunctional growth factor, binds to c-Met, a tyrosine kinase receptor encoded by a proto-oncogene; hypoxia activates the c-met promoter containing HIF-1 binding sites. | KO | Half of all human glioblastomas respond to hypoxia with an induction of c-Met, which can enhance the stimulating effect of SF/HGF on tumor cell migration. |
| Inukai et al. [22] | Japan (2022) | Lab (IV) | Mice and CL | KS-1 | HIF1α | S100A4/NMIIA axis | Following severe hypoxia, S100A4 is upregulated and interacts with NMIIA; this inhibits NMIIA activity and thus derepresses tumor cell migration. | KD | The KD of S100A4 in the glioblastoma cell line KS-1 decreased migration capability, concomitant with decreased Slug expression. |
| Kimura et al. [23] | Italy (2000) | Lab (IV) | CL | A172 and Hep3B | HIF1α | NO and VEGF | The direct involvement of NO in the control of angiogenesis through its regulation of VEGF expression, where HIF1α activity appears to be essential. | DEL | NO-responsive cis-elements are HIF1α binding sites, and an adjacent ancillary sequence is located immediately downstream within the hypoxia-response element (HRE). |
| Li et al. [24] | China (2018) | Lab (IV) | CL | U87 and U251 | HIF1α | BAG3 | Downregulated BAG3 inhibited HIF1α protein through promoting the degradation of HIF1α by HSP70 by the BAG3/HSP70/HIF1α proteasome pathway. | TF | When HIF1α was upregulated, induced by HIF1α plasmid TF based on the downregulation of BAG3, the proliferation inhibition and apoptosis promotion was partially reversed. |
| Mendez et al. [25] | USA (2010) | Lab (C) | Mice and CL | LN308, U87MG, HEK 293T, and GL261 | HIF1α | n/a | HIF1α plays a role in the survival and self-renewal potential of CSCs. | KD | The KD of HIF1α in human and murine glioma cells impairs their migration in vitro and their invasion in vivo. |
| Miska et al. [26] | USA (2019) | Lab (C) | Mice and CL | Biopsy | HIF1α | Foxp3+ T Cells | HIF1α acts as a metabolic switch for Tregs between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression. | KO | The conditional KO of HIF1α in Foxp3+ T Cells inhibits the migration of Tregs to brain tumors in vivo. |
| Mohapatra et al. [27] | Germany (2019) | Lab (IV) | CL | A172 and U-87 MG i LN-18 | HIF1α | Tryptophan-2,3-Dioxygenase (TDO2) | TDO2 in glioblastoma promotes tumor cell motility and suppresses antitumor immune responses by producing Trp metabolites that activate the aryl hydrocarbon receptor (AHR). | KD | The KD of HIF1α restored the expression of TDO2 upon cobalt chloride treatment, confirming that HIF1α controls TDO2 expression. |
| Mongiardi et al. [28] | Italy (2016) | Lab (IV) | CL | U87 | HIF1α | c-MYC | HIF-1 and a deregulated c-MYC in cancer cells cooperatively induce the transcription of genes involved in hypoxic adaptation such as genes regulating metabolic reprogramming and angiogenesis. | TD | MYC inhibition alters the transcriptional response to hypoxia in glioblastoma cells. |
| Nie et al. [29] | China (2012) | Lab (IV) | CL | U87, U251, U118, LN229, and SHG44 | HIF1α | Casein kinase 1α 1 | CK1a is overexpressed in glioblastoma cells, with its levels increasing proportionally with the WHO grade. | TF | Overexpressed CK1a positively regulates autophagy activity through the HIF1α pathway. The inhibition of CK1a might be a potential therapeutic approach for glioblastoma therapy. |
| Noch et al. [30] | United States (2011) | Lab (C) | Mice and CL | U87 and T98-G | HIF1α | Astrocyte-elevated gene-1 (AEG-1) | The hypoxic induction of AEG-1 relies on HIF1α stabilization, with PI3K inhibition disrupting AEG-1 induction by destabilizing HIF1α. | TF | AEG-1 is slightly upregulated following 24 h TF with HIF1α. |
| Pistollato et al. [31] | Italy (2009) | Lab (IV) | CL | Biopsy | HIF1α | Akt/mTOR and BMP2 | Exogenous BMP2, similar to high oxygen exposure, induces the time-dependent activation of the Akt/mTOR pathway in glioblastoma-derived cells. | KD | By silencing HIF1α in glioblastoma cells, a strong differentiation and eventually cell death occurred after 1 week. |
| Qiang et al. [32] | China (2011) | Lab (IV) | CL | U251, SHG44, A172, and C6 | HIF1α | PI3K/Akt and ERK1/2 | PI3K/Akt and ERK1/2 pathways contribute to HIF1α translation in GSCs. | KD | PI3K/Akt and ERK1/2 inhibition partly reduces hypoxia-induced Notch pathway activation and GSC maintenance. |
| Said et al. [33] | Germany (2012) | Lab (IV) | CL | U373, U251, and U87 | HIF1α | ndrg1 N-Myc | Short dsRNA oligonucleotides and iodoacetate inhibit N-Myc downregulated gene 1 protein and mRNA expression in U373 glioblastoma cells by interfering with cellular glycolysis. | KD | Treatment with siRNA and iodoacetate (IAA) in human glioblastoma cell lines led to a nearly complete suppression of NDRG1 expression, highlighting IAA’s role as a glycolysis inhibitor. |
| Sesen et al. [34] | France (2014) | Lab (IV) | CL | LN18, SF767, U87, and U251 | HIF1α and HIF2α | Int6/eIF3e | siInt6 significantly inhibits Int6 mRNA and protein in all glioblastoma cell lines compared to control siRNA. | TF | TF silenced the Int6 gene and protein expression effectively. |
| Rong et al. [35] | United States (2006) | Lab (IV) | CL | U87 and U251 | HIF1α | Egr-1, Sp1, NF-κB, and activator protein-1 (AP-1) | Forced Egr-1 overexpression, but not Sp1, via cDNA TF, increases tissue factor in glioma cells under normoxia (21% O2), while Egr-1 siRNA notably decreases hypoxia-induced tissue factor expression. | TF | The TF of glioma cells with an Sp1 expression plasmid (pSp1, 2.0 μg) for 24 h under normoxia led to a large increase in both nonphosphorylated (bottom band) and phosphorylated (top band) Sp1 protein expression without a concomitant tissue factor expression. |
| Fan et al. [36] | China (2021) | Lab (C) | CL | PN 12,16 and 19, MES23, 27 and 29 | HIF1α | IDH1, TGF-β1, E2F4, and Smad3 | IDH1 mutation activates HIF1α and reduces TGF-β1 expression in proneural GSCs; Smad3 interacts with E2F4 to inhibit the expression of mesenchymal markers. | KD | IDH1 KD elevates HIF1α and decreases TGF-β1 in proneural glioblastoma cells. |
| Voss et al. [37] | USA (2020) | Lab (C) | Mice and CL | HSR-GLIOBLASTOMA1, HSR-040821, HSR-040622, T387, T3691, and T3832 | HIF1α and HIF2α | MBNL1 | MBNL1 expression is highest in glioblastoma defined as MES, inhibited in the hypoxic elements of the tumor and within the MES subgroup, and correlates with better overall patient survival. | KD | Hypoxia suppresses MBNL1 activity in certain tumor-derived neurosphere lines, leading to the increased expression of various gene isoforms that are linked to an ESC-like state. |
| Wang et al. [38] | China (2021) | Lab (C) | Mice and CL | MES02-GSC, MES06-GSC, and MES13-GSC | HIF-1 | PLOD1 | HIF1 can directly induce the expression of PLOD1 under hypoxia. | KO | PLOD1 KO inhibits MES GSC-enriched tumor sphere growth and invasion in vitro, and differentiation in vivo. |
| Bae et al. [39] | South Korea (2021) | Lab (C) | Mice and CL | U87, T98G, H4, U251, immortalized primary human fetal astrocytes, and HMEC-1 | HIF1α | Arrb2 (β-arrestin 2) | Arrb2 interacts with HIF1α and stimulates the ubiquitin-mediated 26S proteasomal degradation of HIF1α by recruiting PHD2 and pVHL. | TF | The overexpression of Arrb2 in glioblastoma cells reduces HIF1α levels, resulting in antitumorigenic effects including suppressed tumor growth and angiogenesis. |
| Feng et al. [40] | China (2019) | Lab (C) | Mice and CL | U251, U87, and HEK293 | HIF1α | ANKDD1A | ANKDD1A inhibits HIF1α activity, decreases its half-life by upregulating FIH1, reduces glucose uptake and lactate production, inhibits glioblastoma autophagy, and induces apoptosis in glioblastoma cells under hypoxia. | TF | Transfected cells had lower glucose uptake and lower LDH. ANKDD1A disturbs the tolerance of glioblastoma cells to hypoxia. |
| Nishikawa et al. [41] | Japan (2021) | Lab (C) | Mice and CL | GSL-1 and GSL-2 | HIF1α and HIF2α | CD44 and OPN | Hypoxia (1% O2) upregulates CD44 expression via the activation of HIF1α. Moderate hypoxia (5% O2) upregulates osteopontin expression via the activation of HIF2α. | KD | The upregulated osteopontin inhibits CD44-promoted GSC migration, invasion, and proliferation. |
| Choksi et al. [42] | USA (2012) | Lab (C) | Mice and CL | TRAF2−/−, wt MEF, A172, IMR-32 and CCF-STTG1 | HIF1α | ATIA | HIF-1 target, ATIA protects cells against TNFα- and hypoxia-induced apoptosis through regulating the function of the mitochondrial antioxidant, thioredoxin-2, and ROS generation. | KD, KO | ATIA KD in glioblastoma cells renders them sensitive to hypoxia-induced apoptosis. |
| Lee et al. [43] | Korea (2022) | Lab (C) | Mice and CL | U251-MG, LN215-MG, CRT-MG, U373-MG, HT-1080 and Panc-1 | HIF1α | Notch1 | HIF1α, induced even in non-hypoxic conditions by cell-to-cell contact, is a critical cue responsible for the malignant characteristics of glioblastoma cells through Notch1 signaling. | TF | Silencing Notch1 signaling with siRNA TF resensitized resistant glioblastoma cells to TMZ and reduced their viability under high-density culture conditions. |
| Katakowski et al. [44] | USA (2016) | Lab (C) | Mice and CL | U87 | HIF1α | miR-9 | miR-9 increases glioma cell migration and decreases proliferation at low densities, but has the opposite effect at high densities. | TF | miR-9 has a biphasic density-dependent effect on glioma cell proliferation. |
| Ji et al. [45] | China (2014) | Lab (C) | Mice and CL | U251 and U87 | HIF1α | Nrf2 | Nrf2 has a role in glioblastoma angiogenesis; human glioblastoma tissues expressing higher Nrf2 levels showed relatively higher microvessel density. | KD | The KD of Nrf2 inhibits glioblastoma angiogenesis by preventing the hypoxia-induced activation of HIF1α. |
| Gauthier et al. [46] | France (2020) | Lab (C) | Mice and CL | TG1N i TG16 GSC | HIF1α | JMY | Post-irradiation, HIF1α induces JMY transcription, promoting GSC migration via its actin nucleation-promoting activity. | KD | The radiation-induced migration of GSCs is associated with the HIF1α-dependent accumulation of JMY in the cytoplasm. |
| Hu et al. [47] | China (2019) | Lab (IV) | CL | U87, U251, T98, LN229, and U118 | HIF1α | miR-576-3p | miR-576-3p’s inhibition of the migration and proangiogenic capacity of hypoxia-induced glioma cells is mediated by HIF1α. | KD, TF | HIF1α KD and miR-576-3p overexpression comparably inhibit migration and angiogenesis in hypoxia-induced glioma cells, with reduced HIF1α expression in miR-576-3p-transfected cells. |
| Ghosh et al. [48] | India (2013) | Lab (IV) | CL | T98G and U87 | HIF1α | TNF-α, β-catenin, and MHC 1 | A TNF-α-induced increase in MHC-I expression and transcriptional activation was concurrent with increased HIF1α, ΝF-κΒ, and β-catenin activities. | KD | The KD of HIF1α and β-catenin abolished TNF-α-induced MHC-I activation, while NF-κB had no effect. |
| Evagelou et al. [49] | Canada (2020) | Lab (IV) | CL | U87 | HIF2α | DDX28 | HIF2α is responsible for regulating eIF4E2-directed translation in hypoxia, whereas DDX28 functions as a negative regulator, hindering HIF2α‘s ability to activate this translation pathway. | KD | eIF4E2 binds to the m7GTP cap structure, enhancing the translation of its target mRNAs, while the repression of HIF2α and eIF4E2 curtails the translation activation of oncogenic mRNAs. |
| Ikemori et al. [50] | Brasil (2014) | Lab (C) | Mice and CL | NG97ht, T98G, and U87G | HIF1α | Galektin-3 (gal-3) | Gal-3 expression shields glioma cells from hypoxia-induced death and facilitates tumor growth in poorly perfused microenvironments. | KD | The KD of Gal-3 enhances cell death in cells deprived of both oxygen and serum. |
| Man et al. [51] | USA (2018) | Lab (C) | Mice and CL | GSCs and non-GSCs | HIF1α | Vasorin | Vasorin prevents TNF-mediated apoptosis, inhibits TGF-beta signaling, and regulates Notch signaling in GSCs within the hypoxic niche. | KD | Vasorin KD reduced proliferation and induced the apoptosis of GSCs. In contrast, Vasorin KD in non-GSCs had little effect on cell viability. |
| Bordji et al. [52] | France (2014) | Lab (IV) | CL | U87, U251MG and GL15 | HIF1α and HIF2α | class III beta-tubulin | HIF2α, not HIF1α, triggers bIII-t expression in hypoxic glioblastoma cells, facilitating tumor cell survival against DNA-targeting and tubulin-binding drugs, and promoting chemoresistance. | TF | HIF2α downregulation inhibits hypoxia-induced BIII-t expression in GL15 and U87 cells, enhancing glioblastoma cell sensitivity to chemotherapy. |
| Maurer et al. [53] | Germany (2019) | Lab (IV) | CL | LNT-229, U87, and T98G | HIF1α | TIGAR | TIGAR gene silencing enhances cell death associated with oxygen restriction. | KD | TIGAR KD enhances cell death under hypoxia and increases sensitivity to ionizing radiation, while also enhancing the effects of TMZ on cell density and clonogenicity. |
| Fan et al. [54] | USA (2014) | Lab (C) | CL | U251 | HIF1α | Profilin-1 | Pfn-1 phosphorylation drives endothelial angiocrine expression, promoting abnormal vascularization and glioblastoma progression via hypoxia-independent HIF1α induction. | KD | HIF1α KD disrupts the angiocrine feed-forward mechanism, normalizing vasculature. |
| Wei et al. [55] | China (2023) | Lab (C) | Mice and CL | U87, U251, and U373 | HIF1α | Beclin-1 | Beclin-1 suppression by 3-MA could reverse radioresistance induced by HIF1A under hypoxia. | KD | HIF1A KD improved glioblastoma radiosensitivity, and silencing Beclin-1 could reverse HIF1A-induced radioresistance under hypoxic conditions. |
| Coma et al. [56] | USA (2011) | Lab (IV) | CL | U87MG and A375SM | HIF1α | NRP2 and SEMA3F | SEMA3F inhibits tumor angiogenesis and metastasis. NRP2 is a receptor expressed by tumor cells that binds both SEMA3F and VEGF. | KD | The repression of NRP2 induced by DFO was hindered by HIF1α siRNA, validating that hypoxia-induced NRP2 repression is reliant on HIF1α. |
| Bao et al. [57] | USA (2018) | Lab (C) | Mice and CL | U251, U87, LN229, and HEK293FT | HIF1α | G9a and GLP | G9a/GLP-mediated K674 methylation decreases HIF1α transcriptional activity. | TF | G9a targets HIF1α, impairing tumorigenesis and glioblastoma cell migration by inhibiting its transcriptional activity and the expression of downstream targets like PTGS1, NDNF, SLC6A3, and Linc01132. |
| Lim et al. [58] | USA (2014) | Lab (C) | Mice and CL | HSR- glioblastoma 1 and JHH- glioblastoma 10 | HIF1α | MCT4 | MCT4 appears to regulate the proliferation, survival, and xenograft implantation/growth of some glioblastoma neurosphere lines. | KD | MCT4 KD reduces CD133+ cells and increases apoptosis, depleting glioblastoma stem-like cells and suppressing HIF transcription independently of lactate. |
| Lei et al. [59] | Taiwan (2023) | Lab (C) | Mice and CL | glioblastoma 8401, U251, glioblastoma04T, glioblastoma 09T, and HUVECs | HIF1α, HIF2α | GPx1 | GPx1 is an antioxidant enzyme detoxifying H2O2 via the binding of HIF1α to GPx1 promoter. Exosomal GPx1 plays a critical role in providing resistance to oxidative stress and radiation. | KD | The inhibitors of GPx1 sensitize vascular endothelial cells to apoptosis triggered by oxidative stress or radiation, potentially restoring the sensitivity of tumor vessels to damage. |
| Joshi et al. [60] | California (2014) | Lab (IV) | CL | LN229-HRE-AP | HIF1α | MDM2 and PTEN-PI3K-AKT axis | HIF1α undergoes hypoxic degradation via the 26 S proteasome, facilitated by MDM2 as the E3 ligase. This process is regulated by the PTEN-PI3K-AKT signaling axis. | KD | The KD of PTEN in LN229-HRE-AP cells boosts HIF1α target gene transcription, while HIF1α degradation occurs under hypoxia. |
| Lulli et al. [61] | Italy (2020) | Lab (C) | Mice and CL | GSC, HNPC, and 293T | HIF1α | miR-370-3p | miR-370-3p functions as a tumor-suppressor, restraining glioma cell growth, migration, and invasion by targeting the lncRNAs NEAT1, HMGA2, and HIF1α. | KD | NEAT1 KD inhibited glioma cell proliferation, invasion, and migration. |
| Jung et al. [62] | USA (2019) | Lab (C) | Mice and CL | SCS from biopsy | HIF1α | NIX | NIX-mediated mitophagy regulates tumor survival in the hypoxic niche of the glioblastoma microenvironment. | KD | The KD of NIX dramatically reduced the expression of stem cell markers and self-renewal by suppressing the RHEB/AKT/HIF signaling cascade. |
| Jin et al. [63] | China (2022) | Lab (C) | Mice and CL | T98G, U87, U118, and U251 | HIF1α | p21 (CDKN1A) | HIF1α binds to the p21 promoter’s HREs, boosting transcription; reciprocally, p21 enhances HIF1α mRNA transcription, sustaining its function during oxygen deficiency. | KD | The KD of HIF1A/p21 pathway inhibited glycolysis by downregulating Glut1 and LDHA and consequently caused the radiosensitivity of glioblastoma cells under hypoxic conditions. |
| Reference | Country (Year) | Study Design | Species | Cell Line(s) | Targeted HIF | Related Factor | Role of HIF and Related Factors | Target/Systematic Therapy | Pharmacological Effects |
|---|---|---|---|---|---|---|---|---|---|
| Nardinocchi et al. [64] | Italy (2010) | Lab (C) | Mice and CL | U373 | HIF1α | VEGF | The results of the luciferase assay showed that the hypoxia-induced as well as the cobalt-induced VEGF-luc activity was strongly inhibited by zinc. | Zinc | Zinc triggers HIF1α proteasomal degradation, potentially serving as a tumor progression inhibitor by suppressing pathways activated by VEGF, MDR1, and Bcl2 target genes, thereby enhancing anticancer therapies. |
| Maugeri et al. [65] | Italy (2021) | Lab (IV) | CL | U87 | HIF1α | PACAP and PAC1R | HIF1α triggers angiogenic cascade via VEGF upregulation. | PACAP | PACAP inhibits VEGF release in the glioblastoma hypoxic microenvironment by reducing new vessel formation. |
| Ma et al. [66] | China (2022) | Lab (C) | Mice and CL | U251 and GL261 | HIF1α | GLUT-1, GLUT-3, and HK2 | The overexpression of HIF1α, GLUT-1, GLUT-3, and HK2 suggests HIF1α correlates with glucose metabolism in tumor tissue. | Acriflavine and PDT | PA group inhibited HIF1α expression and improved PDT efficacy in the treatment of recalcitrant glioblastoma. |
| D’Amico et al. [67] | Italy (2023) | Lab (IV) | Cell culture | U87 and A172 | HIF1α | PACAP and VEGF | ADNP immunoreactivity was detected in most glial cells and its predominant expression in hypoxic areas overexpressing HIF1α. | The active fragment of ANDP—NAP. | ADNP modulated the HIF pathway by reducing VEGF secretion and migration. |
| D’Alessio et al. [68] | Italy (2016) | Lab (IV) | CL | U87, GCSCs, PCSCs, and HUVEC | HIF1α and HIF2α | VEGF, VEGFR1 and VEGFR2 | Angiogenesis-related molecules | Anti-angiogenic therapy | The inhibition of neoangiogenetic events in glioblastoma. |
| Cristofaro et al. [69] | Italy (2020) | Lab (IV) | CL | Glioblastoma GSCs GB7 | HIF1α | M2 | M2 receptor activation by Ape is able to arrest cell proliferation in glioblastoma cell lines. | Ape/M2 agonists | Ape treatment in hypoxic conditions is able to inhibit cell cycle progression. It downregulates the expression of stemness markers and miR-210 levels. |
| Gagner et al. [70] | USA (2017) | Lab (C) | Mice and CL | CT-2A and GL261 | HIF1α | CXCR4 and POL5551 | POL5551 inhibits CXCR4 binding to its ligand, SDF-1α, and reduces hypoxia- and stromal cell-derived factor-1a-mediated migration dose-dependently. | B20-4.1.1 and POL5551 | When combined with B20-4.1.1, POL5551 reduced glioma invasion and the number of tumor-associated MGCs, which promote glioma growth and dissemination. |
| Lin et al. [71] | China (2024) | Lab (C) | Mice and CL | C6 and U251 | HIF1α | n/a | The expression level of HIF1α is closely related to tumor cell proliferation, differentiation, apoptosis, phenotype determination, angiogenesis, energy metabolism, and resistance to therapy. | Borneol and TMZ | Borneol has the potential to enhance the sensitivity of TMZ chemotherapy, with HIF1α being a promising target for enhancing the antitumor effectiveness of TMZ. This association is closely linked to the facilitation of the autophagic degradation of HIF1α. |
| Douglas et al. [72] | USA (2023) | Lab (C) | Mice and CL | U251, D-54MG, U87MG, and CHLA-200. GSC: DB70, DB76, DB77, and DB81, 192, and 83MES | HIF1α | LonP1 and CT-L | LonP1, an ATP-dependent protease, is directly upregulated by HIF1α, with increased expression and CT-L proteasome activities observed in gliomas, correlating with high tumor grade and poor patient survival. | BT317 | BT317 has a dual LonP1 and CT-L inhibition profile and induces increased ROS production and autophagy-dependent cell death in clinically relevant, IDH mutant malignant astrocytoma. |
| Arienti et al. [73] | Italy (2021) | Lab (IV) | CL | G34, G40, G44, and CHME-5 | HIF1α | n/a | The expression of HIF1α stimulates the upregulation of the glycolysis metabolic pathway, boosting ATP production necessary for cell survival and proliferation. | HBO | HBO inhibits cell proliferation, downregulates HIF1α expression, and induces glucose metabolism reprogramming. |
| Lin et al. [74] | USA (2015) | Lab (C) | Mice and CL | U87 and LN229 | HIF1α | IGFBP2 and IGFI | The activation of IGFIR by IGFI and subsequent downstream signaling lead to malignant cell proliferation, motility, and metastasis. | GFBP2-HIF1α targeting | Blocking specific molecular interactions within the insulin signaling pathway could potentially result in a notable decrease in glioblastoma growth. |
| Lund et al. [75] | Denmark (2004) | Lab (IV) | CL | U87 | HIF1α | VEGF and angiopoetin-1, -2, -4 | VEGF protects endothelial cells from apoptosis via Raf activation, while Ang-1 and Ang-2 are essential for angiogenesis, and Ang-4 induces Tie-2 receptor autophosphorylation. | IR | The combinations of radiation therapy and therapy targeting the signaling pathways of VEGF have proven more effective than irradiation alone in animal models. |
| Hofstetter et al. [76] | USA (2012) | Lab (IV) | Cell culture | TSCs (334, 974, and 980) | HIF1α | PP2A | Hypoxia-induced PP2A halts cell proliferation, decreasing metabolic activity, and promotes survival of TSCs in severe hypoxia. | The modulation of PP2A | Possible synergistic effects of chemotherapy with PP2A inhibition. |
| Bi et al. [77] | China (2021) | Lab (IV) | CL | U251 | HIF1α | ARA1 and ARA3 | PSH decreases HIF1α expression via ARA3 inactivation and induces cell cycle arrest via ARA1. | PSH | PSH reduced U251 cell viability via the inhibition of ARA1 and ARA3 expression and further inhibited Akt and 44/42 MAPK phosphorylation, induced apoptosis, and cell cycle arrest. |
| Ma et al. [66] | China (2022) | Lab (C) | Mice and CL | U251 and GL261 | HIF1α | GLUT1, GLUT3, and HK2 | Human glioblastoma tissues showed extensive overexpression of HIF1α, GLUT-1, GLUT-3, and HK2, suggesting HIF1α correlated with glucose metabolism in tumor tissue. | PDT and acriflavine | Acriflavine combined with PDT attenuated the expression of HIF1α, GLUT-1, GLUT-3, and HK2 and improved tumor suppression. |
| Khoei et al. [78] | Iran (2016) | Lab (IV) | Cell culture | U87 | HIF1α | n/a | Hypoxia activates the HIF1α pathway and reduces the sensitivity of tumor cells to radiation and chemotherapeutic drugs. | Res, MX, and IUdR | A combination of MX and Res with IUdR can decrease colony formation ability and increase DNA damage of gamma-ray radiation in 350 mm spheroids. The cytotoxic effect of Rad and therapeutic ratio increases. |
| Liu et al. [79] | China (2020) | Lab (C) | Mice and CL | G422-Glioblastoma | HIF1α | n/a | HIF1α is a mediator in the mechanism of chemotherapy resistance. | RT/TMZ supplemented with mannose | RT/TMZ/Man could offer a disease cure for glioblastoma through metabolically abolishing the HIF-1-mediated resistance. |
| Dačević et al. [80] | Serbia (2013) | Lab (IV) | CL | U87, U87-TxR, NCI-H460, NCI-H460/R, and HaCaT | HIF1α | Pgp, VEGF, and GSH | P-gp activity governs MDR development. GSH is implicated in detoxification and VEGF has a role in tumor angiogenesis and progression. | SF | SF hampers the growth of cancer cells by integrating its phosphorylated derivatives into DNA. Moreover, SF diminishes the levels of HIF1α, which governs the expression of both P-gp and VEGF. As a consequence, SF’s influence on multidrug resistance (MDR) stems from its ability to inhibit the GSH detoxification system. |
| Ishii et al. [81] | Japan (2016) | Lab (IV) | CL | T98G | HIF1α and HIF2α | SOX2 and NANOG | SOX2 and NANOG, transcription factors crucial for embryonic stem cell self-renewal and pluripotency, also play critical roles in glioblastoma tumorigenesis. | The targeting of the peri-necrotic niche | Eradicating glioblastoma cells and overcoming the therapeutic resistance of glioblastomas. |
| Li et al. [82] | China (2023) | Lab (C) | Mice and CL | U251 and U87 | HIF1α | GLUT1 | The HIF-1/GLUT-1 axis enhanced the cytotoxicity of temozolomide in gliomas as a result of PDT treatment, which was influenced by ROS. | TMZ and PDT | Photodynamic therapy boosts the cytotoxic effects of temozolomide on glioblastoma by reshaping anaerobic glycolysis. |
| Bernstock et al. [83] | USA (2017) | Lab (IV) | CL | U251, LN229, Mz18, and SH-SY5Y | HIF1α | SUMO | SUMO maintains cellular function under conditions of stress. | Topotecan | Topotecan reduces the levels of global SUMO conjugation, CDK6, and HIF1α in glioblastoma cells, thereby affecting both the cell cycle and metabolic profile. |
| Tafani et al. [84] | Italy (2011) | Lab (C) | Mice and CL | Biopsy | HIF1α | HK2 and VEGF | After 4 h of hypoxia, there was an elevation in mRNA expression for HIF1α. VEGF mRNA demonstrated an increase during hypoxia treatment, while HK2 mRNA exhibited increases after 4, 24, and 48 h of hypoxia. | Digoxin and acriflavine | The prevention of HIF1α protein synthesis and dimerization. |
| Muh et al. [85] | USA (2014) | Lab (IV) | Mice | U87 and U373 | HIF1α | PTEN-PI3K | This synergistic activity was correlated with a synergistic suppression of HIF1α accumulation under hypoxic conditions in glioma models. | LY294002 and 2ME2 | Drugs demonstrated synergy in blocking HIF1α accumulation in glioblastoma cell lines. |
| Pore et al. [86] | United States (2006) | Lab (C) | Mice and CL | U87 and U251 | HIF1α | PI3K/Akt | Nelfinavir downregulates VEGF and HIF-1 expression through the inactivation of PI3K/Akt pathways. | Nelfinavir and amprenavir | Nelfinavir downregulates VEGF and HIF-1 expression through the inactivation of PI3K/Akt pathways, decreases angiogenesis in vivo, and downregulates HIF1α through the inhibition of protein synthesis. Amprenavir inhibits VEGF and HIF-1 expression in glioblastoma cells but not in normal human astrocytes. |
| Sugimoto et al. [87] | Japan (2017) | Lab (IV) | CL | U87 | n/d | GFAP and CBR1 | Hypoxia decreases the expression of CBR1 and glial fibrillary acidic protein while increasing the expression of VEGF and cyclooxygenase-2. | WIN 55,212-2 | CB engagement induces cell death in U-87 MG cells under normoxic conditions, with CB agonist-induced death being reduced in hypoxic conditions. |
| Lin et al. [88] | Taiwan (2021) | Lab (IV) | CL | U251 | HIF1α | PPARα | Hypoxia-induced HIF1α regulates pH-regulating proteins in glioblastoma. | Fenofibrate | Fenofibrate effectively inhibits hypoxia-induced HIF1α and CA9 expression in glioblastoma by activating HO-1 via AMPK and promoting HIF1α degradation, suggesting its potential as a multi-pathway anti-glioblastoma agent. |
| Reference | Country (Year) | Study Design | Species | Cell Line(s) | Targeted HIF | Related Factor | Role of HIF and Related Factors | Gene Modification | Effect of Gene Modification | Targeted Therapy | Pharmacological Effects |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Huang et al. [90] | China (2018) | Lab (IV) | CL | U87 | HIF1α | PI3K/Akt/mTOR | PI3K/Akt/mTOR/HIF1α pathway is involved in enhancing the migration and invasion of human glioblastoma U87 cells under hypoxia. | TF | The enhancements of the migration and invasion of U87 cells under hypoxia could be suppressed by the mTOR pathway siRNA by targeting HIF1α. | 2-ME, LY294002, rapamycin, and p70S6K siRNA | 2-ME is an HIF1α inhibitor that reduces the migration and invasion of glioblastoma cells. The inhibitors of PI3K/Akt/mTOR, LY294002, and rapamycin, reduced the migration, invasion, and HIF1α protein expression. p70S6K siRNA suppressed the migration, invasion, and HIF1α expression under hypoxia. |
| Chhipa et al. [91] | USA (2018) | Lab (C) | Mice and CL | U87, A172, T98G, and HEK 293T | HIF1α | AMPK (AMPK/CREB1 axis) | By phosphorylating CREB1, AMPK enhances HIF1α and GABPA transcription to support glioblastoma bioenergetics. | KD and KO | Silencing CREB1 decreases HIF1α activity, cell viability, and GSC bioenergetics, while the knockout of AMPKα1 enhances glycolysis and accelerates tumorigenesis. | Bafilomycin | AMPK inhibition reduces GSC viability and has antitumorigenic effects. |
| Pang et al. [92] | USA (2023) | Lab (C) | Mice and CL | 293T | HIF1α | LGMN | LGMN is specifically expressed in TAMs and regulated by HIF1α | KD and KO | BMDMs from HIF1α-mKO mice exhibited aberrantly diminished Lgmn expression levels, while Lgmn-mKD mice displayed a marked extension in survival compared to control mice. | Anti-PD1 | The blockade of the HIF1α-LGMN axis synergizes with anti-PD1 therapy in glioblastoma. |
| Hu et al. [93] | USA (2012) | Lab (C) | Mice and CL | U87, T98G, U251, U138, A172, G55, SF8244, SF8557, and U373 | HIF1α | HIF1α/AMPK | HIF1α and AMPK control hypoxia-induced LC3 changes, while BNIP3 expression depends solely on HIF1α, and p62 degradation occurs independently of both. | KO and TF | The knockdown of the essential autophagy gene ATG7 promotes bevacizumab responsiveness. | BEV and chloroquine | BEV treatment increased BNIP3 expression and hypoxia-driven growth in glioblastoma xenografts, reversed by chloroquine, an autophagy inhibitor. |
| Chou et al. [94] | Taiwan (2012) | Lab (C) | Mice and CL | U87, glioblastoma 8401, and U251 | HIF1α | ABCB1 | Cycling hypoxic stress increases chemoresistance via HIF–1-mediated ABCB1 induction. | KD | When the induction of ABCB1 was inhibited by siRNA, the chemotherapy resistance induced by cycling hypoxic stress decreased. | YC-1 | YC-1 combined with BCNU chemotherapy decreased ABCB1 induction and made therapy more effective. |
| Barliya et al. [95] | Israel (2011) | Lab (IV) | CL | ARPE-19, U87, and RCC-C2VHL−/− | HIF1α | hsp90 | Hsp90 mediates the pathways vital for angiogenesis, cell migration, and invasion. | TF | Hypericin interferes with VEGF promoter activation in tumor cell lines. | Hypericin | The hypericin-induced degradation of hsp90 client proteins compromises the pathways involved in angiogenesis, cell migration, and invasion. |
| Hsieh et al. [96] | Taiwan (2011) | Lab (C) | Mice and CL | glioblastoma 8401 and U87 | HIF-1 | NADPH oxidase subunit 4-mediated reactive oxygen species | Cycling hypoxic stress significantly increases ROS production, HIF-1 activation, and tumor growth. Nox4 is a critical mediator of these processes. | KD | Blocking ROS production through Nox4 shRNA inhibits tumor growth induced by cycling hypoxia or the tumor microenvironment. | Tempol | Tempol treatment inhibits tumor growth induced by cycling hypoxia or the tumor microenvironment. |
| Kannappan et al. [97] | United Kingdom (2022) | Lab (C) | Mice and CL | U87MG, U251MG, and U373MG | HIF1α and HIF2α | NF-kB | NF-kB, HIF1α, and HIF2α induce the expression of key EMT- and metastasis-related genes and promote glioblastoma cell migration and invasion. | TF | The expression of HIF2α mRNA was upregulated by HIF1α transfection but not vice versa. | Disulfiram | Disulfiram inhibits NF-kB activity and targets hypoxia-induced GSCs. It shows selective toxicity to glioblastoma cells, eradicates GSCs, and blocks migration and invasion. |
| Joseph et al. [98] | The Netherlands (2015) | Lab (IV) | CL | U87, SNB75, and U251 | HIF1α and HIF2α | ZEB1 (HIF1α-ZEB1 axis) | HIF1α–ZEB1 signaling axis promotes hypoxia-induced mesenchymal shift and invasion in glioblastoma in a cell line-dependent fashion. | KD | The ShRNA-mediated knockdown of HIF1α, and not HIF2α, prevented hypoxia-induced mesenchymal transition. | Digoxin | Digoxin inhibits HIF1α mRNA translation. |
| Caragher et al. [99] | USA (2019) | Lab (C) | Mice and CL | U251, glioblastoma 43, glioblastoma 12, glioblastoma 5, glioblastoma 6, and glioblastoma 39 | HIF1α and HIF2α | DRD2 | The activation of DRD2 triggers the expression of HIF proteins and enhances the capacity for sphere formation, which serves as an indicator of the GIC state and tumorigenicity. | KD | The SH-RNA-mediated knockdown of DRD2 showed a significant reduction in sphere-forming capacity. | Chlorpromazine | The inhibition of glioblastoma growth by blocking the dopamine signaling pathway. |
| Peng et al. [100] | China (2021) | Lab (C) | Mice and CL | U251 | HIF1α | PDGFD-PDGFRα | Under normoxic or mild-hypoxic conditions, HIF1α binds to the PDGFD proximal promoter and PDGFRA intron enhancers in glioblastoma cells, leading to the induction of their expression. | KD and KO | PDFGRA knockdown extends the survival of xenograft mice, inhibits cell growth and invasion in vitro, and eradicates tumor growth in vivo. | Echinomycin | Echinomycin induces glioblastoma cell apoptosis and effectively inhibits the growth of glioblastoma in vivo by simultaneously targeting the HIF1α-PDGFD/PDGFRα-AKT feedforward pathway. |
| Han et al. [101] | China (2015) | Lab (C) | Mice and CL | U87 and U251 | HIF1α | NF-κB/RelA-PKM2 | NF-κB/RelA is involved in proliferation, anti-apoptosis, angiogenesis, and metastasis, promoting aerobic glycolysis via the transcriptional activation of PKM2. | TF | NF-κB/RelA promotes glioblastoma cell glycolysis depending on PKM2. | Fenofibrate | FF inhibits glioblastoma glycolysis in a dose-related manner depending on PPARα activation. It inhibits the transcriptional activity of NF-κB/RelA and disrupts its association with HIF1α. |
| Dominguez et al. [102] | USA (2013) | Lab (C) | Mice and CL | U251, U87, A375, MDA-MB-231, HeLa, and human fibroblast cell lines | HIF1α | DGKα | DGKα and its product, phosphatidic acid, are associated with multiple oncogenic pathways such as mTOR, HIF1α, and Akt. | KD | In cancer cells, the inhibition of DGKα results in cell toxicity through caspase-mediated apoptosis. The reduced expression of mTOR and HIF1α significantly contributes to the cytotoxic effects observed upon DGKα knockdown and inhibition in cancer. | R50922 and R59949 | Induced caspase-mediated apoptosis in glioblastoma cells and in other cancers, but lacked toxicity in non-cancerous cells. |
| Hsieh et al. [103] | Taiwan (2015) | Lab (C) | Mice and CL | U251, U87, and glioblastoma 8401 | HIF1α and HIF2α | Livin proteins | HIF1α regulates Livin transcription in hypoxia, promoting anti-apoptosis in glioblastoma and enhancing radioresistance and chemoresistance. | KD | The knockdown of Livin suppresses tumor hypoxia-induced TR and generates a synergistic suppression of antitumor growth and tumor cell death. | Cell-permeable peptide TAT-Lp15 | Livin blockage enhances the efficiency of radiation plus temozolomide treatment in glioblastoma xenografts. |
| Ahmed et al. [104] | UK (2018) | Lab (IV) | CL | U251, U87, and SNB219 | HIF1α and HIF2α | CD133 | CD133 is a cell surface marker used to identify glioblastoma cancer stem cells. | KD | HIF1α and HIF2α knockdown led to a reduced CD133 expression. CD133 knockdown increases the sensitivity of glioblastoma cells to cisplatin. | Cisplatin | The hypoxia-induced cisplatin sensitivity of glioblastoma cells may be HIF-independent and may be directly or indirectly induced via CD133 activation. |
| Lee et al. [105] | Korea (2017) | Lab (C) | Mice and CL | Biopsy | HIF1α | ERK1/2 and VEGF | ERK1/2 signaling and VEGF, a HIF1α downstream target, contribute to solid tumor pathogenesis. | TF | DT at clinically relevant concentrations reduces hypoxia-induced HIF1α protein accumulation and downstream signaling pathways. | Digitoxin | DT at clinically achievable concentration functions as an inhibitor of HIF1α. |
| Bar et al. [106] | USA (2010) | Lab (C) | Mice and CL | HSR-glioblastoma 1 and HSR-glioblastoma 2 | HIF1α | CD133 | HIF1α induces CD133 expression and enhances the stem-like tumor subpopulation in hypoxia. | TF | An elevated percentage of CD133 positive cells. | Digoxin | Digoxin suppressed HIF1α protein expression, HIF1α downstream targets, and slowed tumor growth. |
| Chen et al. [107] | China (2015) | Lab (C) | Mice and CL | U251, U87, and glioblastoma 8401 | HIF1α | NF-κB and Bc-xl | Cycling hypoxia mediates Bcl-xL expression via HIF1α or NF-κB activation, which results in chemoresistance. | KD | Bcl-xL knockdown inhibited cycling hypoxia-induced chemoresistance. | Tempol, YC-1, and Bay 11-7082 | The suppression of the cycling hypoxia-mediated Bcl-xL induction. |
| Li et al. [108] | India (2020) | Lab (C) | Mice and CL | U87 and U251 | HIF1α | IDH1-R132H | The overexpression of IDH1-R132H increased the expression of HIF1α and the downregulation of HIF1α suppressed the IDH1-R132H-induced effect on glioblastoma. | KD | The KD of FAT1 inhibited the IDH1-R132H-induced reduction in tumor growth in xenograft mice. | TMZ | The overexpression of IDH1-R132H led to reduced cell proliferation, increased apoptosis, decreased migration and invasion, enhanced TMZ-induced cytotoxicity, and diminished tumor growth in xenograft mice. |
| Ge et al. [109] | China (2018) | Lab (C) | Mice and CL | U87MG and HEK293T | HIF1α | miR-26a | HIF1α/miR-26a axis strengthens the acquisition of TMZ resistance through the prevention of Bax and Bad in mitochondria dysfunction in glioblastoma. | TF | HIF1α serves as a pivotal upstream regulator of miR-26a expression in glioma. | TMZ | miR-26a is an important regulator of TMZ resistance induced by hypoxia, which can effectively protect mitochondria function and reduce apoptosis by targeting bax and bad. |
| Liao et al. [110] | China (2022) | Lab (C) | Mice and CL | U251, U87, A172, GSC11, GSC20, GSC262, GSC267, GSC295, GSC28, GSC284, and GSC627 | HIF1α | PRMT3 | PRMT3 promotes glioblastoma progression by enhancing HIF1α-mediated glycolysis and metabolic rewiring. | KD | The reduced proliferation and migration of glioblastoma cell lines and patient-derived GSC in cell culture and inhibited tumor growth. | SGC707 | The targeting of PRMT3 decreases HIF1α expression and glycolytic rates in glioblastoma cells and inhibits glioblastoma growth. |
| Kioi et al. [111] | California (2010) | Lab (C) | Mice and CL | U251 and U87 | HIF1α | SDF-1/CXCR4 | BMDCs are recruited to tumors through the HIF-1-dependent interaction of SDF-1 and its receptor, CXCR4. | TD | AMD3100 enhanced the radiosensitivity. | AMD3100 | AMD3100 is an inhibitor of SDF-1/CXCR4 interactions, which blocks the vasculogenesis pathway. |
| Boso et al. [112] | Italy (2019) | Lab (IV) | CL | Biopsy | HIF1α | β-catenin/TCF1 | In hypoxic glioblastoma cells, the β-catenin/TCF1 complex recruits HIF1α to promote the transcription of genes associated with neuronal differentiation. | TF | Cells silenced for TCF1 experienced a complete inhibition of their neuronal differentiation potential. | TCF4E | TCF4E possesses inhibitory effects on gene transcription. |
| Reference | Country (Year) | Study Design | Sample (N) | Age | Gender (Male/Female) | Target(s) (Type of HIF) | Findings |
|---|---|---|---|---|---|---|---|
| Chen et al. [114] | China (2019) | Prospective | 42 | 26–76 | 17/25 | CAV1 and HIF1α | HIF1α is more expressed in the nucleus and cytoplasm of neoplastic cells. HIF1α correlated with high CAV1 expression, larger glioblastoma size, and lesser survival time. |
| Bache et al. [115] | Germany (2015) | Retrospective | 41 | Median: 63 | 16/18 | HIF1α, HIF2α, CA9, VEGF, GLUT-1, OPN, survivin, EGFR, hTERT, and OCT4 | HIF2α, CA9, VEGF, hTERT, and OCT4 were higher in glioblastoma than in tumor-free brain tissues; the mRNA expression levels of HIF genes resulted in shorter survival times for patients with glioblastoma; the mRNA expression levels of HIF and stem cell-associated genes are important glioblastoma markers. |
| Erpolat et al. [116] | Turkey (2012) | Retrospective | 79 | Median: 49 | n/d | HIF1α, CA9, and OPN | High levels of cytoplasmic and nuclear HIF1α, CA9, and osteopontin correlated with shorter survival, especially with high hypoxic scores, with high hypoxic score-1 being the main independent negative predictor for survival. |
| Clara et al. [122] | Brazil (2014) | Retrospective | 208 | Median: 56 | 127/81 | HIF1α | HIF1α expression in glioblastoma is correlated with increased vascular density and with VEGF and PDGF-C expression. Nuclear HIF1α and VEGF staining also correlated with survival. |
| Kaynar et al. [117] | Turkey (2008) | Prospective | 26 | Median: 51 | 17/9 | HIF1α | HIF1α levels were elevated in glioblastoma, indicating a role in angiogenesis possibly beyond hypoxia. |
| El-Benhawy et al. [123] | Egypt (2022) | Prospective | 80 | Mean: 49.49 | 58/22 | HIF1α, VEGF, OPN, erythropoietin, caveolin-1, GLUT-1, and LDH | Serum hypoxia biomarkers, including HIF1α, VEGF, and LDH, increased significantly after radiotherapy in patients with glioblastoma, indicating their potential role in tumor progression and treatment response. |
| Nobuyuki et al. [118] | Japan (2004) | Prospective | 60 | Median: 58.7 | 33/27 | HIF1α | HIF1 serves as a hypoxic sensor in tumors like glioblastoma, with its expression level indicating radioresistance and guiding postoperative radiotherapy protocols. |
| Ji et al. [119] | China (2013) | Prospective | 68 | Mean: 48 | 46/22 | HIF1α | High HIF1α expression in glioblastoma correlates with poorer outcomes, including shorter overall and progression-free survival, suggesting its potential as a marker for targeted treatment. |
| Sfifou et al. [120] | Morocco (2021) | Prospective | 22 | Mean: 54 | n/d | HIF1α | Patients with negative HIF1α expression and positive IDH1 expression have a better prognosis, with statistically significant differences observed in overall survival rates, indicating HIF1α as a potential prognostic marker. |
| Potharaju et al. [121] | India (2019) | Prospective | 87 | Median: 55 | 59/28 | HIF1α | The strong nuclear staining of HIF1α was observed in 48% of the samples, correlating with poor prognosis independently. Patients with strong HIF1α and TERT expression had the worst prognosis, indicating HIF1α as a potential prognostic marker in glioblastoma. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begagić, E.; Bečulić, H.; Džidić-Krivić, A.; Kadić Vukas, S.; Hadžić, S.; Mekić-Abazović, A.; Šegalo, S.; Papić, E.; Muchai Echengi, E.; Pugonja, R.; et al. Understanding the Significance of Hypoxia-Inducible Factors (HIFs) in Glioblastoma: A Systematic Review. Cancers 2024, 16, 2089. https://doi.org/10.3390/cancers16112089
Begagić E, Bečulić H, Džidić-Krivić A, Kadić Vukas S, Hadžić S, Mekić-Abazović A, Šegalo S, Papić E, Muchai Echengi E, Pugonja R, et al. Understanding the Significance of Hypoxia-Inducible Factors (HIFs) in Glioblastoma: A Systematic Review. Cancers. 2024; 16(11):2089. https://doi.org/10.3390/cancers16112089
Chicago/Turabian StyleBegagić, Emir, Hakija Bečulić, Amina Džidić-Krivić, Samra Kadić Vukas, Semir Hadžić, Alma Mekić-Abazović, Sabina Šegalo, Emsel Papić, Emmanuel Muchai Echengi, Ragib Pugonja, and et al. 2024. "Understanding the Significance of Hypoxia-Inducible Factors (HIFs) in Glioblastoma: A Systematic Review" Cancers 16, no. 11: 2089. https://doi.org/10.3390/cancers16112089
APA StyleBegagić, E., Bečulić, H., Džidić-Krivić, A., Kadić Vukas, S., Hadžić, S., Mekić-Abazović, A., Šegalo, S., Papić, E., Muchai Echengi, E., Pugonja, R., Kasapović, T., Kavgić, D., Nuhović, A., Juković-Bihorac, F., Đuričić, S., & Pojskić, M. (2024). Understanding the Significance of Hypoxia-Inducible Factors (HIFs) in Glioblastoma: A Systematic Review. Cancers, 16(11), 2089. https://doi.org/10.3390/cancers16112089