
Coupling Kinesin Spindle Protein and Aurora B Inhibition with Apoptosis Induction Enhances Oral Cancer Cell Killing

 ,
, 

 and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Small Molecule Inhibitors
2.2. Cell Culture
2.3. RNA Extraction, cDNA Synthesis, and Real-Time PCR
2.4. Protein Extraction and Western Blotting
2.5. Indirect Immunofluorescence
2.6. MTT Assay
2.7. Apoptosis Detection Using Annexin V/PI Staining
2.8. Mitotic Index Determination
2.9. Time-Lapse Microscopy
2.10. Phase-Contrast and Fluorescence Microscopy Images
2.11. Colony Formation Assay
2.12. Statistical Analysis
3. Results
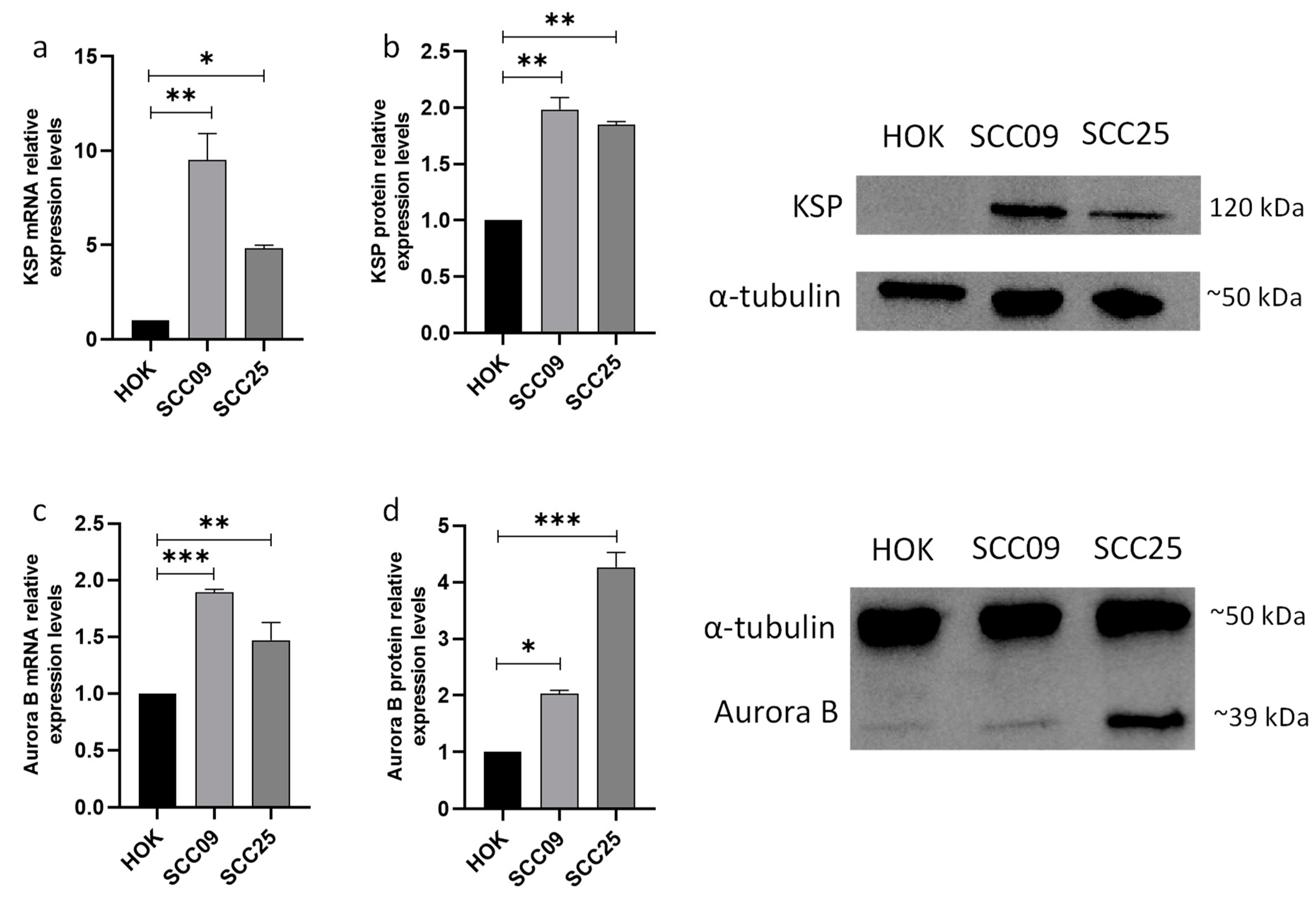
3.1. KSP and Aurora B Proteins Are Overexpressed in Oral Squamous Carcinoma Cells
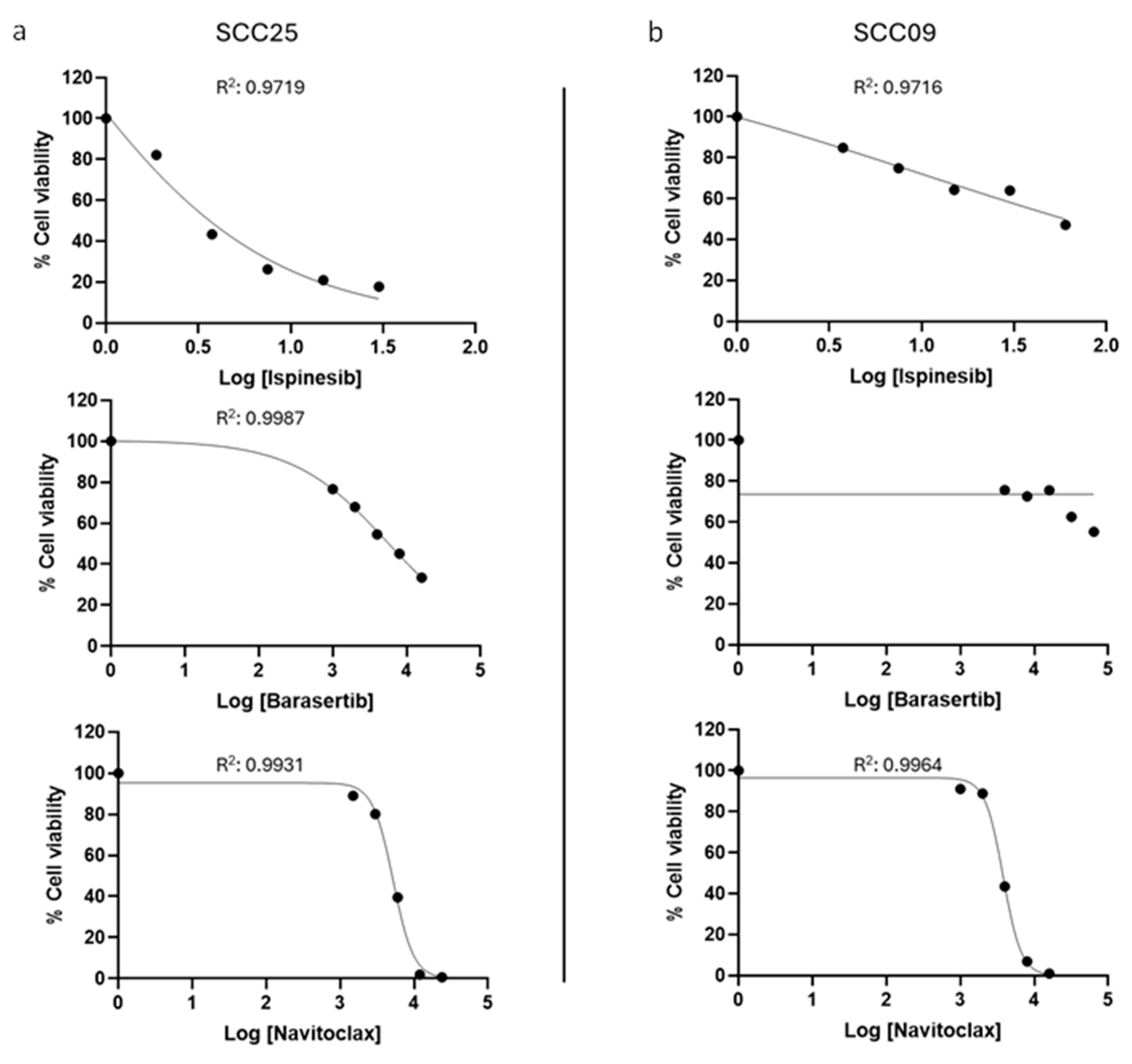
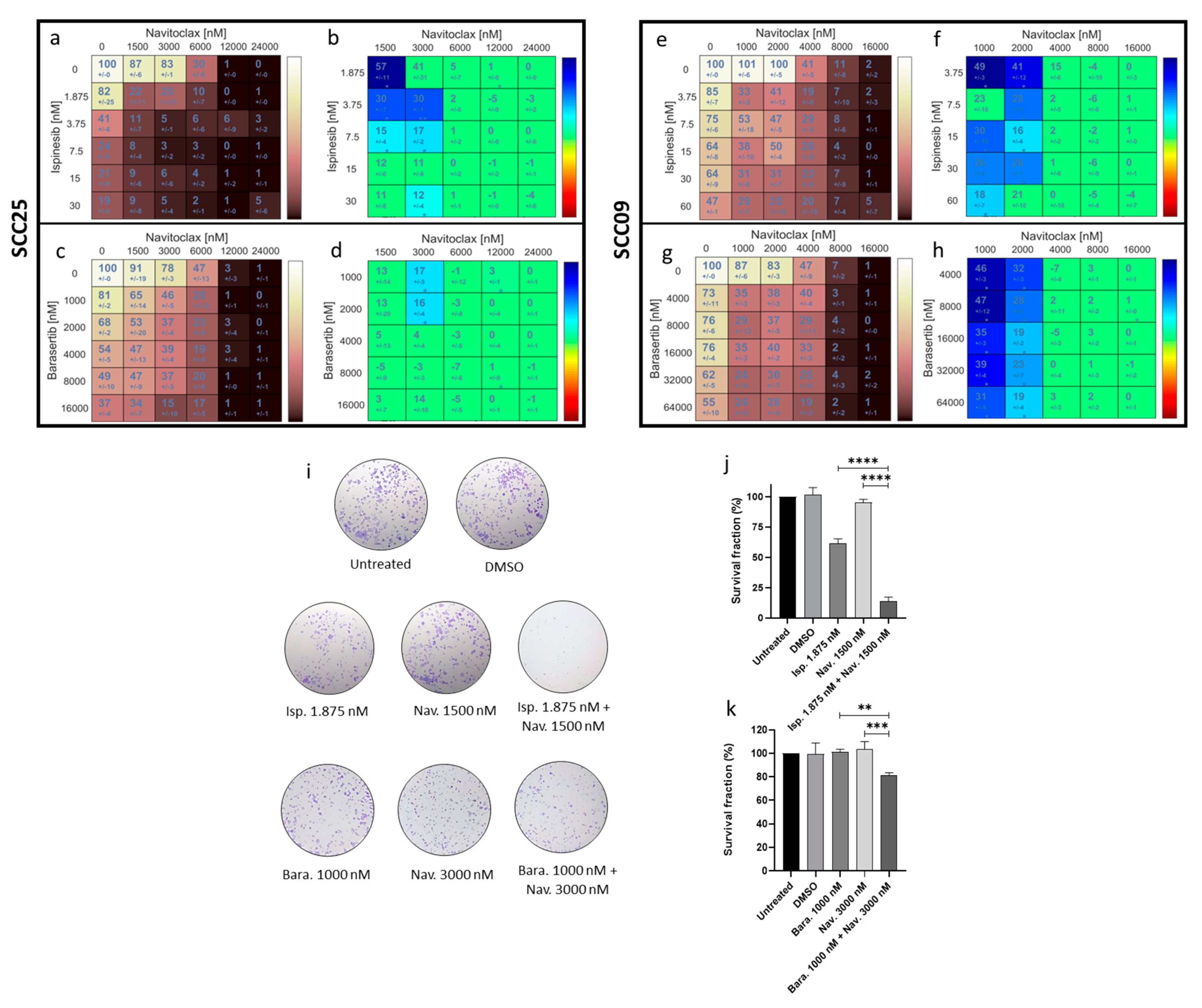
3.2. The Combinatorial Approaches with Ispinesib and Navitoclax and Barasertib with Navitoclax Show Synergistic Effects in Oral Cancer Cells
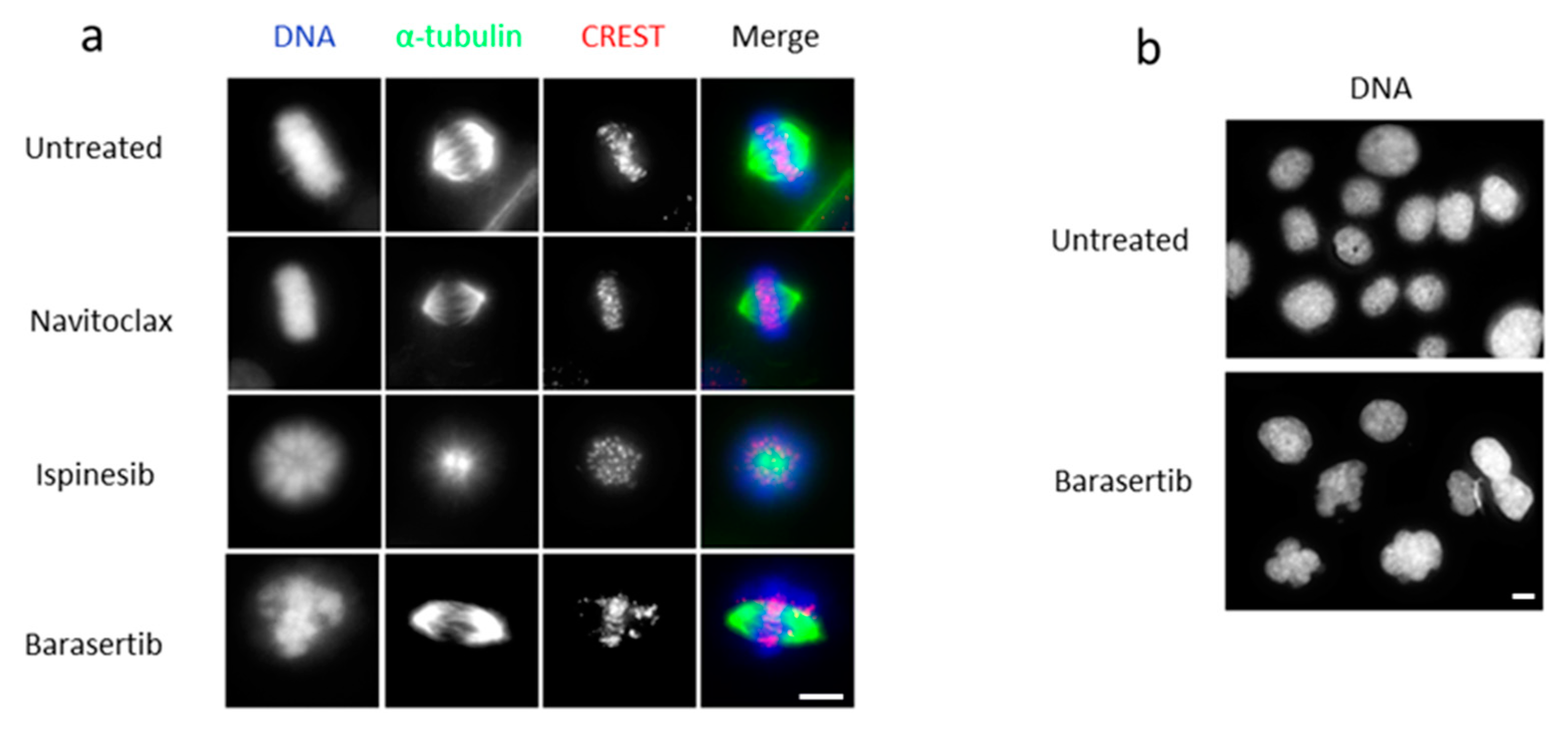
3.3. The Combined Treatment with Ispinesib and Navitoclax Enhances Death in Mitosis in Oral Cancer Cells
3.4. Combining Barasertib-Mediated Aurora B Inhibition with Navitoclax Shifts the Cancer Cell Fate from Post-Slippage Cell Survival to Post-Slippage Cell Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Melo, B.A.dC.; Vilar, L.G.; de Oliveira, N.R.; de Lima, P.O.; Pinheiro, M.d.B.; Domingueti, C.P.; Pereira, M.C. Human Papillomavirus Infection and Oral Squamous Cell Carcinoma—A Systematic Review. Braz. J. Otorhinolaryngol. 2021, 87, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Li, B. STAT3 and Its Targeting Inhibitors in Oral Squamous Cell Carcinoma. Cells 2022, 11, 3131. [Google Scholar] [CrossRef] [PubMed]
- Nandini, D.B.; Rao, R.S.; Hosmani, J.; Khan, S.; Patil, S.; Awan, K.H. Novel Therapies in the Management of Oral Cancer: An Update. Disease-a-Month 2020, 66, 101036. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lu, Y.; Nannapaneni, S.; Griffith, C.C.; Steuer, C.; Qian, G.; Wang, X.; Chen, Z.; Patel, M.; El-Deiry, M.; et al. Combinatorial Approaches Targeting the EGFR Family and C-Met in SCCHN. Oral Oncol. 2021, 112, 105074. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; Agrawal, N.; Gooi, Z. Lip and Oral Cavity Squamous Cell Carcinoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Guigay, J.; Tahara, M.; Licitra, L.; Keilholz, U.; Friesland, S.; Witzler, P.; Mesía, R. The Evolving Role of Taxanes in Combination With Cetuximab for the Treatment of Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck: Evidence, Advantages, and Future Directions. Front. Oncol. 2019, 9, 668. [Google Scholar] [CrossRef] [PubMed]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-Targeting Agents and Their Impact on Cancer Treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics 2021, 13, 1011. [Google Scholar] [CrossRef]
- Yu, W.-X.; Li, Y.-K.; Xu, M.-F.; Xu, C.-J.; Chen, J.; Wei, Y.-L.; She, Z.-Y. Kinesin-5 Eg5 Is Essential for Spindle Assembly, Chromosome Stability and Organogenesis in Development. Cell Death Discov. 2022, 8, 490. [Google Scholar] [CrossRef]
- Bartoli, K.M.; Jakovljevic, J.; Woolford, J.L.; Saunders, W.S. Kinesin Molecular Motor Eg5 Functions during Polypeptide Synthesis. Mol. Biol. Cell 2011, 22, 3420–3430. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.J.; Wadsworth, P. Kinesin-5 Regulation and Function in Mitosis. Trends Cell Biol. 2019, 29, 66–79. [Google Scholar] [CrossRef]
- Shao, Y.-Y.; Sun, N.-Y.; Jeng, Y.-M.; Wu, Y.-M.; Hsu, C.; Hsu, C.-H.; Hsu, H.-C.; Cheng, A.-L.; Lin, Z.-Z. Eg5 as a Prognostic Biomarker and Potential Therapeutic Target for Hepatocellular Carcinoma. Cells 2021, 10, 1698. [Google Scholar] [CrossRef]
- Portella, G.; Passaro, C.; Chieffi, P. Aurora B: A New Prognostic Marker and Therapeutic Target in Cancer. Curr. Med. Chem. 2011, 18, 482–496. [Google Scholar] [CrossRef]
- Qi, G.; Ogawa, I.; Kudo, Y.; Miyauchi, M.; Siriwardena, B.S.M.S.; Shimamoto, F.; Tatsuka, M.; Takata, T. Aurora-B Expression and Its Correlation with Cell Proliferation and Metastasis in Oral Cancer. Virchows Arch. 2007, 450, 297–302. [Google Scholar] [CrossRef]
- Roy, B.; Han, S.J.Y.; Fontan, A.N.; Jema, S.; Joglekar, A.P. Aurora B Phosphorylates Bub1 to Promote Spindle Assembly Checkpoint Signaling. Curr. Biol. 2022, 32, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.A.; Siu, L.L.; Chen, E.X.; Hotte, S.J.; Chia, S.; Schwarz, J.K.; Pond, G.R.; Johnson, C.; Colevas, A.D.; Synold, T.W.; et al. Phase II Study of Ispinesib in Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck. Investig. New Drugs 2008, 26, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Bélanger, K.; Rao, S.C.; Petrella, T.M.; Tozer, R.G.; Wood, L.; Savage, K.J.; Eisenhauer, E.A.; Synold, T.W.; Wainman, N.; et al. A Phase II Study of Ispinesib (SB-715992) in Patients with Metastatic or Recurrent Malignant Melanoma: A National Cancer Institute of Canada Clinical Trials Group Trial. Investig. New Drugs 2008, 26, 249–255. [Google Scholar] [CrossRef]
- Boss, D.S.; Witteveen, P.O.; van der Sar, J.; Lolkema, M.P.; Voest, E.E.; Stockman, P.K.; Ataman, O.; Wilson, D.; Das, S.; Schellens, J.H. Clinical Evaluation of AZD1152, an i.v. Inhibitor of Aurora B Kinase, in Patients with Solid Malignant Tumors. Ann. Oncol. 2011, 22, 431–437. [Google Scholar] [CrossRef]
- Helfrich, B.A.; Kim, J.; Gao, D.; Chan, D.C.; Zhang, Z.; Tan, A.-C.; Bunn, P.A. Barasertib (AZD1152), a Small Molecule Aurora B Inhibitor, Inhibits the Growth of SCLC Cell Lines In Vitro and In Vivo. Mol. Cancer Ther. 2016, 15, 2314–2322. [Google Scholar] [CrossRef]
- Exertier, P.; Javerzat, S.; Wang, B.; Franco, M.; Herbert, J.; Platonova, N.; Winandy, M.; Pujol, N.; Nivelles, O.; Ormenese, S.; et al. Impaired Angiogenesis and Tumor Development by Inhibition of the Mitotic Kinesin Eg5. Oncotarget 2013, 4, 2302–2316. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic Slippage: An Old Tale with a New Twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Suleimenov, M.; Bekbayev, S.; Ten, M.; Suleimenova, N.; Tlegenova, M.; Nurmagambetova, A.; Kauanova, S.; Vorobjev, I. Bcl-XL Activity Influences Outcome of the Mitotic Arrest. Front. Pharmacol. 2022, 13, 933112. [Google Scholar] [CrossRef]
- Alam, M.; Mishra, R. Bcl-XL Expression and Regulation in the Progression, Recurrence, and Cisplatin Resistance of Oral Cancer. Life Sci. 2021, 280, 119705. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Chakraborty, R.; Ranganathan, S. Proliferation and Apoptosis Pathways and Factors in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 1562. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kayano, T.; Takagi, M. Dysregulated Expression of Bcl-2 and Bax in Oral Carcinomas: Evidence of Post-Transcriptional Control. J. Oral Pathol. Med. 2000, 29, 63–69. [Google Scholar] [CrossRef]
- Sharma, A.; Boise, L.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, Y.; Huang, H.-C.; Mitchison, T.J. Navitoclax (ABT-263) Accelerates Apoptosis during Drug-Induced Mitotic Arrest by Antagonizing Bcl-XL. Cancer Res. 2011, 71, 4518–4526. [Google Scholar] [CrossRef]
- Tan, N.; Malek, M.; Zha, J.; Yue, P.; Kassees, R.; Berry, L.; Fairbrother, W.J.; Sampath, D.; Belmont, L.D. Navitoclax Enhances the Efficacy of Taxanes in Non–Small Cell Lung Cancer Models. Clin. Cancer Res. 2011, 17, 1394–1404. [Google Scholar] [CrossRef]
- Wang, C.; Huang, S.-B.; Yang, M.-C.; Lin, Y.-T.; Chu, I.-H.; Shen, Y.-N.; Chiu, Y.-H.; Hung, S.-H.; Kang, L.; Hong, Y.-R.; et al. Combining Paclitaxel with ABT-263 Has a Synergistic Effect on Paclitaxel Resistant Prostate Cancer Cells. PLoS ONE 2015, 10, e0120913. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.; Novais, P.; Henriques, A.C.; Carvalho-Tavares, J.; Silva, P.M.A.; Bousbaa, H. Navitoclax Enhances the Therapeutic Effects of PLK1 Targeting on Lung Cancer Cells in 2D and 3D Culture Systems. Pharmaceutics 2022, 14, 1209. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.M.A.; Ribeiro, N.; Lima, R.T.; Andrade, C.; Diogo, V.; Teixeira, J.; Florindo, C.; Tavares, Á.; Vasconcelos, M.H.; Bousbaa, H. Suppression of Spindly Delays Mitotic Exit and Exacerbates Cell Death Response of Cancer Cells Treated with Low Doses of Paclitaxel. Cancer Lett. 2017, 394, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Pedro, M.; Ferreira, M.M.; Cidade, H.; Kijjoa, A.; Bronze-da-Rocha, E.; Nascimento, M.S.J. Artelastin Is a Cytotoxic Prenylated Flavone That Disturbs Microtubules and Interferes with DNA Replication in MCF-7 Human Breast Cancer Cells. Life Sci. 2005, 77, 293–311. [Google Scholar] [CrossRef]
- Tsuda, Y.; Iimori, M.; Nakashima, Y.; Nakanishi, R.; Ando, K.; Ohgaki, K.; Kitao, H.; Saeki, H.; Oki, E.; Maehara, Y. Mitotic Slippage and the Subsequent Cell Fates after Inhibition of Aurora B during Tubulin-Binding Agent–Induced Mitotic Arrest. Sci. Rep. 2017, 7, 16762. [Google Scholar] [CrossRef]
- Marxer, M.; Ma, H.T.; Man, W.Y.; Poon, R.Y.C. P53 Deficiency Enhances Mitotic Arrest and Slippage Induced by Pharmacological Inhibition of Aurora Kinases. Oncogene 2014, 33, 3550–3560. [Google Scholar] [CrossRef]
- Gurden, M.D.; Anderhub, S.J.; Faisal, A.; Linardopoulos, S. Aurora B Prevents Premature Removal of Spindle Assembly Checkpoint Proteins from the Kinetochore: A Key Role for Aurora B in Mitosis. Oncotarget 2018, 9, 19525–19542. [Google Scholar] [CrossRef] [PubMed]
- Salmela, A.-L.; Pouwels, J.; Mäki-Jouppila, J.; Kohonen, P.; Toivonen, P.; Kallio, L.; Kallio, M. Novel Pyrimidine-2,4-Diamine Derivative Suppresses the Cell Viability and Spindle Assembly Checkpoint Activity by Targeting Aurora Kinases. Carcinogenesis 2013, 34, 436–445. [Google Scholar] [CrossRef]
- Li, J.; Yan, Z.; Li, H.; Shi, Q.; Ahire, V.; Zhang, S.; Nimishetti, N.; Yang, D.; Allen, T.D.; Zhang, J. The Phytochemical Scoulerine Inhibits Aurora Kinase Activity to Induce Mitotic and Cytokinetic Defects. J. Nat. Prod. 2021, 84, 2312–2320. [Google Scholar] [CrossRef]
- Montero, P.H.; Patel, S.G. Cancer of the Oral Cavity. Surg. Oncol. Clin. N. Am. 2015, 24, 491–508. [Google Scholar] [CrossRef]
- Pannone, G.; Hindi, S.A.H.; Santoro, A.; Sanguedolce, F.; Rubini, C.; Cincione, R.I.; De Maria, S.; Tortorella, S.; Rocchetti, R.; Cagiano, S.; et al. Aurora B Expression as a Prognostic Indicator and Possibile Therapeutic Target in Oral Squamous Cell Carcinoma. Int. J. Immunopathol. Pharmacol. 2011, 24, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Daigo, K.; Takano, A.; Manh, T.; Yoshitake, Y.; Shinohara, M.; Tohnai, I.; Murakami, Y.; Maegawa, J.; Daigo, Y. Characterization of KIF11 as a Novel Prognostic Biomarker and Therapeutic Target for Oral Cancer. Int. J. Oncol. 2018, 52, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Coon, J.; Kingsley, K. Assessment of MicroRNA (MiR)-365 Effects on Oral Squamous Carcinoma Cell Line Phenotypes. Biomolecules 2021, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.J. The Proliferation Rate Paradox in Antimitotic Chemotherapy. Mol. Biol. Cell 2012, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef]
- Pinto, B.; Silva, J.P.N.; Silva, P.M.A.; Barbosa, D.J.; Sarmento, B.; Tavares, J.C.; Bousbaa, H. Maximizing Anticancer Response with MPS1 and CENPE Inhibition Alongside Apoptosis Induction. Pharmaceutics 2023, 16, 56. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (nM) | ||
|---|---|---|
| Drugs|Cell Line | SCC25 | SCC09 |
| Navitoclax | 5197.0 ± 364.0 | 3754.0 ± 237.0 |
| Ispinesib | 3.4 ± 0.5 | 58.9 ± 3.2 |
| Barasertib | 5580.0 ± 664.0 | >64,000.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, J.P.N.; Pinto, B.; Monteiro, L.; Silva, P.M.A.; Bousbaa, H. Coupling Kinesin Spindle Protein and Aurora B Inhibition with Apoptosis Induction Enhances Oral Cancer Cell Killing. Cancers 2024, 16, 2014. https://doi.org/10.3390/cancers16112014
Silva JPN, Pinto B, Monteiro L, Silva PMA, Bousbaa H. Coupling Kinesin Spindle Protein and Aurora B Inhibition with Apoptosis Induction Enhances Oral Cancer Cell Killing. Cancers. 2024; 16(11):2014. https://doi.org/10.3390/cancers16112014
Chicago/Turabian StyleSilva, João P. N., Bárbara Pinto, Luís Monteiro, Patrícia M. A. Silva, and Hassan Bousbaa. 2024. "Coupling Kinesin Spindle Protein and Aurora B Inhibition with Apoptosis Induction Enhances Oral Cancer Cell Killing" Cancers 16, no. 11: 2014. https://doi.org/10.3390/cancers16112014
APA StyleSilva, J. P. N., Pinto, B., Monteiro, L., Silva, P. M. A., & Bousbaa, H. (2024). Coupling Kinesin Spindle Protein and Aurora B Inhibition with Apoptosis Induction Enhances Oral Cancer Cell Killing. Cancers, 16(11), 2014. https://doi.org/10.3390/cancers16112014