




Immune Regulation and Immune Therapy in Melanoma: Review with Emphasis on CD155 Signalling

 , and
, and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Current Melanoma Patient Immunotherapy Strategies
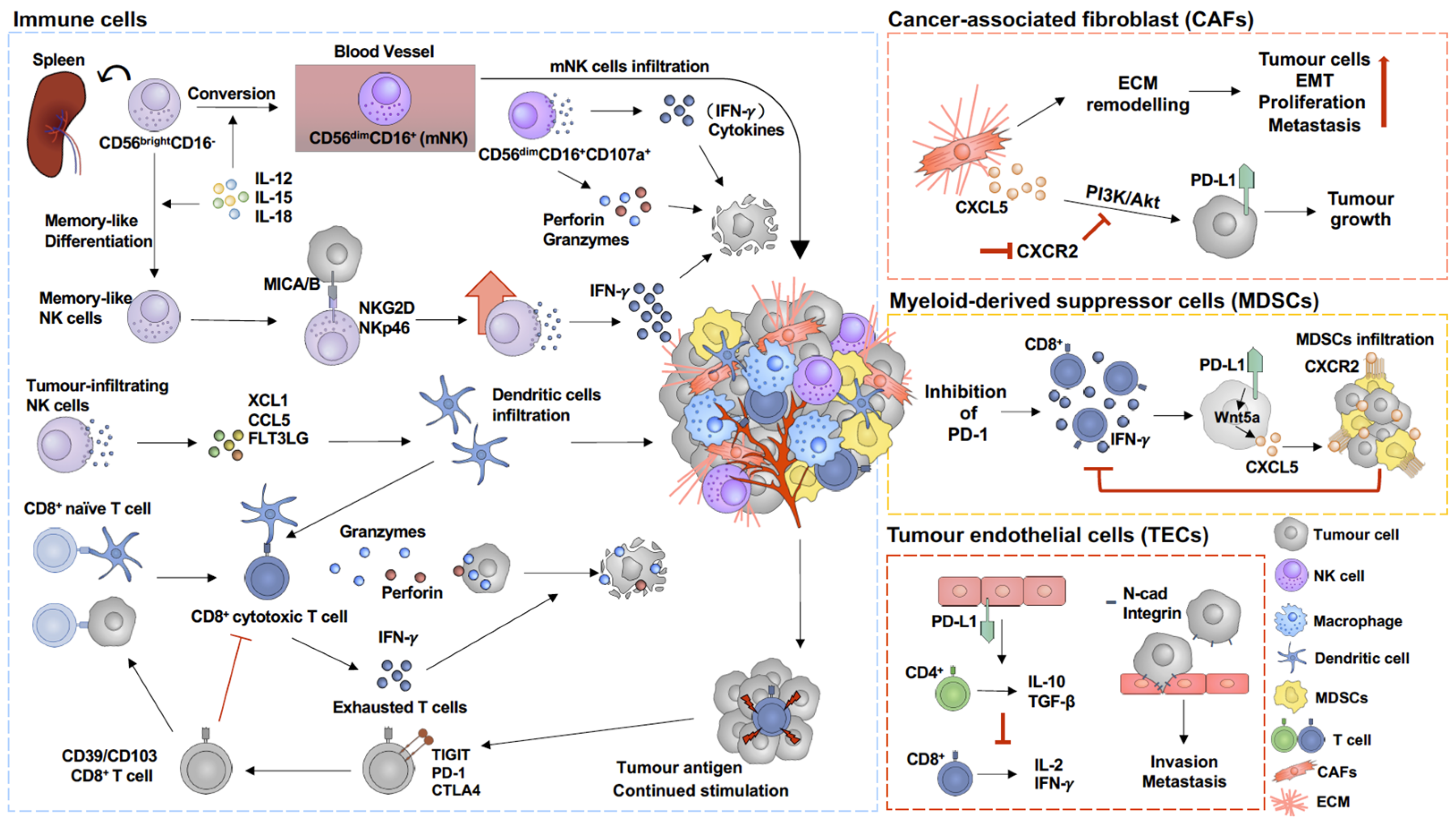
3. Immunoregulation Mechanisms within the Melanoma Tumour Microenvironment
3.1. Tumour Endothelial Cells (TECs)
3.2. Cancer-Associated Fibroblast (CAF)
3.3. Myeloid-Derived Suppressor Cells (MDSCs)
3.4. Cytotoxic T Cells (CD8+ T Cells)
3.5. Natural Killer Cells (NK Cells)
4. Processes of Resistance to Immunotherapy Responses
4.1. Tertiary Lymphoid Structures (TLSs)
4.2. Therapy-Resistant Cancer Cells
4.3. Cytoskeleton Remodelling
4.4. Immune Responses
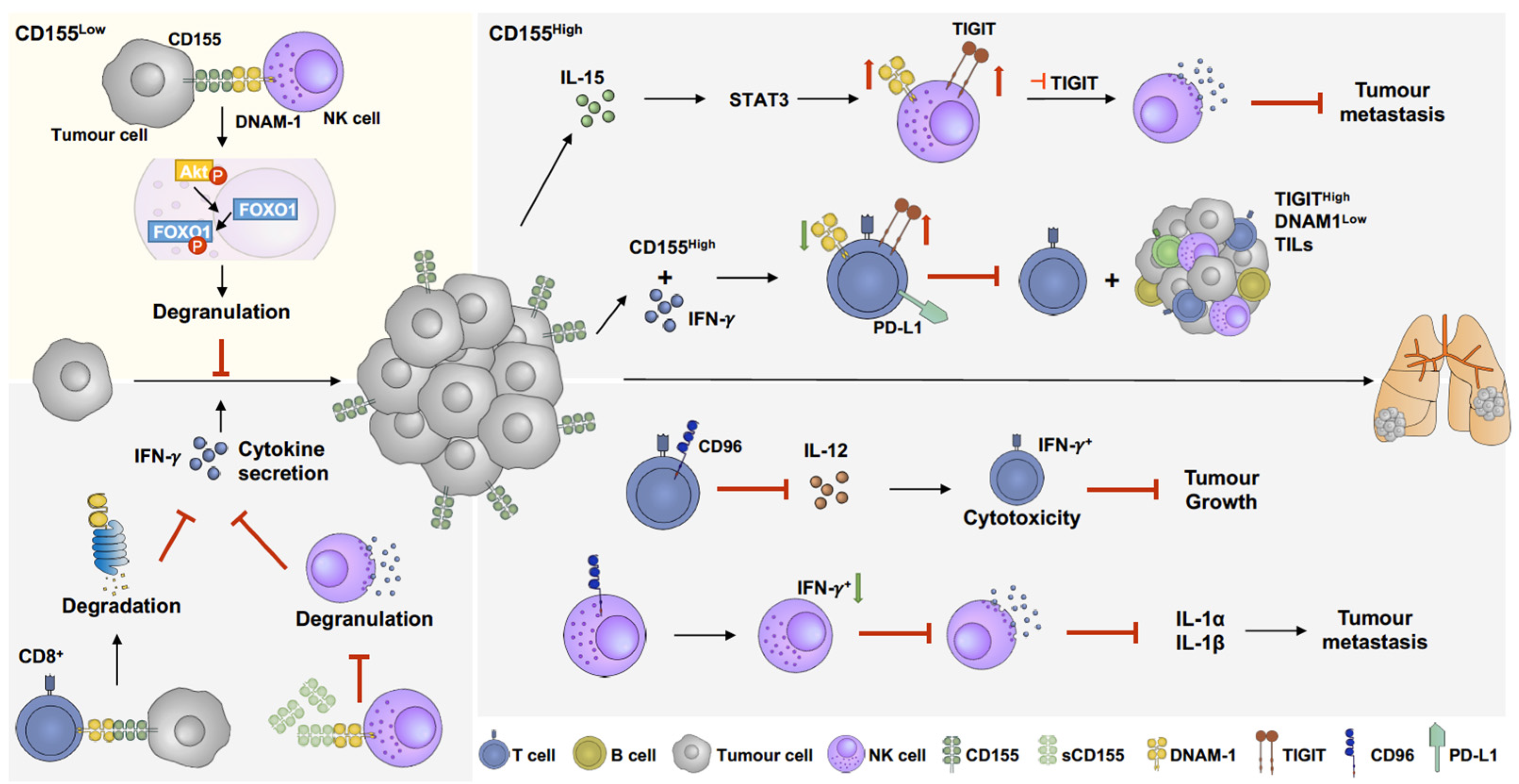
5. The Role of CD155 in the Melanoma Microenvironment and Its Potential as Immunotherapy Target
5.1. DNAM-1(CD226)
5.2. T-Cell Immunoreceptor with Ig and ITIM Domains (TIGIT)
5.3. CD96 (TACTILE)
6. Overall Summary
Author Contributions
Funding
Conflicts of Interest
References
- Liu-Smith, F.; Jia, J.; Zheng, Y. UV-Induced Molecular Signaling Differences in Melanoma and Non-melanoma Skin Cancer. Adv. Exp. Med. Biol. 2017, 996, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Karimkhani, C.; Green, A.C.; Nijsten, T.; Weinstock, M.A.; Dellavalle, R.P.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Green, A.C.; Williams, G.M.; Logan, V.; Strutton, G.M. Reduced melanoma after regular sunscreen use: Randomized trial follow-up. J. Clin. Oncol. 2011, 29, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Raimondi, S.; Suppa, M.; Gandini, S. Melanoma Epidemiology and Sun Exposure. Acta Derm. Venereol. 2020, 100, adv00136. [Google Scholar] [CrossRef] [PubMed]
- Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. Int. J. Mol. Sci. 2021, 22, 6395. [Google Scholar] [CrossRef]
- O’Neill, C.H.; Scoggins, C.R. Melanoma. J. Surg. Oncol. 2019, 120, 873–881. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef]
- Ng, M.F.; Simmons, J.L.; Boyle, G.M. Heterogeneity in Melanoma. Cancers 2022, 14, 3030. [Google Scholar] [CrossRef] [PubMed]
- Nassar, K.W.; Tan, A.C. The mutational landscape of mucosal melanoma. Semin. Cancer Biol. 2020, 61, 139–148. [Google Scholar] [CrossRef]
- Scolyer, R.A.; Long, G.V.; Thompson, J.F. Evolving concepts in melanoma classification and their relevance to multidisciplinary melanoma patient care. Mol. Oncol. 2011, 5, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Faramarzi, S.; Ghafouri-Fard, S. Melanoma: A prototype of cancer-testis antigen-expressing malignancies. Immunotherapy 2017, 9, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Braeuer, R.R.; Watson, I.R.; Wu, C.J.; Mobley, A.K.; Kamiya, T.; Shoshan, E.; Bar-Eli, M. Why is melanoma so metastatic? Pigment. Cell Melanoma Res. 2014, 27, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Ralli, M.; Botticelli, A.; Visconti, I.C.; Angeletti, D.; Fiore, M.; Marchetti, P.; Lambiase, A.; de Vincentiis, M.; Greco, A. Immunotherapy in the Treatment of Metastatic Melanoma: Current Knowledge and Future Directions. J. Immunol. Res. 2020, 2020, 9235638. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef]
- Willsmore, Z.N.; Coumbe, B.G.T.; Crescioli, S.; Reci, S.; Gupta, A.; Harris, R.J.; Chenoweth, A.; Chauhan, J.; Bax, H.J.; McCraw, A.; et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur. J. Immunol. 2021, 51, 544–556. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Uhara, H.; Kiyohara, Y.; Uehara, J.; Fujisawa, Y.; Takenouchi, T.; Otsuka, M.; Uchi, H.; Fukushima, S.; Minami, H.; Hatsumichi, M.; et al. Five-year survival with nivolumab in previously untreated Japanese patients with advanced or recurrent malignant melanoma. J. Dermatol. 2021, 48, 592–599. [Google Scholar] [CrossRef]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tille, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.R.; Readler, J.M.; Sharma, P.; Excoffon, K. Poliovirus Receptor: More than a simple viral receptor. Virus Res. 2017, 242, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Inozume, T.; Kawazu, M.; Ueno, T.; Nagasaki, J.; Tanji, E.; Honobe, A.; Ohnuma, T.; Kawamura, T.; Umeda, Y.; et al. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed tumor microenvironment. J. Immunother. Cancer 2021, 9, e003134. [Google Scholar] [CrossRef]
- Lepletier, A.; Madore, J.; O’Donnell, J.S.; Johnston, R.L.; Li, X.Y.; McDonald, E.; Ahern, E.; Kuchel, A.; Eastgate, M.; Pearson, S.A.; et al. Tumor CD155 Expression Is Associated with Resistance to Anti-PD1 Immunotherapy in Metastatic Melanoma. Clin. Cancer Res. 2020, 26, 3671–3681. [Google Scholar] [CrossRef]
- Ito, M.; Mimura, K.; Nakajima, S.; Saito, K.; Min, A.K.T.; Okayama, H.; Saito, M.; Momma, T.; Saze, Z.; Ohtsuka, M.; et al. Immune escape mechanism behind resistance to anti-PD-1 therapy in gastrointestinal tract metastasis in malignant melanoma patients with multiple metastases. Cancer Immunol. Immunother. 2022, 71, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Aguilera, A.R.; Sundarrajan, A.; Corvino, D.; Stannard, K.; Krumeich, S.; Das, I.; Lima, L.G.; Meza Guzman, L.G.; Li, K.; et al. CD155 on Tumor Cells Drives Resistance to Immunotherapy by Inducing the Degradation of the Activating Receptor CD226 in CD8(+) T Cells. Immunity 2020, 53, 805–823.e15. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Enk, A.H. TIGIT-CD155 Interactions in Melanoma: A Novel Co-Inhibitory Pathway with Potential for Clinical Intervention. J. Investig. Dermatol. 2016, 136, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B., II. Contribution to the Knowledge of Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef]
- Lindenmann, J.; Burke, D.C.; Isaacs, A. Studies on the production, mode of action and properties of interferon. Br. J. Exp. Pathol. 1957, 38, 551–562. [Google Scholar]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of Patients with Metastatic Melanoma with Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. JNCI J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Vesely, M.D.; Chen, L. Normalization Cancer Immunotherapy for Melanoma. J. Investig. Dermatol. 2020, 140, 1134–1142. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991, 174, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Weber, J. Overcoming immunologic tolerance to melanoma: Targeting CTLA-4 with ipilimumab (MDX-010). Oncologist 2008, 13 (Suppl. 4), 16–25. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Carlino, M.S.; McNeil, C.; Ribas, A.; Grob, J.-J.; Schachter, J.; Nyakas, M.; Kee, D.; Petrella, T.M.; Blaustein, A.; et al. Seven-Year Follow-Up of the Phase III KEYNOTE-006 Study: Pembrolizumab Versus Ipilimumab in Advanced Melanoma. J. Clin. Oncol. 2023, 41, 3998–4003. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Patel, S.P.; Othus, M.; Chen, Y.; Wright, G.P.; Yost, K.J.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.D.; Fecher, L.A.; Truong, T.-G.; et al. Neoadjuvant–Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N. Engl. J. Med. 2023, 388, 813–823. [Google Scholar] [CrossRef]
- Amaral, T.; Seeber, O.; Mersi, E.; Sanchez, S.; Thomas, I.; Meiwes, A.; Forschner, A.; Leiter, U.; Eigentler, T.; Keim, U.; et al. Primary Resistance to PD-1-Based Immunotherapy-A Study in 319 Patients with Stage IV Melanoma. Cancers 2020, 12, 1027. [Google Scholar] [CrossRef]
- VanderWalde, A.; Bellasea, S.L.; Kendra, K.L.; Khushalani, N.I.; Campbell, K.M.; Scumpia, P.O.; Kuklinski, L.F.; Collichio, F.; Sosman, J.A.; Ikeguchi, A.; et al. Ipilimumab with or without nivolumab in PD-1 or PD-L1 blockade refractory metastatic melanoma: A randomized phase 2 trial. Nat. Med. 2023, 29, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Arance, A.; Cruz-Merino, L.d.l.; Petrella, T.M.; Jamal, R.; Ny, L.; Carneiro, A.; Berrocal, A.; Márquez-Rodas, I.; Spreafico, A.; Atkinson, V.; et al. Phase II LEAP-004 Study of Lenvatinib Plus Pembrolizumab for Melanoma with Confirmed Progression on a Programmed Cell Death Protein-1 or Programmed Death Ligand 1 Inhibitor Given as Monotherapy or in Combination. J. Clin. Oncol. 2023, 41, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C.; Zimmer, L.; Sickmann, T.; Eigentler, T.K.; Meier, F.; Mohr, P.; Pukrop, T.; Roesch, A.; Vordermark, D.; Wendl, C.; et al. Medical Needs and Therapeutic Options for Melanoma Patients Resistant to Anti-PD-1-Directed Immune Checkpoint Inhibition. Cancers 2023, 15, 3448. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Lacina, L.; Kodet, O.; Dvorankova, B.; Szabo, P.; Smetana, K., Jr. Ecology of melanoma cell. Histol. Histopathol. 2018, 33, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Onoe, T.; Yoshida, T.; Yamashita, Y.; Tanaka, Y.; Ohdan, H. Tumor Endothelial Cell-Mediated Antigen-Specific T-cell Suppression via the PD-1/PD-L1 Pathway. Mol. Cancer Res. 2020, 18, 1427–1440. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Chang, Y.C.; Wu, J.W.; Wang, C.W.; Jang, A.C. Hippo Signaling-Mediated Mechanotransduction in Cell Movement and Cancer Metastasis. Front. Mol. Biosci. 2019, 6, 157. [Google Scholar] [CrossRef]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Haass, N.K.; Smalley, K.S.; Li, L.; Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment. Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Liesche, C.; van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandstrom, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef] [PubMed]
- Marin, N.D.; Krasnick, B.A.; Becker-Hapak, M.; Conant, L.; Goedegebuure, S.P.; Berrien-Elliott, M.M.; Robbins, K.J.; Foltz, J.A.; Foster, M.; Wong, P.; et al. Memory-like Differentiation Enhances NK Cell Responses to Melanoma. Clin. Cancer Res. 2021, 27, 4859–4869. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Miller, J.S. Promoting T and NK cell attack: Preserving tumor MICA/B by vaccines. Cell Res. 2022, 32, 961–962. [Google Scholar] [CrossRef]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2020, 11, 622509. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Hochheiser, K.; Gyorki, D.E.; Gebhardt, T. Tumor reactivity of CD8(+) T cells favors acquisition of dysfunctional states in human melanoma. Immunol. Cell Biol. 2021, 99, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Sobierajska, K.; Ciszewski, W.M.; Sacewicz-Hofman, I.; Niewiarowska, J. Endothelial Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Rous-Whipple Award Lecture. How tumors make bad blood vessels and stroma. Am. J. Pathol. 2003, 162, 1747–1757. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Benedicto, A.; Hernandez-Unzueta, I.; Sanz, E.; Marquez, J. Ocoxin Increases the Antitumor Effect of BRAF Inhibition and Reduces Cancer Associated Fibroblast-Mediated Chemoresistance and Protumoral Activity in Metastatic Melanoma. Nutrients 2021, 13, 686. [Google Scholar] [CrossRef]
- Tsang, M.; Quesnel, K.; Vincent, K.; Hutchenreuther, J.; Postovit, L.M.; Leask, A. Insights into Fibroblast Plasticity: Cellular Communication Network 2 Is Required for Activation of Cancer-Associated Fibroblasts in a Murine Model of Melanoma. Am. J. Pathol. 2020, 190, 206–221. [Google Scholar] [CrossRef]
- Kawamoto, H.; Minato, N. Myeloid cells. Int. J. Biochem. Cell Biol. 2004, 36, 1374–1379. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Singh, L.; Muise, E.S.; Bhattacharya, A.; Grein, J.; Javaid, S.; Stivers, P.; Zhang, J.; Qu, Y.; Joyce-Shaikh, B.; Loboda, A.; et al. ILT3 (LILRB4) Promotes the Immunosuppressive Function of Tumor-Educated Human Monocytic Myeloid-Derived Suppressor Cells. Mol. Cancer Res. 2021, 19, 702–716. [Google Scholar] [CrossRef]
- Weber, J.; Gibney, G.; Kudchadkar, R.; Yu, B.; Cheng, P.; Martinez, A.J.; Kroeger, J.; Richards, A.; McCormick, L.; Moberg, V.; et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol. Res. 2016, 4, 345–353. [Google Scholar] [CrossRef]
- Douglass, S.M.; Fane, M.E.; Sanseviero, E.; Ecker, B.L.; Kugel, C.H., 3rd; Behera, R.; Kumar, V.; Tcyganov, E.N.; Yin, X.; Liu, Q.; et al. Myeloid-Derived Suppressor Cells Are a Major Source of Wnt5A in the Melanoma Microenvironment and Depend on Wnt5A for Full Suppressive Activity. Cancer Res. 2021, 81, 658–670. [Google Scholar] [CrossRef]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J. Hematol. Oncol. 2019, 12, 84. [Google Scholar] [CrossRef]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5(+) Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Ma, P.; Beatty, P.L.; McKolanis, J.; Brand, R.; Schoen, R.E.; Finn, O.J. Circulating Myeloid Derived Suppressor Cells (MDSC) That Accumulate in Premalignancy Share Phenotypic and Functional Characteristics with MDSC in Cancer. Front. Immunol. 2019, 10, 1401. [Google Scholar] [CrossRef]
- Joseph, R.; Soundararajan, R.; Vasaikar, S.; Yang, F.; Allton, K.L.; Tian, L.; den Hollander, P.; Isgandarova, S.; Haemmerle, M.; Mino, B.; et al. CD8(+) T cells inhibit metastasis and CXCL4 regulates its function. Br. J. Cancer 2021, 125, 176–189. [Google Scholar] [CrossRef]
- Payne, A.S.; Cornelius, L.A. The role of chemokines in melanoma tumor growth and metastasis. J. Investig. Dermatol. 2002, 118, 915–922. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef]
- Chen, X.; Chen, R.; Jin, R.; Huang, Z. The role of CXCL chemokine family in the development and progression of gastric cancer. Int. J. Clin. Exp. Pathol. 2020, 13, 484–492. [Google Scholar] [PubMed]
- Paczek, S.; Lukaszewicz-Zajac, M.; Gryko, M.; Mroczko, P.; Kulczynska-Przybik, A.; Mroczko, B. CXCL-8 in Preoperative Colorectal Cancer Patients: Significance for Diagnosis and Cancer Progression. Int. J. Mol. Sci. 2020, 21, 2040. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.H.; Benner, B.; Savardekar, H.; Lapurga, G.; Good, L.; Abood, D.; Nagle, E.; Duggan, M.; Stiff, A.; DiVincenzo, M.J.; et al. Effect of Immune Checkpoint Blockade on Myeloid-Derived Suppressor Cell Populations in Patients with Melanoma. Front. Immunol. 2021, 12, 740890. [Google Scholar] [CrossRef]
- Sapir, Y.; Vitenshtein, A.; Barsheshet, Y.; Zohar, Y.; Wildbaum, G.; Karin, N. A fusion protein encoding the second extracellular domain of CCR5 arrests chemokine-induced cosignaling and effectively suppresses ongoing experimental autoimmune encephalomyelitis. J. Immunol. 2010, 185, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, V.; Ziccheddu, G.; Macchia, I.; La Sorsa, V.; Peschiaroli, F.; Buccione, C.; Sistigu, A.; Sanchez, M.; Andreone, S.; D’Urso, M.T.; et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. Oncoimmunology 2017, 6, e1317420. [Google Scholar] [CrossRef]
- Lim, H.X.; Choi, S.; Cho, D.; Kim, T.S. IL-33 inhibits the differentiation and immunosuppressive activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Immunol. Cell Biol. 2017, 95, 99–107. [Google Scholar] [CrossRef]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.D.; Ely, K.H.; Woodland, D.L. Differential contributions of central and effector memory T cells to recall responses. J. Exp. Med. 2005, 202, 123–133. [Google Scholar] [CrossRef]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Lopez, J.A.; Susanto, O.; Jenkins, M.R.; Lukoyanova, N.; Sutton, V.R.; Law, R.H.; Johnston, A.; Bird, C.H.; Bird, P.I.; Whisstock, J.C.; et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013, 121, 2659–2668. [Google Scholar] [CrossRef]
- D’Angelo, M.E.; Bird, P.I.; Peters, C.; Reinheckel, T.; Trapani, J.A.; Sutton, V.R. Cathepsin H is an additional convertase of pro-granzyme B. J. Biol. Chem. 2010, 285, 20514–20519. [Google Scholar] [CrossRef]
- Tsukumo, S.I.; Yasutomo, K. Regulation of CD8(+) T Cells and Antitumor Immunity by Notch Signaling. Front. Immunol. 2018, 9, 101. [Google Scholar] [CrossRef]
- Hodge, G.; Barnawi, J.; Jurisevic, C.; Moffat, D.; Holmes, M.; Reynolds, P.N.; Jersmann, H.; Hodge, S. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-gamma by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin. Exp. Immunol. 2014, 178, 79–85. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef]
- Webb, J.R.; Milne, K.; Nelson, B.H. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 926–935. [Google Scholar] [CrossRef]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpreville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef]
- Pena-Asensio, J.; Calvo, H.; Torralba, M.; Miquel, J.; Sanz-de-Villalobos, E.; Larrubia, J.R. Anti-PD-1/PD-L1 Based Combination Immunotherapy to Boost Antigen-Specific CD8(+) T Cell Response in Hepatocellular Carcinoma. Cancers 2021, 13, 1922. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Loo, K.; Pauli, M.L.; Sanchez-Rodriguez, R.; Sandoval, P.M.; Taravati, K.; Tsai, K.; Nosrati, A.; Nardo, L.; Alvarado, M.D.; et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Investig. 2016, 126, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, A.; Liu, X.; Han, S.; Sun, Y.; Zhang, J.; Guo, L.; Zhang, Y. Blocking TIGIT/CD155 signalling reverses CD8(+) T cell exhaustion and enhances the antitumor activity in cervical cancer. J. Transl. Med. 2022, 20, 280. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Q.; Wang, Z.; Zhang, H.; Zeng, H.; Huang, Q.; Chen, Y.; Jiang, W.; Lin, Z.; Qu, Y.; et al. Intratumoral TIGIT(+) CD8(+) T-cell infiltration determines poor prognosis and immune evasion in patients with muscle-invasive bladder cancer. J. Immunother. Cancer 2020, 8, e000978. [Google Scholar] [CrossRef] [PubMed]
- Ortaldo, J.R.; Oldham, R.K.; Cannon, G.C.; Herberman, R.B. Specificity of natural cytotoxic reactivity of normal human lymphocytes against a myeloid leukemia cell line. J. Natl. Cancer Inst. 1977, 59, 77–82. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Bi, J.; Wang, X. Molecular Regulation of NK Cell Maturation. Front. Immunol. 2020, 11, 1945. [Google Scholar] [CrossRef] [PubMed]
- Angelo, L.S.; Banerjee, P.P.; Monaco-Shawver, L.; Rosen, J.B.; Makedonas, G.; Forbes, L.R.; Mace, E.M.; Orange, J.S. Practical NK cell phenotyping and variability in healthy adults. Immunol. Res. 2015, 62, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Poli, A.; Cuapio, A.; Briquemont, B.; Iserentant, G.; Ollert, M.; Zimmer, J. Human CD56bright NK Cells: An Update. J. Immunol. 2016, 196, 2923–2931. [Google Scholar] [CrossRef]
- Crinier, A.; Milpied, P.; Escaliere, B.; Piperoglou, C.; Galluso, J.; Balsamo, A.; Spinelli, L.; Cervera-Marzal, I.; Ebbo, M.; Girard-Madoux, M.; et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 2018, 49, 971–986.e5. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 2017, 47, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.; Hintzen, G.; Kemper, A.; Beul, K.; Kempf, S.; Behrens, G.; Sykora, K.W.; Schmidt, R.E. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur. J. Immunol. 2001, 31, 3121–3127. [Google Scholar] [CrossRef] [PubMed]
- Bjorkstrom, N.K.; Riese, P.; Heuts, F.; Andersson, S.; Fauriat, C.; Ivarsson, M.A.; Bjorklund, A.T.; Flodstrom-Tullberg, M.; Michaelsson, J.; Rottenberg, M.E.; et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 2010, 116, 3853–3864. [Google Scholar] [CrossRef] [PubMed]
- Cohnen, A.; Chiang, S.C.; Stojanovic, A.; Schmidt, H.; Claus, M.; Saftig, P.; Janssen, O.; Cerwenka, A.; Bryceson, Y.T.; Watzl, C. Surface CD107a/LAMP-1 protects natural killer cells from degranulation-associated damage. Blood 2013, 122, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Amand, M.; Iserentant, G.; Poli, A.; Sleiman, M.; Fievez, V.; Sanchez, I.P.; Sauvageot, N.; Michel, T.; Aouali, N.; Janji, B.; et al. Human CD56(dim)CD16(dim) Cells As an Individualized Natural Killer Cell Subset. Front. Immunol. 2017, 8, 699. [Google Scholar] [CrossRef] [PubMed]
- Mujal, A.M.; Delconte, R.B.; Sun, J.C. Natural Killer Cells: From Innate to Adaptive Features. Annu. Rev. Immunol. 2021, 39, 417–447. [Google Scholar] [CrossRef]
- Kamimura, Y.; Lanier, L.L. Homeostatic control of memory cell progenitors in the natural killer cell lineage. Cell Rep. 2015, 10, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Tsutsui, H.; Yoshimoto, T.; Adachi, O.; Yoshida, N.; Kishimoto, T.; Okamura, H.; Nakanishi, K.; Akira, S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 1998, 8, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Chaix, J.; Tessmer, M.S.; Hoebe, K.; Fuseri, N.; Ryffel, B.; Dalod, M.; Alexopoulou, L.; Beutler, B.; Brossay, L.; Vivier, E.; et al. Cutting edge: Priming of NK cells by IL-18. J. Immunol. 2008, 181, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, H.; Diao, Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef] [PubMed]
- Cozar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021, 11, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Sconocchia, G.; Eppenberger, S.; Spagnoli, G.C.; Tornillo, L.; Droeser, R.; Caratelli, S.; Ferrelli, F.; Coppola, A.; Arriga, R.; Lauro, D.; et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology 2014, 3, e952197. [Google Scholar] [CrossRef] [PubMed]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, L.F.; Lu, Y.; Luoma, A.; Ito, Y.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Yuan, G.C.; Wucherpfennig, K.W. Discovery of specialized NK cell populations infiltrating human melanoma metastases. JCI Insight 2019, 4, e133103. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Ji, S.; Lee, J.; Lee, E.S.; Kim, D.H.; Sin, J.I. B16 melanoma control by anti-PD-L1 requires CD8+ T cells and NK cells: Application of anti-PD-L1 Abs and Trp2 peptide vaccines. Hum. Vaccin. Immunother. 2021, 17, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Cappello, S.; Sung, H.M.; Ickes, C.; Gibhardt, C.S.; Vultur, A.; Bhat, H.; Hu, Z.; Brafford, P.; Denger, A.; Stejerean-Todoran, I.; et al. Protein Signatures of NK Cell-Mediated Melanoma Killing Predict Response to Immunotherapies. Cancer Res. 2021, 81, 5540–5554. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma Cells Control Antimelanoma CTL Responses via Interaction between TIGIT and CD155 in the Effector Phase. J. Investig. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Briukhovetska, D.; Suarez-Gosalvez, J.; Voigt, C.; Markota, A.; Giannou, A.D.; Schubel, M.; Jobst, J.; Zhang, T.; Dorr, J.; Markl, F.; et al. T cell-derived interleukin-22 drives the expression of CD155 by cancer cells to suppress NK cell function and promote metastasis. Immunity 2023, 56, 143–161.e11. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, eabf9419. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhou, J.; Xiao, Y.; Ming, J.; Zhou, J.; Dong, F.; Zhou, X.; Xu, Z.; Zhao, X.; Lei, P.; et al. CD20(+)CD22(+)ADAM28(+) B Cells in Tertiary Lymphoid Structures Promote Immunotherapy Response. Front. Immunol. 2022, 13, 865596. [Google Scholar] [CrossRef] [PubMed]
- Sautes-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Chelvanambi, M.; Fecek, R.J.; Taylor, J.L.; Storkus, W.J. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J. Immunother. Cancer 2021, 9, e001906. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Davies, M.A.; Tanese, K.; Roszik, J.; Shin-Sim, M.; Bassett, R.L., Jr.; Milton, D.R.; Woodman, S.E.; Prieto, V.G.; Gershenwald, J.E.; et al. Inflammatory Marker Testing Identifies CD74 Expression in Melanoma Tumor Cells, and Its Expression Associates with Favorable Survival for Stage III Melanoma. Clin. Cancer Res. 2016, 22, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.R.; Azevedo, R.A.; Mousdell, S.; Resende-Lara, P.T.; Ireland, L.; Santos, A.; Girola, N.; Cunha, R.; Schmid, M.C.; Polonelli, L.; et al. Blockade of MIF-CD74 Signalling on Macrophages and Dendritic Cells Restores the Antitumour Immune Response Against Metastatic Melanoma. Front. Immunol. 2018, 9, 1132. [Google Scholar] [CrossRef] [PubMed]
- de Azevedo, R.A.; Shoshan, E.; Whang, S.; Markel, G.; Jaiswal, A.R.; Liu, A.; Curran, M.A.; Travassos, L.R.; Bar-Eli, M. MIF inhibition as a strategy for overcoming resistance to immune checkpoint blockade therapy in melanoma. Oncoimmunology 2020, 9, 1846915. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Seijo, A.; Garcia-Martinez, E.; Barrio-Alonso, C.; Parra-Blanco, V.; Aviles-Izquierdo, J.A.; Sanchez-Mateos, P.; Samaniego, R. Activin A Sustains the Metastatic Phenotype of Tumor-Associated Macrophages and Is a Prognostic Marker in Human Cutaneous Melanoma. J. Investig. Dermatol. 2022, 142, 653–661.e2. [Google Scholar] [CrossRef] [PubMed]
- Pinjusic, K.; Dubey, O.A.; Egorova, O.; Nassiri, S.; Meylan, E.; Faget, J.; Constam, D.B. Activin-A impairs CD8 T cell-mediated immunity and immune checkpoint therapy response in melanoma. J. Immunother. Cancer 2022, 10, e004533. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.H.; Lee, J.K.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Barreno, A.; Orgaz, J.L. Cytoskeletal Remodelling as an Achilles’ Heel for Therapy Resistance in Melanoma. Cells 2022, 11, 518. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Crosas-Molist, E.; Sadok, A.; Perdrix-Rosell, A.; Maiques, O.; Rodriguez-Hernandez, I.; Monger, J.; Mele, S.; Georgouli, M.; Bridgeman, V.; et al. Myosin II Reactivation and Cytoskeletal Remodeling as a Hallmark and a Vulnerability in Melanoma Therapy Resistance. Cancer Cell 2020, 37, 85–103.e9. [Google Scholar] [CrossRef]
- Wu, L.Y.; Han, C.L.; Lin, H.H.; Tang, M.J. Ha-Ras(V12)-Induced Multilayer Cellular Aggregates Is Mediated by Rac1 Activation Rather Than YAP Activation. Biomedicines 2022, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Dalle, S.; Monet, M.A.; Ligier, M.; Boespflug, A.; Pommier, R.M.; de la Fouchardiere, A.; Perier-Muzet, M.; Depaepe, L.; Barnault, R.; et al. ZEB1-mediated melanoma cell plasticity enhances resistance to MAPK inhibitors. EMBO Mol. Med. 2016, 8, 1143–1161. [Google Scholar] [CrossRef]
- Plaschka, M.; Benboubker, V.; Grimont, M.; Berthet, J.; Tonon, L.; Lopez, J.; Le-Bouar, M.; Balme, B.; Tondeur, G.; de la Fouchardiere, A.; et al. ZEB1 transcription factor promotes immune escape in melanoma. J. Immunother. Cancer 2022, 10, e003484. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gomez, H.; Bastholt, L.; et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 204. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; de Almeida, P.; Manieri, N.; de Almeida Nagata, D.; Wu, T.D.; Harden Bowles, K.; Arumugam, V.; Schartner, J.; Cubas, R.; Mittman, S.; et al. CD226 regulates natural killer cell antitumor responses via phosphorylation-mediated inactivation of transcription factor FOXO1. Proc. Natl. Acad. Sci. USA 2018, 115, E11731–E11740. [Google Scholar] [CrossRef]
- Okumura, G.; Iguchi-Manaka, A.; Murata, R.; Yamashita-Kanemaru, Y.; Shibuya, A.; Shibuya, K. Tumor-derived soluble CD155 inhibits DNAM-1-mediated antitumor activity of natural killer cells. J. Exp. Med. 2020, 217, e20191290. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Ka, M.; Pagliano, O.; Menna, C.; Ding, Q.; DeBlasio, R.; Sanders, C.; Hou, J.; Li, X.Y.; Ferrone, S.; et al. IL15 Stimulation with TIGIT Blockade Reverses CD155-mediated NK-Cell Dysfunction in Melanoma. Clin. Cancer Res. 2020, 26, 5520–5533. [Google Scholar] [CrossRef]
- Mittal, D.; Lepletier, A.; Madore, J.; Aguilera, A.R.; Stannard, K.; Blake, S.J.; Whitehall, V.L.J.; Liu, C.; Bettington, M.L.; Takeda, K.; et al. CD96 Is an Immune Checkpoint That Regulates CD8(+) T-cell Antitumor Function. Cancer Immunol. Res. 2019, 7, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Baury, B.; Masson, D.; McDermott, B.M., Jr.; Jarry, A.; Blottiere, H.M.; Blanchardie, P.; Laboisse, C.L.; Lustenberger, P.; Racaniello, V.R.; Denis, M.G. Identification of secreted CD155 isoforms. Biochem. Biophys. Res. Commun. 2003, 309, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; O’Farrell, S.; Clayberger, C.; Krensky, A.M. Identification and molecular cloning of tactile. A novel human T cell activation antigen that is a member of the Ig gene superfamily. J. Immunol. 1992, 148, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Fuhrman, C.A.; Yeh, W.I.; Seay, H.R.; Saikumar Lakshmi, P.; Chopra, G.; Zhang, L.; Perry, D.J.; McClymont, S.A.; Yadav, M.; Lopez, M.C.; et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J. Immunol. 2015, 195, 145–155. [Google Scholar] [CrossRef]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-Induced Killer Cells as Pharmacological Tools for Cancer Immunotherapy. Front. Immunol. 2017, 8, 774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhao, W.; Li, H.; Chen, Y.; Tian, H.; Li, L.; Zhang, L.; Gao, C.; Zheng, J. Immunoreceptor TIGIT inhibits the cytotoxicity of human cytokine-induced killer cells by interacting with CD155. Cancer Immunol. Immunother. 2016, 65, 305–314. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, L.F.; Smyth, M.J.; Martinet, L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol. 2014, 92, 237–244. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Jarry, A.; Baury, B.; Blanchardie, P.; Laboisse, C.; Lustenberger, P.; Denis, M.G. Overexpression of the CD155 gene in human colorectal carcinoma. Gut 2001, 49, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.G.; Kiessling, R.; Malmberg, K.J. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 2009, 183, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Clinical significance of CD155 expression in human pancreatic cancer. Anticancer Res. 2015, 35, 2287–2297. [Google Scholar]
- Deng, Y.; Kerdiles, Y.; Chu, J.; Yuan, S.; Wang, Y.; Chen, X.; Mao, H.; Zhang, L.; Zhang, J.; Hughes, T.; et al. Transcription factor Foxo1 is a negative regulator of natural killer cell maturation and function. Immunity 2015, 42, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef]
- Iguchi-Manaka, A.; Okumura, G.; Kojima, H.; Cho, Y.; Hirochika, R.; Bando, H.; Sato, T.; Yoshikawa, H.; Hara, H.; Shibuya, A.; et al. Increased Soluble CD155 in the Serum of Cancer Patients. PLoS ONE 2016, 11, e0152982. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, S.; Dheeraj; Basak, M.; Chitkara, D.; Mittal, A. Surface functionalization of exosomes for target-specific delivery and in vivo imaging & tracking: Strategies and significance. J. Control. Release 2020, 326, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cai, X.; Liu, F. CD96 as a Potential Diagnostic Biomarker and New Target for Skin Cutaneous Melanoma. Contrast Media Mol. Imaging 2022, 2022, 6409376. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Madore, J.; Li, X.Y.; Smyth, M.J. Tumor intrinsic and extrinsic immune functions of CD155. Semin. Cancer Biol. 2020, 65, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Kucan Brlic, P.; Lenac Rovis, T.; Cinamon, G.; Tsukerman, P.; Mandelboim, O.; Jonjic, S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell. Mol. Immunol. 2019, 16, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Kakunaga, S.; Ikeda, W.; Shingai, T.; Fujito, T.; Yamada, A.; Minami, Y.; Imai, T.; Takai, Y. Enhancement of serum- and platelet-derived growth factor-induced cell proliferation by Necl-5/Tage4/poliovirus receptor/CD155 through the Ras-Raf-MEK-ERK signaling. J. Biol. Chem. 2004, 279, 36419–36425. [Google Scholar] [CrossRef]
- Sloan, K.E.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; Roy, J.E.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer 2004, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Das, I.; Lepletier, A.; Addala, V.; Bald, T.; Stannard, K.; Barkauskas, D.; Liu, J.; Aguilera, A.R.; Takeda, K.; et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J. Clin. Investig. 2018, 128, 2613–2625. [Google Scholar] [CrossRef]
- Beasley, G.M.; Nair, S.K.; Farrow, N.E.; Landa, K.; Selim, M.A.; Wiggs, C.A.; Jung, S.H.; Bigner, D.D.; True Kelly, A.; Gromeier, M.; et al. Phase I trial of intratumoral PVSRIPO in patients with unresectable, treatment-refractory melanoma. J. Immunother. Cancer 2021, 9, e002203. [Google Scholar] [CrossRef]
- Rasco, D.; Dumbrava, E.; Sharma, M.; Shepard, D.; Vaena, D.; Fleming, G.; Chmielowski, B.; Hamilton, E.; Sullivan, R.; Papadopoulos, K.; et al. 659 COM701 plus nivolumab demonstrates preliminary antitumor activity and immune modulation of tumor microenvironment in patients with metastatic MSS-CRC and liver metastases. J. Immunother. Cancer 2022, 10, A690. [Google Scholar] [CrossRef]
- Obeidat, A.; Atieh, A.; Vitenshtein, A.; Cinamon, G.; Paz, K.; Roviš, T.; Brilc, P.; Hirsl, L.; Mandelboim, O.; Jonjic, S.; et al. 474 First-in-class anti-PVR mAb NTX1088 restores expression of DNAM1 and augments antitumor immunity. J. Immunother. Cancer 2022, 10, A494. [Google Scholar] [CrossRef]
- Long, G.V.; Eggermont, A.M.; Gershenwald, J.E.; Schadendorf, D.; Ascierto, P.A.; Dummer, R.; Hauschild, A.; Carlino, M.S.; Ribas, A.; Robert, C.; et al. KEYVIBE-010: Adjuvant coformulated vibostolimab with pembrolizumab versus adjuvant pembrolizumab in patients with high-risk stage II-IV melanoma. J. Clin. Oncol. 2023, 41, TPS9611. [Google Scholar] [CrossRef]
- Dummer, R.; Long, G.V.; Pavlick, A.; Postow, M.; Ribas, A.; Robert, C.; Scolyer, R.A.; Taube, J.; Tetzlaff, M.; Liao, J.; et al. 426 MK-3475-U02: Phase 1/2 study of investigational agents with or without pembrolizumab versus pembrolizumab monotherapy in melanoma. J. Immunother. Cancer 2020, 8, A259. [Google Scholar] [CrossRef]
- Niu, J.; Maurice-Dror, C.; Lee, D.H.; Kim, D.W.; Nagrial, A.; Voskoboynik, M.; Chung, H.C.; Mileham, K.; Vaishampayan, U.; Rasco, D.; et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer. Ann. Oncol. 2022, 33, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Rasco, D.W.; Vaena, D.A.; Fleming, G.F.; Dumbrava, E.E.; Yeku, O.O.; Sharma, M.; Papadopoulos, K.P.; Sullivan, R.J.; Gaillard, S.; Adewoye, A.H.; et al. Preliminary antitumor activity of the combination of COM701 + BMS-986207 + nivolumab in patients with recurrent, metastatic MSS endometrial cancer. J. Clin. Oncol. 2023, 41, 5595. [Google Scholar] [CrossRef]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Treatment | Target | Cancer Type | Clinical Trial |
|---|---|---|---|
| COM701 | CD155 | advanced solid tumour | NCT03667716 [212] |
| NTX-1088 | CD155 | advanced solid tumour | NCT05378425 [213] |
| Vibostolimab | TIGIT | melanoma | KEYVIBE-010 [214,215] |
| advanced solid tumour | KEYVIBE-001 [216] | ||
| BMS-986207 | TIGIT | endometrial cancer | NCT04570839 [217] |
| Tiragolumab | TIGIT | non-small-cell lung cancer | CITYSCAPE [218] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.-Y.; Park, S.-H.; Jakobsson, H.; Shackleton, M.; Möller, A. Immune Regulation and Immune Therapy in Melanoma: Review with Emphasis on CD155 Signalling. Cancers 2024, 16, 1950. https://doi.org/10.3390/cancers16111950
Wu L-Y, Park S-H, Jakobsson H, Shackleton M, Möller A. Immune Regulation and Immune Therapy in Melanoma: Review with Emphasis on CD155 Signalling. Cancers. 2024; 16(11):1950. https://doi.org/10.3390/cancers16111950
Chicago/Turabian StyleWu, Li-Ying, Su-Ho Park, Haakan Jakobsson, Mark Shackleton, and Andreas Möller. 2024. "Immune Regulation and Immune Therapy in Melanoma: Review with Emphasis on CD155 Signalling" Cancers 16, no. 11: 1950. https://doi.org/10.3390/cancers16111950
APA StyleWu, L.-Y., Park, S.-H., Jakobsson, H., Shackleton, M., & Möller, A. (2024). Immune Regulation and Immune Therapy in Melanoma: Review with Emphasis on CD155 Signalling. Cancers, 16(11), 1950. https://doi.org/10.3390/cancers16111950