
Exploring Gut Microbiome Composition and Circulating Microbial DNA Fragments in Patients with Stage II/III Colorectal Cancer: A Comprehensive Analysis
 ,
,  , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Patients’ Characteristics
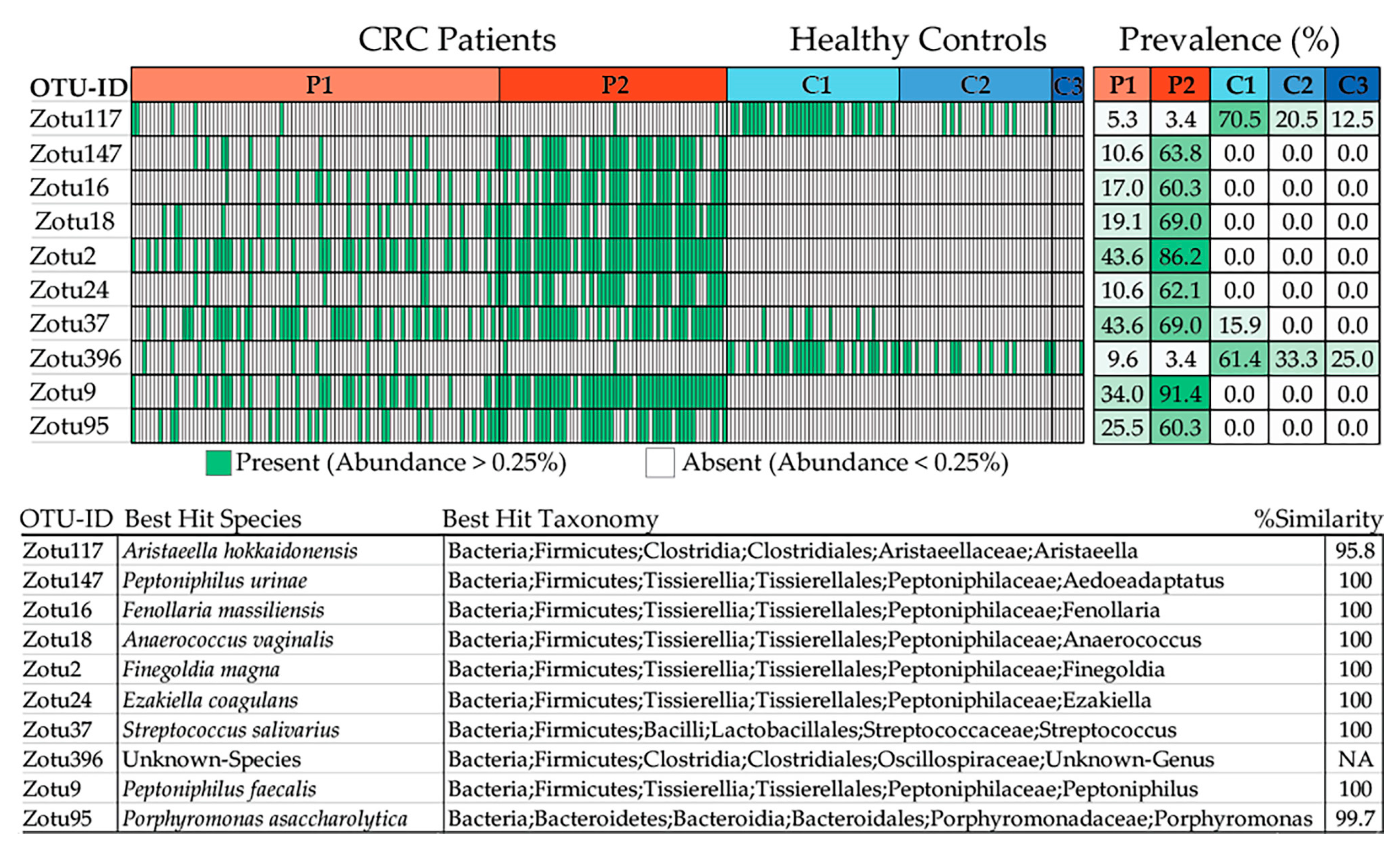
2.2. Taxa Refinement and Downstream Analysis
2.2.1. Normalization
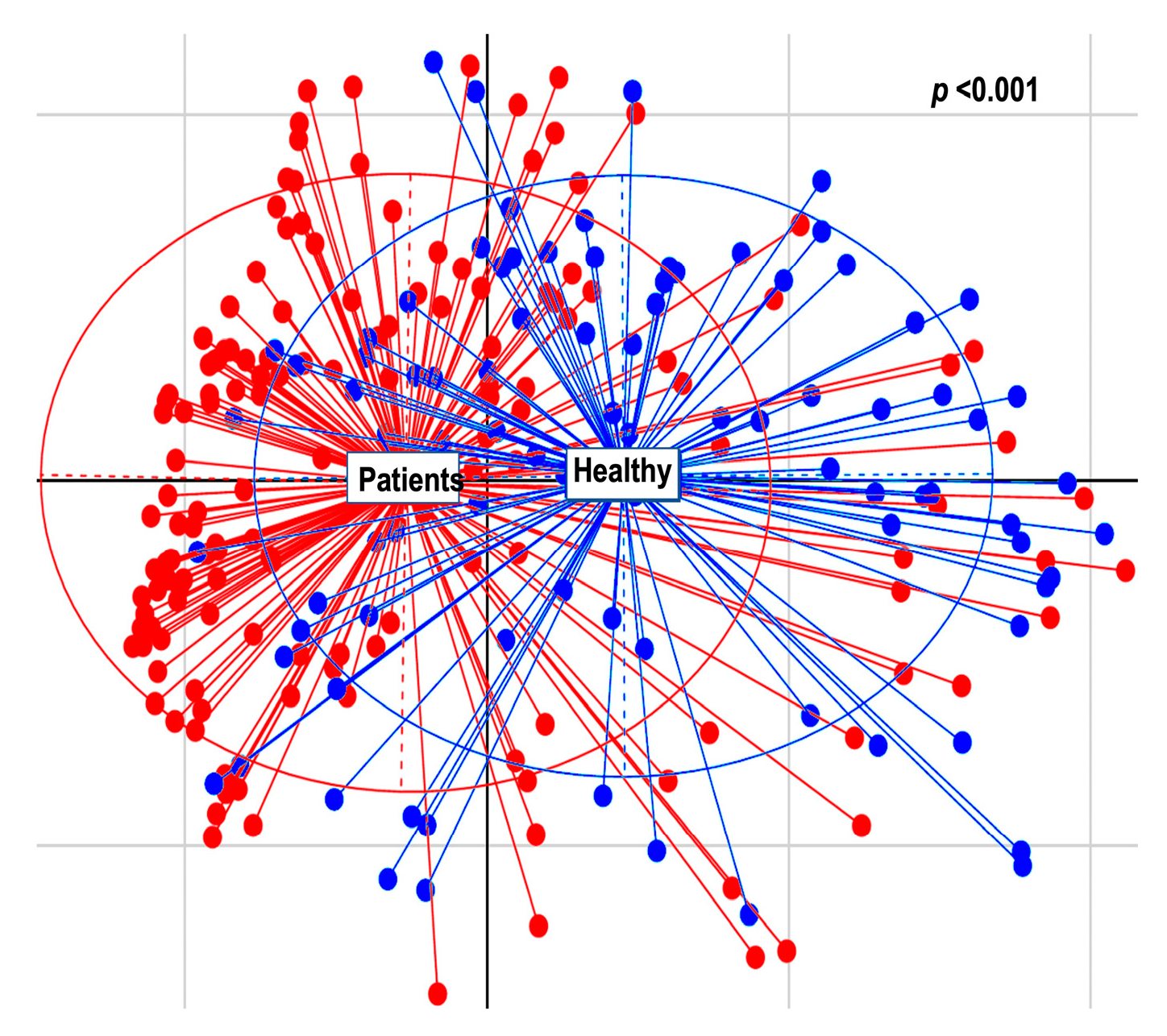
2.2.2. Alpha and Beta Diversity Calculation
2.2.3. Taxonomic Binning
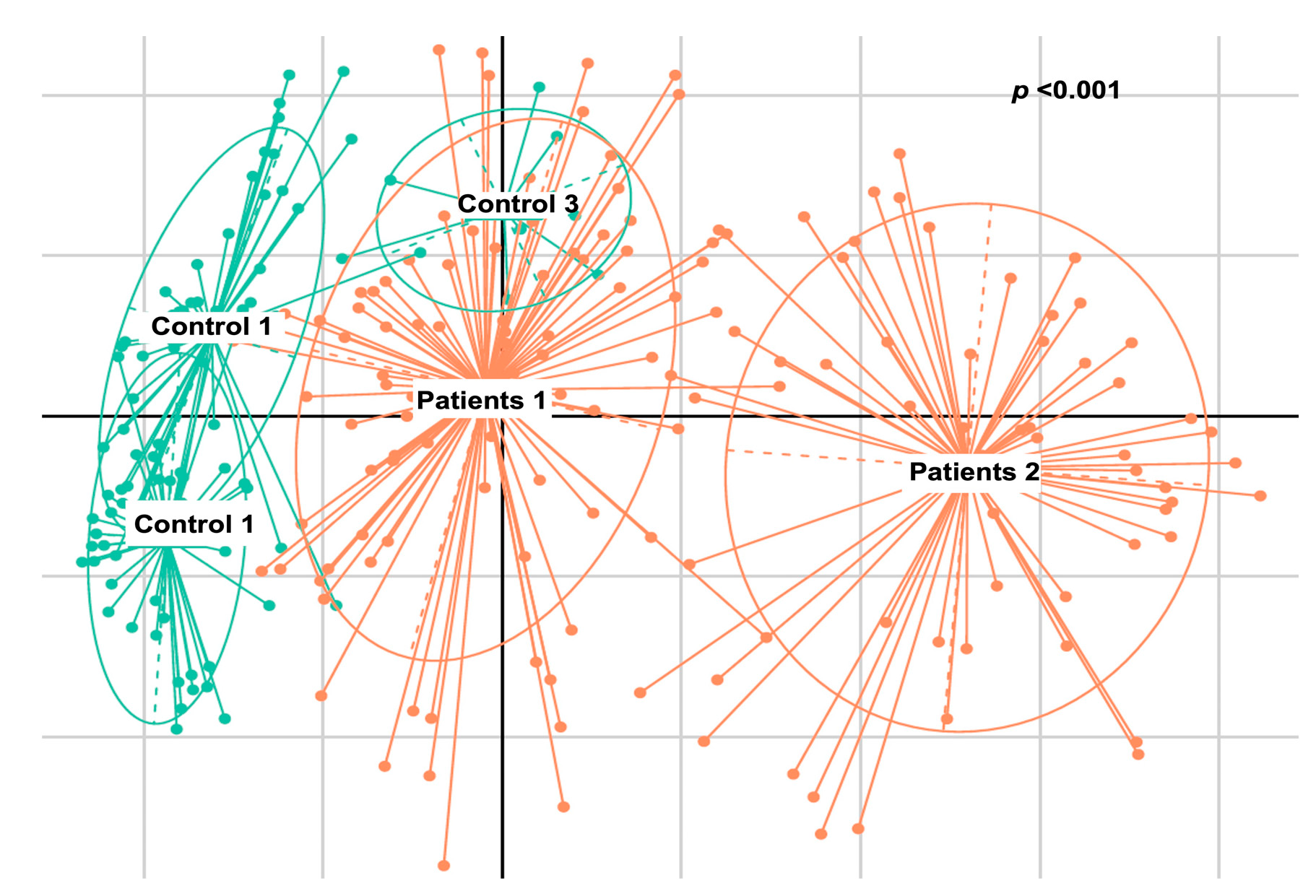
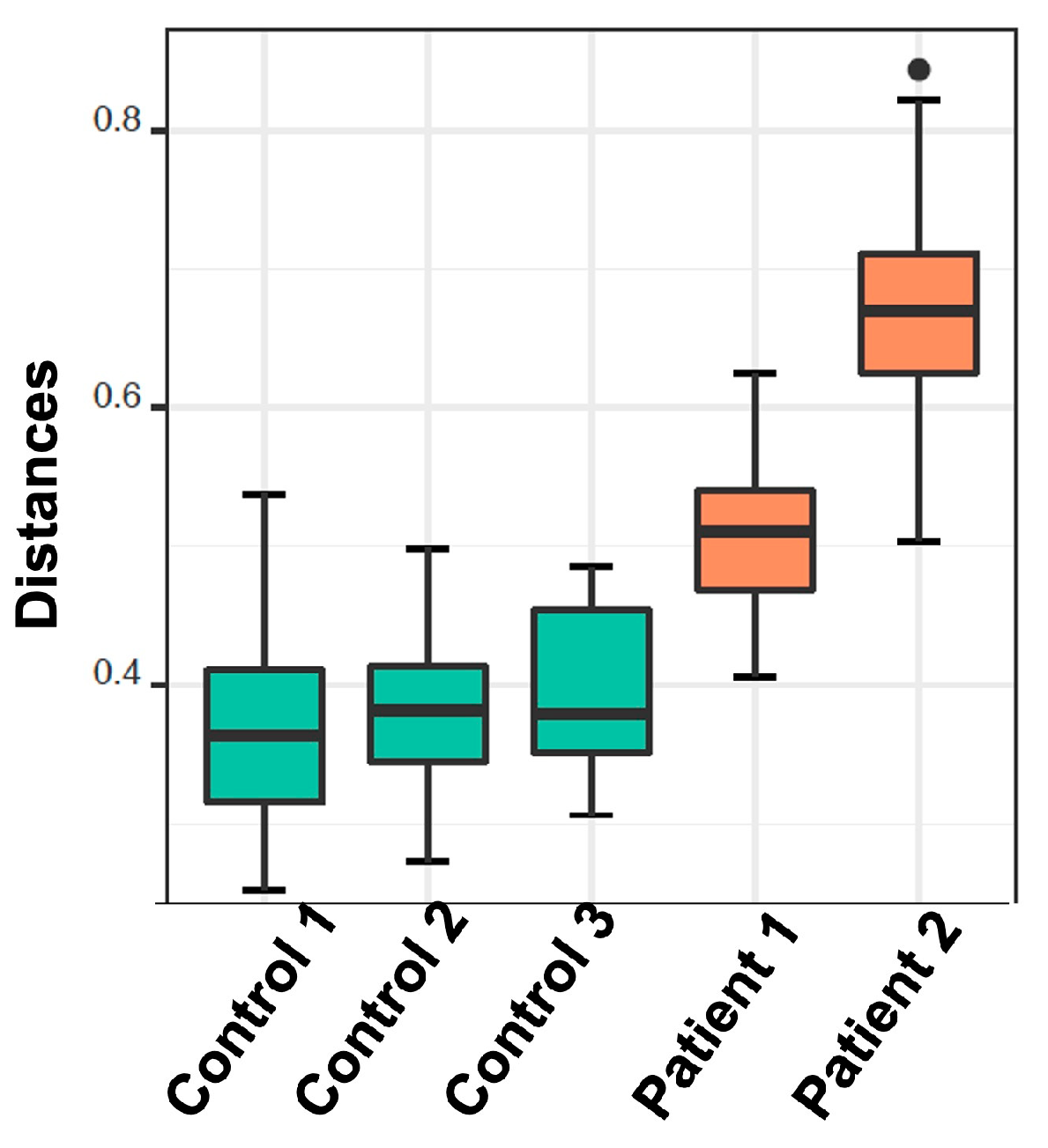
2.2.4. De Novo Clustering Analysis
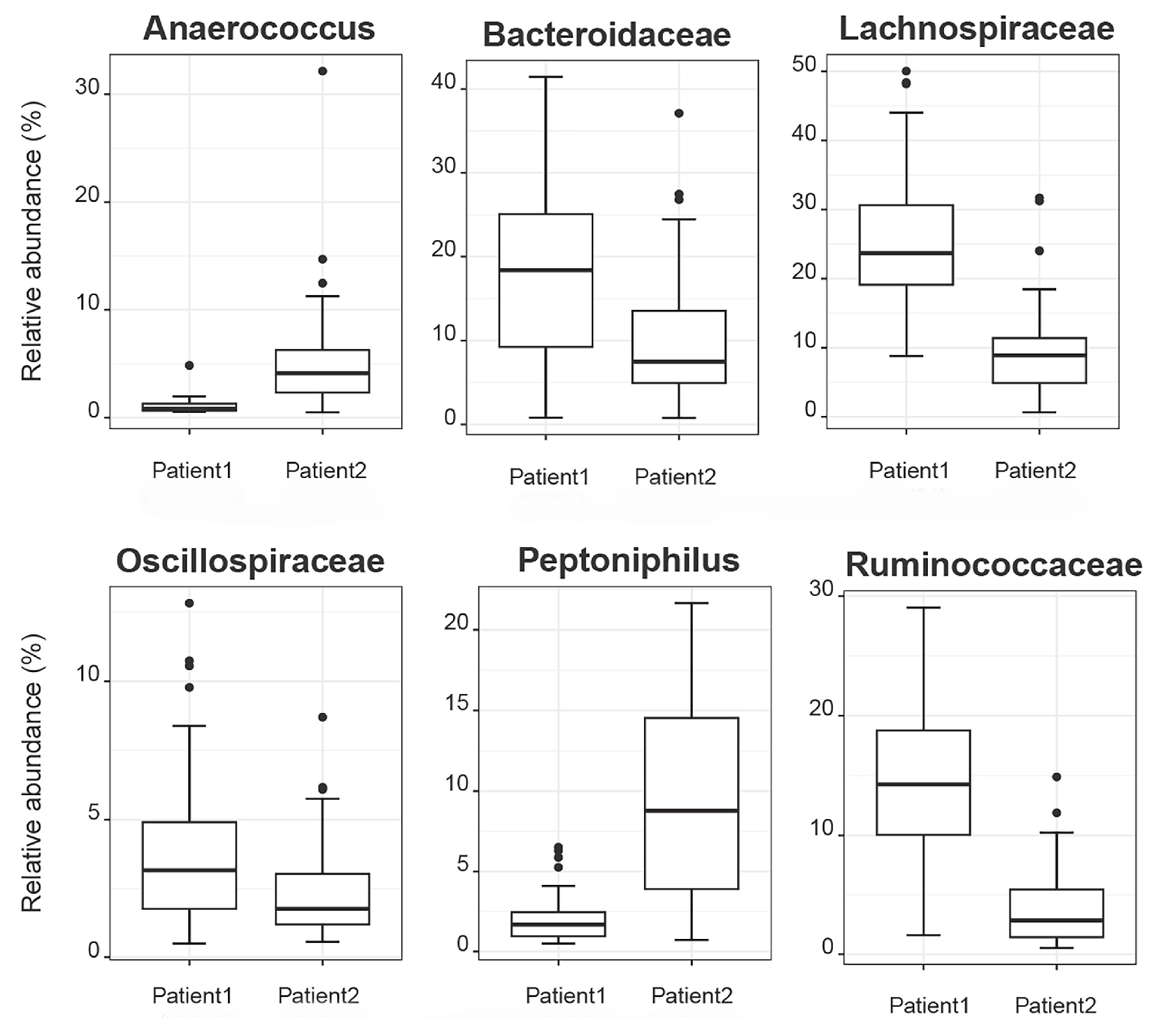
2.2.5. Correlation of De Novo Clustering with Microbial Fragment Detection
3. Discussion
4. Materials and Methods
4.1. Patients and Healthy Controls
4.2. Blood Samples and Microbial DNA Fragment Amplification
4.3. Fecal Samples
4.4. 16S rRNA Sequencing Library Preparation and Microbiome Analysis
4.5. Amplicon Sequence Analysis
4.6. Taxa Refinement and Downstream Analysis
4.7. Bioinformatic and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kohne, C.H.; Schoffski, P.; Wilke, H.; Kaufer, C.; Andreesen, R.; Ohl, U.; Klaasen, U.; Westerhausen, M.; Hiddemann, W.; Schott, G.; et al. Effective biomodulation by leucovorin of high-dose infusion fluorouracil given as a weekly 24-h infusion: Results of a randomized trial in patients with advanced colorectal cancer. J. Clin. Oncol. 1998, 16, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Compton, C.; Fenoglio-Preiser, C.M.; Pettigrew, N.; Fielding, L.P. American Joint Committee on Cancer Prognostic Factors Consensus Conference: Colorectal Working Group. Cancer 2000, 88, 1739–1757. [Google Scholar] [CrossRef]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite Instability Testing and Its Role in the Management of Colorectal Cancer. Curr. Treat. Options Oncol. 2015, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Argente-Pla, M.; Perez-Lazaro, A.; Martinez-Millana, A.; Del Olmo-Garcia, M.I.; Espi-Reig, J.; Beneyto-Castello, I.; Lopez-Andujar, R.; Merino-Torres, J.F. Simultaneous Pancreas Kidney Transplantation Improves Cardiovascular Autonomic Neuropathy with Improved Valsalva Ratio as the Most Precocious Test. J. Diabetes Res. 2020, 2020, 7574628. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, P.D.; Shanahan, F.; Clune, Y.; Collins, J.K.; O’Sullivan, G.C.; O’Riordan, M.; Holmes, E.; Wang, Y.; Marchesi, J.R. Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ. Microbiol. 2008, 10, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Irrazabal, T.; Belcheva, A.; Girardin, S.E.; Martin, A.; Philpott, D.J. The multifaceted role of the intestinal microbiota in colon cancer. Mol. Cell 2014, 54, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Reddick, L.E.; Alto, N.M. Bacteria fighting back: How pathogens target and subvert the host innate immune system. Mol. Cell 2014, 54, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Messaritakis, I.; Koulouridi, A.; Boukla, E.; Sfakianaki, M.; Vogiatzoglou, K.; Karagianni, M.; Gouvas, N.; Tsiaoussis, J.; Xynos, E.; Athanasakis, E.; et al. Investigation of Microbial Translocation, TLR and VDR Gene Polymorphisms, and Recurrence Risk in Stage III Colorectal Cancer Patients. Cancers 2022, 14, 4407. [Google Scholar] [CrossRef]
- Messaritakis, I.; Vogiatzoglou, K.; Tsantaki, K.; Ntretaki, A.; Sfakianaki, M.; Koulouridi, A.; Tsiaoussis, J.; Mavroudis, D.; Souglakos, J. The Prognostic Value of the Detection of Microbial Translocation in the Blood of Colorectal Cancer Patients. Cancers 2020, 12, 1058. [Google Scholar] [CrossRef]
- Koulouridi, A.; Messaritakis, I.; Theodorakis, E.; Chondrozoumaki, M.; Sfakianaki, M.; Gouvas, N.; Tsiaoussis, J.; Mavroudis, D.; Tzardi, M.; Souglakos, J. Detection of Circulating Tumor Cells and Microbial DNA Fragments in Stage III Colorectal Cancer Patients under Three versus Six Months of Adjuvant Treatment. Cancers 2021, 13, 3552. [Google Scholar] [CrossRef] [PubMed]
- Koulouris, A.; Tsagkaris, C.; Messaritakis, I.; Gouvas, N.; Sfakianaki, M.; Trypaki, M.; Spyrou, V.; Christodoulakis, M.; Athanasakis, E.; Xynos, E.; et al. Resectable Colorectal Cancer: Current Perceptions on the Correlation of Recurrence Risk, Microbiota and Detection of Genetic Mutations in Liquid Biopsies. Cancers 2021, 13, 3522. [Google Scholar] [CrossRef] [PubMed]
- Laukens, D.; Brinkman, B.M.; Raes, J.; De Vos, M.; Vandenabeele, P. Heterogeneity of the gut microbiome in mice: Guidelines for optimizing experimental design. FEMS Microbiol. Rev. 2016, 40, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Markman, J.L.; Shiao, S.L. Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest. Oncol. 2015, 6, 208–223. [Google Scholar] [CrossRef] [PubMed]
- Sadrekarimi, H.; Gardanova, Z.R.; Bakhshesh, M.; Ebrahimzadeh, F.; Yaseri, A.F.; Thangavelu, L.; Hasanpoor, Z.; Zadeh, F.A.; Kahrizi, M.S. Emerging role of human microbiome in cancer development and response to therapy: Special focus on intestinal microflora. J. Transl. Med. 2022, 20, 301. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; He, C.; Li, X.; Cai, Y.; Hu, J.; Liao, Y.; Zhao, J.; Xia, L.; He, W.; Liu, L.; et al. Gut microbiota dysbiosis worsens the severity of acute pancreatitis in patients and mice. J. Gastroenterol. 2019, 54, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Alhhazmi, A.A.; Alhamawi, R.M.; Almisned, R.M.; Almutairi, H.A.; Jan, A.A.; Kurdi, S.M.; Almutawif, Y.A.; Mohammed-Saeid, W. Gut Microbial and Associated Metabolite Markers for Colorectal Cancer Diagnosis. Microorganisms 2023, 11, 2037. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. mBio 2013, 4, e00692-13. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef]
- Gagniere, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Probert, H.M.; Loo, J.V.; Rastall, R.A.; Roberfroid, M.B. Dietary modulation of the human colonic microbiota: Updating the concept of prebiotics. Nutr. Res. Rev. 2004, 17, 259–275. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Gonzalez-Mercado, V.J.; Lim, J.; Yu, G.; Penedo, F.; Pedro, E.; Bernabe, R.; Tirado-Gomez, M.; Aouizerat, B. Co-Occurrence of Symptoms and Gut Microbiota Composition Before Neoadjuvant Chemotherapy and Radiation Therapy for Rectal Cancer: A Proof of Concept. Biol. Res. Nurs. 2021, 23, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jia, Y.; Wen, L.; Mu, W.; Wu, X.; Liu, T.; Liu, X.; Fang, J.; Luan, Y.; Chen, P.; et al. Correction: Porphyromonas Gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res. 2022, 82, 2196. [Google Scholar] [CrossRef] [PubMed]
- Tarashi, S.; Siadat, S.D.; Ahmadi Badi, S.; Zali, M.; Biassoni, R.; Ponzoni, M.; Moshiri, A. Gut Bacteria and their Metabolites: Which One Is the Defendant for Colorectal Cancer? Microorganisms 2019, 7, 561. [Google Scholar] [CrossRef]
- Abbas, M.; Gaia, N.; Buchs, N.C.; Delaune, V.; Girard, M.; Andrey, D.O.; Meyer, J.; Schrenzel, J.; Ris, F.; Harbarth, S.; et al. Changes in the gut bacterial communities in colon cancer surgery patients: An observational study. Gut Pathog. 2022, 14, 2. [Google Scholar] [CrossRef]
- Fenner, L.; Roux, V.; Ananian, P.; Raoult, D. Alistipes finegoldii in blood cultures from colon cancer patients. Emerg. Infect. Dis. 2007, 13, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Kwong, T.N.Y.; Chow, T.C.; Luk, A.K.C.; Dai, R.Z.W.; Nakatsu, G.; Lam, T.Y.T.; Zhang, L.; Wu, J.C.Y.; Chan, F.K.L.; et al. Quantitation of faecal Fusobacterium improves faecal immunochemical test in detecting advanced colorectal neoplasia. Gut 2017, 66, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Zheng, S.; Li, M.; Xu, C.; Jia, D.; Qi, Y.; Hou, T.; Wang, L.; Wang, B.; et al. Fusobacterium nucleatum promotes colorectal cancer cells adhesion to endothelial cells and facilitates extravasation and metastasis by inducing ALPK1/NF-kappaB/ICAM1 axis. Gut Microbes 2022, 14, 2038852. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Feng, Y.Q.; Lyu, N.; Wang, D.; Yu, W.D.; Hu, Y.F. Fusobacterium nucleatum promotes colon cancer progression by changing the mucosal microbiota and colon transcriptome in a mouse model. World J. Gastroenterol. 2022, 28, 1981–1995. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Angulo, M.; Ayala-Madrigal, M.L.; Moreno-Ortiz, J.M.; Peregrina-Sandoval, J.; Garcia-Ayala, F.D. Microbiota composition and its impact on DNA methylation in colorectal cancer. Front. Genet. 2023, 14, 1037406. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Wang, H.; Tao, Y.; Luo, K.; Ye, J.; Ran, S.; Guan, Z.; Wang, Y.; Hu, H.; Huang, R. Fusobacterium nucleatum and colorectal cancer: From phenomenon to mechanism. Front. Cell Infect. Microbiol. 2022, 12, 1020583. [Google Scholar] [CrossRef]
- Serna, G.; Ruiz-Pace, F.; Hernando, J.; Alonso, L.; Fasani, R.; Landolfi, S.; Comas, R.; Jimenez, J.; Elez, E.; Bullman, S.; et al. Fusobacterium nucleatum persistence and risk of recurrence after preoperative treatment in locally advanced rectal cancer. Ann. Oncol. 2020, 31, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Bergonzini, A.; Lopez Chiloeches, M.; Paparouna, E.; Butter, D.; Theodorou, S.D.P.; Haykal, M.M.; Boutet-Robinet, E.; Tebaldi, T.; Wakeham, A.; et al. Influence of the microenvironment on modulation of the host response by typhoid toxin. Cell Rep. 2021, 35, 108931. [Google Scholar] [CrossRef]
- Lyu, W.N.; Lin, M.C.; Shen, C.Y.; Chen, L.H.; Lee, Y.H.; Chen, S.K.; Lai, L.C.; Chuang, E.Y.; Lou, P.J.; Tsai, M.H. An Oral Microbial Biomarker for Early Detection of Recurrence of Oral Squamous Cell Carcinoma. ACS Infect. Dis. 2023, 9, 1783–1792. [Google Scholar] [CrossRef]
- Sun, L.; Ke, X.; Guan, A.; Jin, B.; Qu, J.; Wang, Y.; Xu, X.; Li, C.; Sun, H.; Xu, H.; et al. Intratumoural microbiome can predict the prognosis of hepatocellular carcinoma after surgery. Clin. Transl. Med. 2023, 13, e1331. [Google Scholar] [CrossRef] [PubMed]
- Debelius, J.W.; Engstrand, L.; Matussek, A.; Brusselaers, N.; Morton, J.T.; Stenmarker, M.; Olsen, R.S. The Local Tumor Microbiome Is Associated with Survival in Late-Stage Colorectal Cancer Patients. Microbiol. Spectr. 2023, 11, e0506622. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Wang, N.; Han, W.; Ban, M.; Sun, T.; Xu, J. Vaginal and tumor microbiomes in gynecological cancer (Review). Oncol. Lett. 2023, 25, 153. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Jiang, R.; Chen, W.; Liu, Y.; Wang, G.; Gong, X.; Wang, Y. Novel prognostic indicator combining inflammatory indicators and tumor markers for gastric cancer. World J. Surg. Oncol. 2023, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Sharma, N.; Bharmjeet; Das, A. Unraveling the intricate relationship: Influence of microbiome on the host immune system in carcinogenesis. Cancer Rep. 2023, 6, e1892. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, X.; Wang, Y.; Kaur, B.; Han, H.; Yu, J. The Notch signaling pathway: A potential target for cancer immunotherapy. J. Hematol. Oncol. 2023, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Amit, U.; Facciabene, A.; Ben-Josef, E. Radiation Therapy and the Microbiome; More Than a Gut Feeling. Cancer J. 2023, 29, 84–88. [Google Scholar] [CrossRef]
- Luu, M.; Schutz, B.; Lauth, M.; Visekruna, A. The Impact of Gut Microbiota-Derived Metabolites on the Tumor Immune Microenvironment. Cancers 2023, 15, 1588. [Google Scholar] [CrossRef]
- Dai, Z.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.; Chen, Z.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef]
- Zhuang, Y.P.; Zhou, H.L.; Chen, H.B.; Zheng, M.Y.; Liang, Y.W.; Gu, Y.T.; Li, W.T.; Qiu, W.L.; Zhou, H.G. Gut microbiota interactions with antitumor immunity in colorectal cancer: From understanding to application. Biomed. Pharmacother. 2023, 165, 115040. [Google Scholar] [CrossRef] [PubMed]
- Pantalone, M.R.; Martin Almazan, N.; Lattanzio, R.; Taher, C.; De Fabritiis, S.; Valentinuzzi, S.; Bishehsari, F.; Mahdavinia, M.; Verginelli, F.; Rahbar, A.; et al. Human cytomegalovirus infection enhances 5-lipoxygenase and cycloxygenase-2 expression in colorectal cancer. Int. J. Oncol. 2023, 63, 116. [Google Scholar] [CrossRef]
- Fan, J.Q.; Zhao, W.F.; Lu, Q.W.; Zha, F.R.; Lv, L.B.; Ye, G.L.; Gao, H.L. Fecal microbial biomarkers combined with multi-target stool DNA test improve diagnostic accuracy for colorectal cancer. World J. Gastrointest. Oncol. 2023, 15, 1424–1435. [Google Scholar] [CrossRef]
- Gao, W.; Gao, X.; Zhu, L.; Gao, S.; Sun, R.; Feng, Z.; Wu, D.; Liu, Z.; Zhu, R.; Jiao, N. Multimodal metagenomic analysis reveals microbial single nucleotide variants as superior biomarkers for early detection of colorectal cancer. Gut Microbes 2023, 15, 2245562. [Google Scholar] [CrossRef]
- Shahbazi, T.; Bakhshi, B.; Sadegh Fazeli, M.; Derakhshan-Nezhadc, E. Bacterial biomarkers: New aspects of colorectal tumors diagnosis: Reality or fantasy. Eur. J. Cancer Prev. 2023, 32, 485–497. [Google Scholar] [CrossRef]
- Bassis, C.M.; Moore, N.M.; Lolans, K.; Seekatz, A.M.; Weinstein, R.A.; Young, V.B.; Hayden, M.K.; for the CDC Prevention Epicenters Program. Comparison of stool versus rectal swab samples and storage conditions on bacterial community profiles. BMC Microbiol. 2017, 17, 78. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 2016, 6, 33721. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R. UCHIME, USEARCH software and documentation. 2011. Available online: http://drive5.com/usearch/ (accessed on 20 August 2023).
- Reitmeier, S.; Hitch, T.C.A.; Treichel, N.; Fikas, N.; Hausmann, B.; Ramer-Tait, A.E.; Neuhaus, K.; Berry, D.; Haller, D.; Lagkouvardos, I.; et al. Handling of spurious sequences affects the outcome of high-throughput of 16S rRNA gene amplicon profiling. ISME Commun. 2021, 1, 31. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.megasoftware.net (accessed on 12 September 2023).
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Fischer, S.; Kumar, N.; Clavel, T. Rhea: A transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ 2017, 5, e2836. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Jost, L. Partitioning diversity into independent alpha and beta components. Ecology 2007, 88, 2427–2439. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Minchin, P.R. An evaluation of the relative robustness of techniques for ecological ordination. Vegetatio 1987, 69, 89–107. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.; Rousseeuw, P. Finding Groups in Data: An Introduction to Cluster Analysis. 1990. Available online: https://www.researchgate.net/publication/220695963_Finding_Groups_in_Data_An_Introduction_To_Cluster_Analysis (accessed on 10 October 2023).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA | Gene Target | Detection | No (%) |
|---|---|---|---|
| Microbial DNA fragments | 16S rRNA | Positive | 71 (50.0%) |
| Negative | 71 (50.0%) | ||
| E. coli | Positive | 35 (24.6%) | |
| Negative | 107 (75.4%) | ||
| B. fragilis | Positive | 46 (32.4%) | |
| Negative | 96 (67.6%) | ||
| C. albicans | Positive | 76 (53.5%) | |
| Negative | 66 (46.5%) |
| Taxonomy | Patients (N = 142) | % |
|---|---|---|
| Bacteria; Firmicutes; Clostridia; Peptostreptococcales-Tissierellales; Peptoniphilus | 85 | 59.9 |
| Bacteria; Firmicutes; Clostridia; Peptostreptococcales-Tissierellales; Fenollaria | 51 | 35.9 |
| Bacteria; Firmicutes; Clostridia; Peptostreptococcales-Tissierellales; Anaerococcus | 58 | 40.8 |
| Bacteria; Firmicutes; Clostridia; Peptostreptococcales-Tissierellales; Finegoldia | 91 | 64.1 |
| Bacteria; Firmicutes; Clostridia; Peptostreptococcales-Tissierellales; Ezakiella | 46 | 32.4 |
| Bacteria; Bacteroidota; Bacteroidia; Bacteroidales; Porphyromonadaceae; Porphyromonas | 59 | 41.5 |
| Groups | p-Value |
|---|---|
| Control 1–Control 3 | 0.3081 |
| Control 1–Control 2 | 0.3292 |
| Control 2–Control 3 | 0.5913 |
| Patient 1–Control 1 | 0.0000 |
| Patient 1–Control 2 | 0.0000 |
| Patient 2–Control 1 | 0.0000 |
| Patient 2–Control 2 | 0.0000 |
| Patient 2–Control 3 | 0.0000 |
| Patient 1–Control 3 | 0.0006 |
| Patient 1–Patient 2 | 0.0000 |
| Group 1 (N = 89) | Group 2 (N = 53) | p-Value | ||
|---|---|---|---|---|
| Gender | Males | 60 (68.2%) | 34 (65.4%) | 0.733 |
| Females | 28 (31.8%) | 18 (34.6%) | ||
| Age | ≥70 | 15 (33.3%) | 9 (36.0%) | 0.822 |
| <70 | 30 (66.7%) | 16 (64.0) | ||
| 16S rRNA | Positive | 37 (42.0%) | 34 (65.4%) | 0.008 |
| Negative | 51 (58.0%) | 18 (34.6%) | ||
| E. coli | Positive | 22 (25.0%) | 13 (25.0%) | 1.000 |
| Negative | 66 (75.0%) | 39 (75.0%) | ||
| B. fragilis | Positive | 31 (35.2%) | 15 (28.8%) | 0.437 |
| Negative | 57 (64.8%) | 37 (71.2%) | ||
| C. albicans | Positive | 40 (45.5%) | 35 (67.3%) | 0.012 |
| Negative | 48 (54.5%) | 17 (32.7%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messaritakis, I.; Koulouris, A.; Boukla, E.; Vogiatzoglou, K.; Lagkouvardos, I.; Intze, E.; Sfakianaki, M.; Chondrozoumaki, M.; Karagianni, M.; Athanasakis, E.; et al. Exploring Gut Microbiome Composition and Circulating Microbial DNA Fragments in Patients with Stage II/III Colorectal Cancer: A Comprehensive Analysis. Cancers 2024, 16, 1923. https://doi.org/10.3390/cancers16101923
Messaritakis I, Koulouris A, Boukla E, Vogiatzoglou K, Lagkouvardos I, Intze E, Sfakianaki M, Chondrozoumaki M, Karagianni M, Athanasakis E, et al. Exploring Gut Microbiome Composition and Circulating Microbial DNA Fragments in Patients with Stage II/III Colorectal Cancer: A Comprehensive Analysis. Cancers. 2024; 16(10):1923. https://doi.org/10.3390/cancers16101923
Chicago/Turabian StyleMessaritakis, Ippokratis, Andreas Koulouris, Eleni Boukla, Konstantinos Vogiatzoglou, Ilias Lagkouvardos, Evangelia Intze, Maria Sfakianaki, Maria Chondrozoumaki, Michaela Karagianni, Elias Athanasakis, and et al. 2024. "Exploring Gut Microbiome Composition and Circulating Microbial DNA Fragments in Patients with Stage II/III Colorectal Cancer: A Comprehensive Analysis" Cancers 16, no. 10: 1923. https://doi.org/10.3390/cancers16101923
APA StyleMessaritakis, I., Koulouris, A., Boukla, E., Vogiatzoglou, K., Lagkouvardos, I., Intze, E., Sfakianaki, M., Chondrozoumaki, M., Karagianni, M., Athanasakis, E., Xynos, E., Tsiaoussis, J., Christodoulakis, M., Flamourakis, M. E., Tsagkataki, E. S., Giannikaki, L., Chliara, E., Mavroudis, D., Tzardi, M., & Souglakos, J. (2024). Exploring Gut Microbiome Composition and Circulating Microbial DNA Fragments in Patients with Stage II/III Colorectal Cancer: A Comprehensive Analysis. Cancers, 16(10), 1923. https://doi.org/10.3390/cancers16101923