
Novel Endocrine Therapeutic Opportunities for Estrogen Receptor-Positive Ovarian Cancer—What Can We Learn from Breast Cancer?


Abstract
Simple Summary
Abstract
1. Introduction
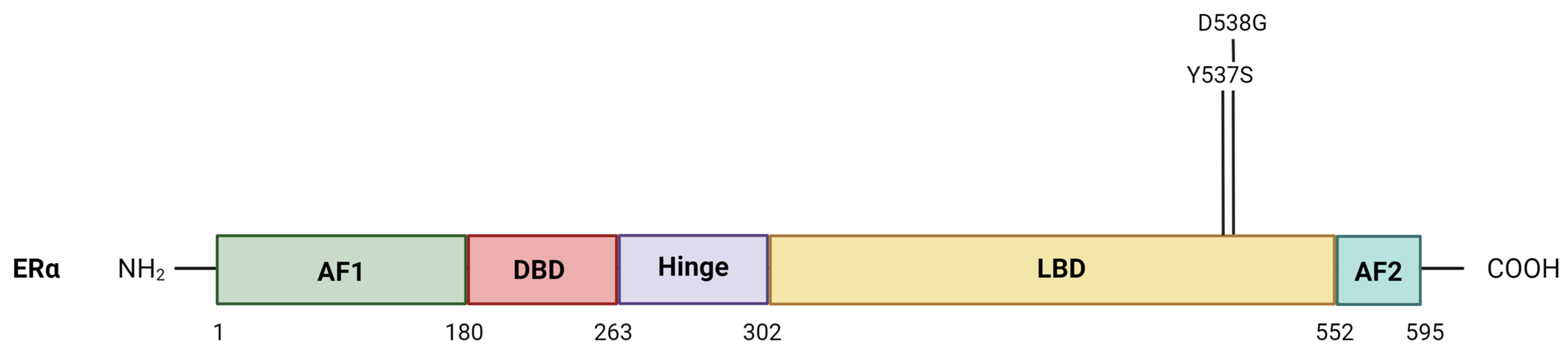
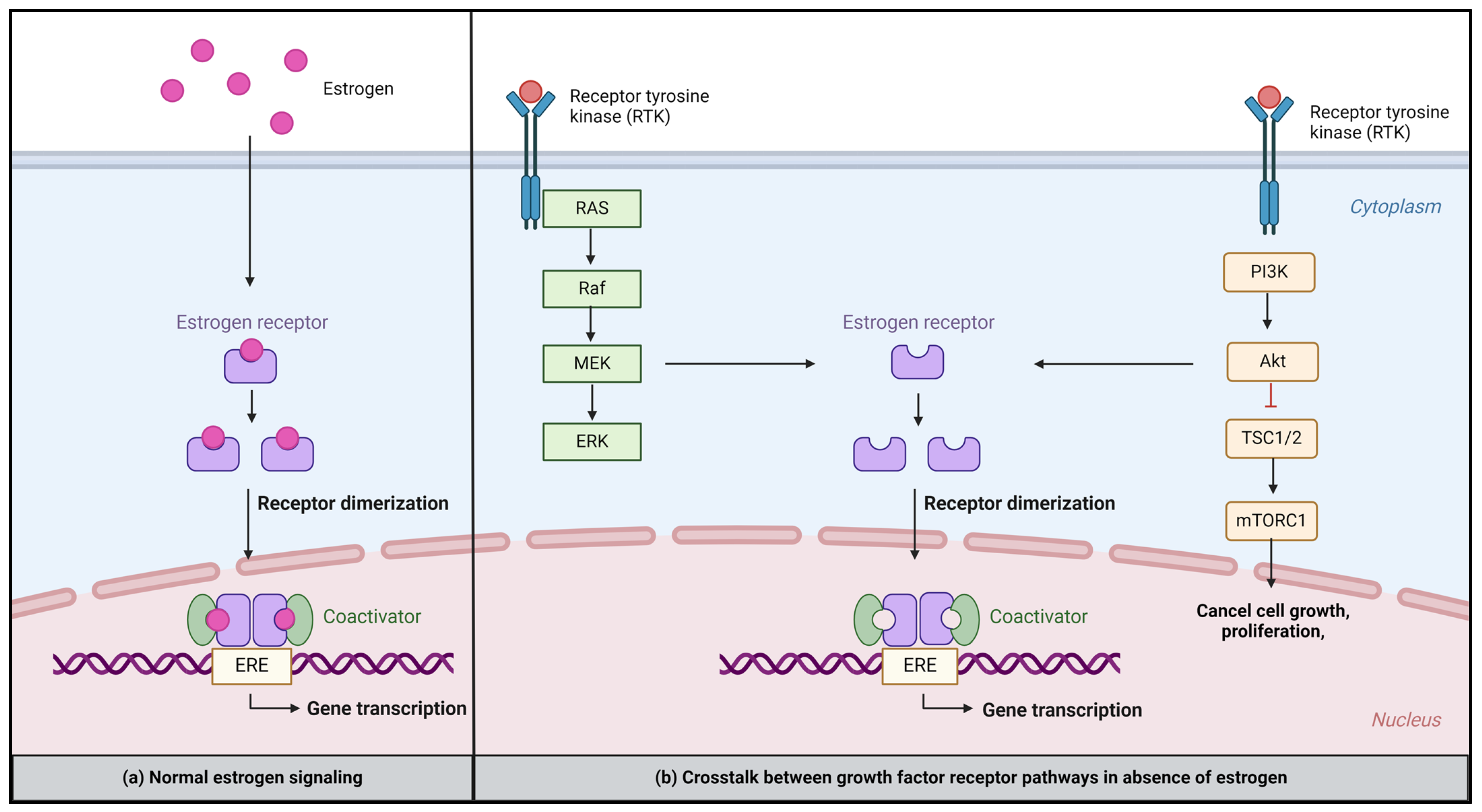
2. Mechanisms of Estrogen Receptor Signaling
3. Possible Mechanisms of Endocrine Resistance
3.1. ESR1 Alterations
3.1.1. Loss of ER Expression
3.1.2. Mutations in the ESR1 Gene
- Ligand-independent activation
- Activation of alternative signaling pathways
3.2. Crosstalk between ER and Growth Factor Signaling Pathways
- Growth factor receptor pathways
3.3. Epigenetic Modification
4. Novel Potential Therapeutic Strategies
4.1. Combination Therapies with Molecularly Targeted Agents
4.1.1. CDK4/6 Inhibitors Combinations
4.1.2. MEK Inhibitor Combinations
4.1.3. PI3K/mTOR Inhibitors Combinations
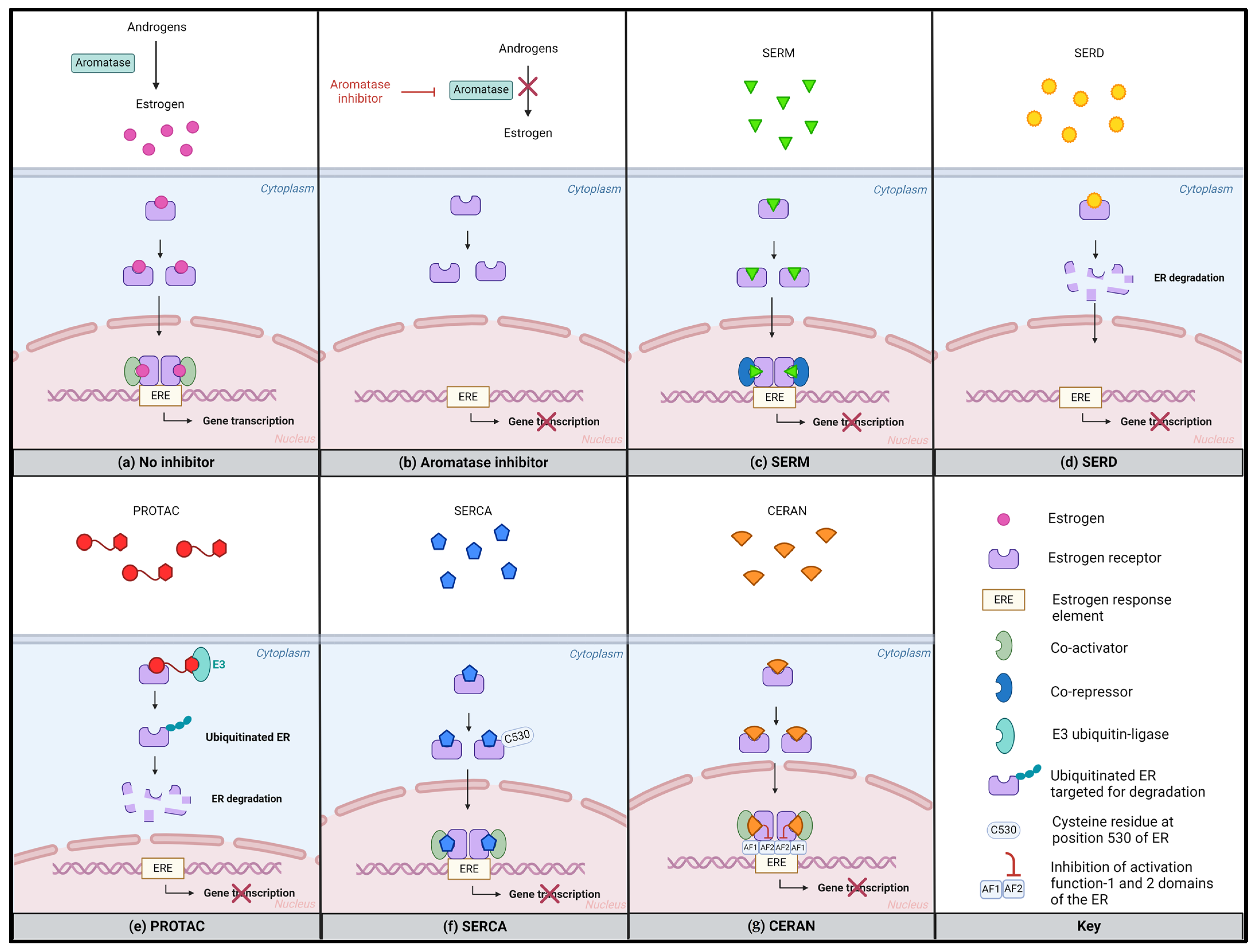
5. Next-Generation SERMs and SERDs to Overcome Endocrine Resistance

{kind=link}
{kind=link}
{kind=link}
| Drug(s) | Mode of Action | Clinical Trial | Phase | Indication | Design | Primary Endpoint | Reference |
|---|---|---|---|---|---|---|---|
| Giredestrant combination therapy | SERD | persevERA NCT04546009 | III | ER+HER2- advanced or metastatic breast cancer, first-line | Giredestrant combined with palbociclib vs. palbociclib + letrozole | PFS, as determined by the investigator according to RECIST v1.1 | Turner et al. [89] |
| Camizestrant combination therapy | SERD | SERENA-4 NCT04711252 | III | ER+/HER2- advanced or metastatic breast cancer, first-line | Camizestrant combined with palbociclib vs. palbociclib + aromatase inhibitor (anastrozole) | PFS, as determined by the investigator according to RECIST v1.1 | Im et al. [90] |
| Giredestrant combination therapy | SERD | evERA NCT05306340 | III | ER+/HER2- advanced or metastatic breast cancer, who received previous treatment with a CDK4/6i and ET | Giredestrant combined with everolimus vs. physician’s choice of endocrine therapy + everolimus | PFS, as determined by the investigator according to RECIST v1.1 | Mayer et al. [91] |
| Giredestrant combination therapy | SERD | MORPHEUS NCT04802759 | Ib/II | ER+/HER2- advanced or metastatic breast cancer, second- or third-line | Giredestrant alone or combined with abemaciclib, palbociclib, ribociclib, ipatasertib, inavolisib, everolimus, or samuraciclib | ORR, as determined by the investigator according to RECIST v1.1 | Oliveira et al. [92] |
| Camizestrant combination therapy | SERD | SERENA-6 NCT04964934 | III | ER+/HER2-, ESR1 mutated, advanced or metastatic breast cancer on first-line aromatase inhibitor + CDK4/6i before disease progression | Switch to camizestrant combined with CDK4/6 inhibitor vs. continue current treatment with aromatase inhibitor + CDK4/6 inhibitor | PFS, as determined by the investigator according to RECIST v1.1 | Turner et al. [93] |
| Imlunestrant monotherapy or combination therapy | SERD | EMBER-3 NCT04975308 | III | ER+/HER2- advanced or metastatic breast cancer, who received previous treatment with ET, +/− CDKi | Imlunestrant monotherapy vs. combined with abemaciclib vs. investigator’s choice of endocrine therapy | PFS, as determined by the investigator according to RECIST v1.1 | Jhaveri et al. [94] |
| Drug(s) | Mode of Action | Clinical Trial | Phase | Indication | Design | Primary Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Rintodestrant combination therapy | SERD | NCT03455270 | I | ER+/HER2- advanced or metastatic breast cancer, 2nd-line or more | Rintodestrant monotherapy vs. combined with palbociclib | Dose-limiting toxicity Recommended phase 2 dose | Adreano et al. [95] |
| ZN-c5 combination therapy | SERD | NCT04514159 | Ib | ER+/HER2- advanced or metastatic breast cancer, 2nd-line or more, no prior CDK4/6i | ZN-c5 combined with abemaciclib | Maximum tolerated dose Recommended phase 2 dose | Keogh et al. [96] |
| ZN-c5 monotherapy or combination therapy | SERD | NCT03560531 | I/II | ER+/HER2- advanced or metastatic breast cancer, 2nd-line or more | ZN-c5 monotherapy or combined with palbociclib | Maximum tolerated dose Recommended phase 2 dose | Abramson et al. [97] |
| Borestrant monotherapy and combination therapy | SERD | ENZENO NCT04669587 | I/II | ER+/HER2- advanced or metastatic breast cancer at any line | Borestrant monotherapy and combined with palbociclib | Safety and tolerability | NA |
| D-0502 monotherapy and combination therapy | SERD | NCT03471663 | I | ER+/HER2- advanced or metastatic breast cancer, 2nd-line or more | D-0502 monotherapy and combined with palbociclib | Safety and tolerability | Osborne et al. [98] |
| ARV-471 monotherapy and combination therapy | PROTAC | NCT04072952 | I/II | ER+/HER2- advanced or metastatic breast cancer, 2nd-line or more | ARV-471 monotherapy and combined with palbociclib | Safety and tolerability | Hamilton et al. [86] |
| OP-1250 monotherapy | CERAN | NCT04505826 | I/II | ER+/HER2- advanced or metastatic breast cancer, 2nd-line or more | OP-1250 monotherapy | Safety and tolerability | Hamilton et al. [88] |
| H3B-6545 combination therapy | SERCA | NCT04288089 | Ib | ER+/HER2- advanced or metastatic breast cancer, 3rd-line or more | H3b-6545 combined with palbociclib | Maximum tolerated dose Recommended phase 2 dose | Johnston et al. [87] |
6. Future Directions
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schmeler, K.M.; Gershenson, D.M. Low-Grade Serous Ovarian Cancer: A Unique Disease. Curr. Oncol. Rep. 2008, 10, 519–523. [Google Scholar] [CrossRef]
- Zwimpfer, T.A.; Tal, O.; Geissler, F.; Coelho, R.; Rimmer, N.; Jacob, F.; Heinzelmann-Schwarz, V. Low Grade Serous Ovarian Cancer—A Rare Disease with Increasing Therapeutic Options. Cancer Treat. Rev. 2023, 112, 102497. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Sun, C.C.; Bodurka, D.; Coleman, R.L.; Lu, K.H.; Sood, A.K.; Deavers, M.; Malpica, A.L.; Kavanagh, J.J. Recurrent Low-Grade Serous Ovarian Carcinoma Is Relatively Chemoresistant. Gynecol. Oncol. 2009, 114, 48–52. [Google Scholar] [CrossRef]
- Grabowski, J.P.; Harter, P.; Heitz, F.; Pujade-Lauraine, E.; Reuss, A.; Kristensen, G.; Ray-Coquard, I.; Heitz, J.; Traut, A.; Pfisterer, J.; et al. Operability and Chemotherapy Responsiveness in Advanced Low-Grade Serous Ovarian Cancer. An Analysis of the AGO Study Group Metadatabase. Gynecol. Oncol. 2016, 140, 457–462. [Google Scholar] [CrossRef]
- Grisham, R.N.; Slomovitz, B.M.; Andrews, N.; Banerjee, S.; Brown, J.; Carey, M.S.; Chui, H.; Coleman, R.L.; Fader, A.N.; Gaillard, S.; et al. Low-Grade Serous Ovarian Cancer: Expert Consensus Report on the State of the Science. Int. J. Gynecol. Cancer 2023, 33, 1331–1344. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Sun, C.C.; Lu, K.H.; Coleman, R.L.; Sood, A.K.; Malpica, A.; Deavers, M.T.; Silva, E.G.; Bodurka, D.C. Clinical Behavior of Stage II–IV Low-Grade Serous Carcinoma of the Ovary. Obstet. Gynecol. 2006, 108, 361–368. [Google Scholar] [CrossRef]
- Wong, K.K.; Lu, K.H.; Malpica, A.; Bodurka, D.C.; Shvartsman, H.S.; Schmandt, R.E.; Thornton, A.D.; Deavers, M.T.; Silva, E.G.; Gershenson, D.M. Significantly Greater Expression of ER, PR, and ECAD in Advanced-Stage Low-Grade Ovarian Serous Carcinoma as Revealed by Immunohistochemical Analysis. Int. J. Gynecol. Pathol. 2007, 26, 404–409. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast Cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Lei, J.T.; Anurag, M.; Haricharan, S.; Gou, X.; Ellis, M.J. Endocrine Therapy Resistance: New Insights. Breast 2019, 48, S26. [Google Scholar] [CrossRef]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4). Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine Resistance in Breast Cancer—An Overview and Update. Mol. Cell Endocrinol. 2015, 418, 220. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Sun, C.C.; Iyer, R.B.; Malpica, A.L.; Kavanagh, J.J.; Bodurka, D.C.; Schmeler, K.; Deavers, M. Hormonal Therapy for Recurrent Low-Grade Serous Carcinoma of the Ovary or Peritoneum. Gynecol. Oncol. 2012, 125, 661–666. [Google Scholar] [CrossRef]
- Tang, M.; O’Connell, R.L.; Amant, F.; Beale, P.; McNally, O.; Sjoquist, K.M.; Grant, P.; Davis, A.; Sykes, P.; Mileshkin, L.; et al. PARAGON: A Phase II Study of Anastrozole in Patients with Estrogen Receptor-Positive Recurrent/Metastatic Low-Grade Ovarian Cancers and Serous Borderline Ovarian Tumors. Gynecol. Oncol. 2019, 154, 531–538. [Google Scholar] [CrossRef]
- Van Meurs, H.S.; Van Lonkhuijzen, L.R.C.W.; Limpens, J.; Van Der Velden, J.; Buist, M.R. Hormone Therapy in Ovarian Granulosa Cell Tumors: A Systematic Review. Gynecol. Oncol. 2014, 134, 196–205. [Google Scholar] [CrossRef]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen Signaling and Its Potential as a Target for Therapy in Ovarian Cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef]
- Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef]
- Kozieł, M.J.; Piastowska-Ciesielska, A.W. Estrogens, Estrogen Receptors and Tumor Microenvironment in Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 14673. [Google Scholar] [CrossRef]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front. Endocrinol. Lausanne 2021, 12, 599586. [Google Scholar] [CrossRef]
- McIntyre, J.B.; Rambau, P.F.; Chan, A.; Yap, S.; Morris, D.; Nelson, G.S.; Köbel, M. Molecular Alterations in Indolent, Aggressive and Recurrent Ovarian Low-Grade Serous Carcinoma. Histopathology 2017, 70, 347–358. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 Ligand-Binding Domain Mutations in Hormone-Resistant Breast Cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Brett, J.O.; Spring, L.M.; Bardia, A.; Wander, S.A. ESR1 Mutation as an Emerging Clinical Biomarker in Metastatic Hormone Receptor-Positive Breast Cancer. Breast Cancer Res. 2021, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.G.; Bae, S.J.; Kim, Y.; Ji, J.H.; Chu, C.; Kim, D.; Lee, J.; Cha, Y.J.; Lee, K.A.; Jeong, J. Primary Endocrine Resistance of ER+ Breast Cancer with ESR1 Mutations Interrogated by Droplet Digital PCR. NPJ Breast Cancer 2022, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Zundelevich, A.; Dadiani, M.; Kahana-Edwin, S.; Itay, A.; Sella, T.; Gadot, M.; Cesarkas, K.; Farage-Barhom, S.; Saar, E.G.; Eyal, E.; et al. ESR1 Mutations Are Frequent in Newly Diagnosed Metastatic and Loco-Regional Recurrence of Endocrine-Treated Breast Cancer and Carry Worse Prognosis. Breast Cancer Res. 2020, 22, 16. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, S.L.; Andreano, K.J.; Gay, L.M.; Steiner, M.; Jorgensen, M.S.; Davidson, B.A.; Havrilesky, L.J.; Alvarez Secord, A.; Valea, F.A.; Colon-Otero, G.; et al. Constitutively Active ESR1 Mutations in Gynecologic Malignancies and Clinical Response to Estrogen-Receptor Directed Therapies. Gynecol. Oncol. 2019, 154, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Stover, E.H.; Feltmate, C.; Berkowitz, R.S.; Lindeman, N.I.; Matulonis, U.A.; Konstantinopoulos, P.A. Targeted Next-Generation Sequencing Reveals Clinically Actionable BRAF and ESR1 Mutations in Low-Grade Serous Ovarian Carcinoma. JCO Precis. Oncol. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Shiino, S.; Kinoshita, T.; Yoshida, M.; Jimbo, K.; Asaga, S.; Takayama, S.; Tsuda, H. Prognostic Impact of Discordance in Hormone Receptor Status Between Primary and Recurrent Sites in Patients with Recurrent Breast Cancer. Clin. Breast Cancer 2016, 16, e133–e140. [Google Scholar] [CrossRef] [PubMed]
- Viedma-RodríGuez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Esparza-Garrido, R.R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms Associated with Resistance to Tamoxifen in Estrogen Receptor-Positive Breast Cancer (Review). Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of Naturally Occurring ESR1 Mutations in Breast Cancer Cell Lines Modelling Endocrine Resistance. Nat. Commun. 2017, 8, 1865. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 Mutations—A Mechanism for Acquired Endocrine Resistance in Breast Cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Fader, A.N.; Bergstrom, J.; Jernigan, A.; Tanner, E.J.; Roche, K.L.; Stone, R.L.; Levinson, K.L.; Ricci, S.; Wethingon, S.; Wang, T.L.; et al. Primary Cytoreductive Surgery and Adjuvant Hormonal Monotherapy in Women with Advanced Low-Grade Serous Ovarian Carcinoma: Reducing Overtreatment without Compromising Survival? Gynecol. Oncol. 2017, 147, 85–91. [Google Scholar] [CrossRef]
- Stergiopoulou, D.; Markou, A.; Giannopoulou, L.; Buderath, P.; Balgkouranidou, I.; Xenidis, N.; Kakolyris, S.; Kasimir-Bauer, S.; Lianidou, E. Detection of ESR1 Mutations in Primary Tumors and Plasma Cell-Free DNA in High-Grade Serous Ovarian Carcinoma Patients. Cancers 2022, 14, 3790. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, J.; Zhang, L.; Luo, Y.; Zhao, Z.; Li, M. Clinical Implications of Monitoring ESR1 Mutations by Circulating Tumor DNA in Estrogen Receptor Positive Metastatic Breast Cancer: A Pilot Study. Transl. Oncol. 2020, 13, 321. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.M.; Anglesio, M.S.; Ryland, G.L.; Sharma, R.; Chiew, Y.E.; Rowley, S.M.; Doyle, M.A.; Li, J.; Blake Gilks, C.; Moss, P.; et al. Molecular Profiling of Low Grade Serous Ovarian Tumours Identifies Novel Candidate Driver Genes. Oncotarget 2015, 6, 37663. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Arnold, J.M.; George, J.; Tinker, A.V.; Tothill, R.; Waddell, N.; Simms, L.; Locandro, B.; Fereday, S.; Traficante, N.; et al. Mutation of ERBB2 Provides a Novel Alternative Mechanism for the Ubiquitous Activation of RAS-MAPK in Ovarian Serous Low Malignant Potential Tumors. Mol Cancer Res. 2008, 6, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Raheem, F.; Karikalan, S.A.; Batalini, F.; El Masry, A.; Mina, L. Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 16198. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496. [Google Scholar] [CrossRef]
- Gershenson, D.M.; Sun, C.C.; Westin, S.N.; Eyada, M.; Cobb, L.P.; Nathan, L.C.; Sood, A.K.; Malpica, A.; Hillman, R.T.; Wong, K.K. The Genomic Landscape of Low-Grade Serous Ovarian/Peritoneal Carcinoma and Its Impact on Clinical Outcomes. Gynecol. Oncol. 2022, 165, 560–567. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/MTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W. Endocrine Resistance: What Do We Know? Am. Soc. Clin. Oncol. Educ. Book 2013, 33, e37–e42. [Google Scholar] [CrossRef]
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of Phosphatidylinositol-3 Kinase Promotes Escape from Hormone Dependence in Estrogen Receptor–Positive Human Breast Cancer. J. Clin. Investig. 2010, 120, 2406. [Google Scholar] [CrossRef]
- Beltrame, L.; Di Marino, M.; Fruscio, R.; Calura, E.; Chapman, B.; Clivio, L.; Sina, F.; Mele, C.; Iatropoulos, P.; Grassi, T.; et al. Profiling Cancer Gene Mutations in Longitudinal Epithelial Ovarian Cancer Biopsies by Targeted Next-Generation Sequencing: A Retrospective Study. Ann. Oncol. 2015, 26, 1363–1371. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Kurman, R.J.; Nakayama, K.; Velculescu, V.E.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; Shih, I.M. Low-Grade Serous Carcinomas of the Ovary Contain Very Few Point Mutations. J. Pathol. 2012, 226, 413. [Google Scholar] [CrossRef]
- Van Nieuwenhuysen, E.; Busschaert, P.; Laenen, A.; Moerman, P.; Han, S.N.; Neven, P.; Lambrechts, D.; Vergote, I. Loss of 1p36.33 Frequent in Low-Grade Serous Ovarian Cancer. Neoplasia 2019, 21, 582–590. [Google Scholar] [CrossRef]
- Arend, R.C.; Davis, A.M.; Chimiczewski, P.; O’Malley, D.M.; Provencher, D.; Vergote, I.; Ghamande, S.; Birrer, M.J. EMR 20006-012: A Phase II Randomized Double-Blind Placebo Controlled Trial Comparing the Combination of Pimasertib (MEK Inhibitor) with SAR245409 (PI3K Inhibitor) to Pimasertib Alone in Patients with Previously Treated Unresectable Borderline or Low Grade Ovarian Cancer. Gynecol. Oncol. 2020, 156, 301–307. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic Mechanisms in Breast Cancer Therapy and Resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]
- Dimitrakopoulos, F.I.; Kottorou, A.; Tzezou, A. Endocrine Resistance and Epigenetic Reprogramming in Estrogen Receptor Positive Breast Cancer. Cancer Lett. 2021, 517, 55–65. [Google Scholar] [CrossRef]
- Soleimani Dodaran, M.; Borgoni, S.; Sofyall, E.; Verschure, P.J.; Wiemann, S.; Moerland, P.D.; Van Kampen, A.H.C. Candidate Methylation Sites Associated with Endocrine Therapy Resistance in ER+/HER2- Breast Cancer. BMC Cancer 2020, 20, 676. [Google Scholar] [CrossRef]
- Lin, X.; Li, J.; Yin, G.; Zhao, Q.; Elias, D.; Lykkesfeldt, A.E.; Stenvang, J.; Brünner, N.; Wang, J.; Yang, H.; et al. Integrative Analyses of Gene Expression and DNA Methylation Profiles in Breast Cancer Cell Line Models of Tamoxifen-Resistance Indicate a Potential Role of Cells with Stem-like Properties. Breast Cancer Res. 2013, 15, R119. [Google Scholar] [CrossRef]
- Stone, A.; Zotenko, E.; Locke, W.J.; Korbie, D.; Millar, E.K.A.; Pidsley, R.; Stirzaker, C.; Graham, P.; Trau, M.; Musgrove, E.A.; et al. DNA Methylation of Oestrogen-Regulated Enhancers Defines Endocrine Sensitivity in Breast Cancer. Nat. Commun. 2015, 6, 7758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cui, Y. Dysregulation of DNA Methylation Patterns May Identify Patients with Breast Cancer Resistant to Endocrine Therapy: A Predictive Classifier Based on Differentially Methylated Regions. Oncol. Lett. 2019, 18, 1287–1303. [Google Scholar] [CrossRef] [PubMed]
- Muluhngwi, P.; Klinge, C.M. Roles for MiRNAs in Endocrine Resistance in Breast Cancer. Endocr. Relat. Cancer 2015, 22, R279. [Google Scholar] [CrossRef] [PubMed]
- Muluhngwi, P.; Klinge, C.M. Identification of MiRNAs as Biomarkers for Acquired Endocrine Resistance in Breast Cancer. Mol. Cell Endocrinol. 2017, 456, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Klein, P.; Tiersten, A.; Sparano, J.A. An Emerging Generation of Endocrine Therapies in Breast Cancer: A Clinical Perspective. npj Breast Cancer 2023, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of Aromatase Inhibitor Resistance. Nat. Rev. Cancer 2015, 15, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Minden, A. Current Molecular Combination Therapies Used for the Treatment of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 11046. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Cosio, S. Therapeutic Approach to Low-Grade Serous Ovarian Carcinoma: State of Art and Perspectives of Clinical Research. Cancers 2020, 12, 1336. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 Final PFS: A Randomized Study of Abemaciclib as Initial Therapy for Advanced Breast Cancer. NPJ Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women with HR+/HER2-Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortes, J.; Dieras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2- Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2022, 386, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Winterhoff, B.; Kolarova, T.; Qi, J.; Manivong, K.; Dering, J.; Yang, G.; Chalukya, M.; Wang, H.J.; Anderson, L.; et al. Expression of P16 and Retinoblastoma Determines Response to CDK4/6 Inhibition in Ovarian Cancer. Clin. Cancer Res. 2011, 17, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Dall’acqua, A.; Bartoletti, M.; Masoudi-Khoram, N.; Sorio, R.; Puglisi, F.; Belletti, B.; Baldassarre, G. Inhibition of CDK4/6 as Therapeutic Approach for Ovarian Cancer Patients: Current Evidences and Future Perspectives. Cancers 2021, 13, 3035. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Hendrickson, A.E.W.; Jatoi, A.; Burton, J.K.; Paroly, J.; Glaspy, J.A.; Dowdy, S.C.; Slamon, D.J. A Multicenter Open-Label Phase II Study of the Efficacy and Safety of Palbociclib a Cyclin-Dependent Kinases 4 and 6 Inhibitor in Patients with Recurrent Ovarian Cancer. J. Clin. Oncol. 2016, 34, 5557. [Google Scholar] [CrossRef]
- Colon-Otero, G.; Zanfagnin, V.; Hou, X.; Foster, N.R.; Asmus, E.J.; Wahner Hendrickson, A.; Jatoi, A.; Block, M.S.; Langstraat, C.L.; Glaser, G.E.; et al. Phase II Trial of Ribociclib and Letrozole in Patients with Relapsed Oestrogen Receptor-Positive Ovarian or Endometrial Cancers. ESMO Open 2020, 5, e000926. [Google Scholar] [CrossRef] [PubMed]
- Ottenbourgs, T.; Van Gorp, T.; Kridelka, F.; Baert, T.; Denys, H.; Selle, F.; Baas, I.; Van Rompuy, A.-S.; Lambrechts, D.; Van Nieuwenhuysen, E.; et al. A Phase II, Multicenter, Open-Label Study of Abemaciclib and Letrozole in Patients with Estrogen Receptor-Positive Rare Ovarian Cancer: ALEPRO Trial. Int. J. Gynecol. Cancer 2024, 34, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, D.M.; Miller, A.; Brady, W.E.; Paul, J.; Carty, K.; Rodgers, W.; Millan, D.; Coleman, R.L.; Moore, K.N.; Banerjee, S.; et al. Trametinib versus Standard of Care in Patients with Recurrent Low-Grade Serous Ovarian Cancer (GOG 281/LOGS): An International, Randomised, Open-Label, Multicentre, Phase 2/3 Trial. Lancet 2022, 399, 541–553. [Google Scholar] [CrossRef]
- Tholander, B.; Koliadi, A.; Botling, J.; Dahlstrand, H.; Von Heideman, A.; Ahlström, H.; Öberg, K.; Ullenhag, G.J. Complete Response with Combined BRAF and MEK Inhibition in BRAF Mutated Advanced Low-Grade Serous Ovarian Carcinoma. Ups J. Med. Sci. 2020, 125, 325. [Google Scholar] [CrossRef] [PubMed]
- Bussies, P.L.; Schlumbrecht, M. Dual Fulvestrant-Trametinib Therapy in Recurrent Low-Grade Serous Ovarian Cancer. Oncologist 2020, 25, e1124–e1126. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K.; Winterhalder, R.; Mamot, C.; Hasler-Strub, U.; Rochlitz, C.; Mueller, A.; Berset, C.; Wiliders, H.; Perey, L.; Rudolf, C.B.; et al. Fulvestrant with or without Selumetinib, a MEK 1/2 Inhibitor, in Breast Cancer Progressing after Aromatase Inhibitor Therapy: A Multicentre Randomised Placebo-Controlled Double-Blind Phase II Trial, SAKK 21/08. Eur. J. Cancer 2015, 51, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.Y.; Rodriguez-Gabin, A.; Samaweera, L.; Hazan, R.; Goldberg, G.L.; Horwitz, S.B.; McDaid, H.M. Exploiting MEK Inhibitor-Mediated Activation of ERα for Therapeutic Intervention in ER-Positive Ovarian Carcinoma. PLoS ONE 2013, 8, e54103. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; Van Cutsem, E.; Pérez-García, J.; Stathis, A.; Britten, C.D.; et al. A Phase Ib Dose-Escalation Study of the Oral Pan-PI3K Inhibitor Buparlisib (BKM120) in Combination with the Oral MEK1/2 Inhibitor Trametinib (GSK1120212) in Patients with Selected Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Skolariki, A.; D’Costa, J.; Little, M.; Lord, S. Role of PI3K/Akt/MTOR Pathway in Mediating Endocrine Resistance: Concept to Clinic. Explor. Target Antitumor. Ther. 2022, 3, 172. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating Tumour DNA Analysis to Direct Therapy in Advanced Breast Cancer (PlasmaMATCH): A Multicentre, Multicohort, Phase 2a, Platform Trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.R.; Wander, S.A.; Hamilton, E.; Razavi, P.; Bardia, A. Next-Generation Selective Estrogen Receptor Degraders and Other Novel Endocrine Therapies for Management of Metastatic Hormone Receptor-Positive Breast Cancer: Current and Emerging Role. Ther. Adv. Med. Oncol. 2022, 14, 17588359221113694. [Google Scholar] [CrossRef]
- Goetz, M.P.; Bagegni, N.A.; Batist, G.; Brufsky, A.; Cristofanilli, M.A.; Damodaran, S.; Daniel, B.R.; Fleming, G.F.; Gradishar, W.J.; Graff, S.L.; et al. Lasofoxifene versus Fulvestrant for ER+/HER2- Metastatic Breast Cancer with an ESR1 Mutation: Results from the Randomized, Phase II ELAINE 1 Trial. Ann. Oncol. 2023, 34, 1141–1151. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (Oral Selective Estrogen Receptor Degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results from the Randomized Phase III EMERALD Trial. J. Clin. Oncol. 2022, 40, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Chan, A.; Petrakova, K.; Delaloge, S.; Campone, M.; Iwata, H.; Peddi, P.F.; Kaufman, P.A.; De Kermadec, E.; Liu, Q.; et al. AMEERA-3: Randomized Phase II Study of Amcenestrant (Oral Selective Estrogen Receptor Degrader) Versus Standard Endocrine Monotherapy in Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer. J. Clin. Oncol. 2023, 41, 4014–4024. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Lim, E.; Gregor, M.C.M.; Shivhare, M.; Ross, G.; Patre, M.; Roncoroni, L.; Louka, M.; Sohn, J. AcelERA Breast Cancer (BC): Phase II Study Evaluating Efficacy and Safety of Giredestrant (GDC-9545) versus Physician’s Choice of Endocrine Monotherapy in Patients (Pts) with Estrogen Receptor-Positive, HER2-Negative (ER+/HER2-) Locally Advanced or Metastatic Breast Cancer (LA/MBC). J. Clin. Oncol. 2021, 39, TPS1100. [Google Scholar] [CrossRef]
- Martin Jimenez, M.; Lim, E.; Chavez Mac Gregor, M.; Bardia, A.; Wu, J.; Zhang, Q.; Nowecki, Z.; Cruz, F.; Safin, R.; Kim, S.-B.; et al. 211MO Giredestrant (GDC-9545) vs Physician Choice of Endocrine Monotherapy (PCET) in Patients (Pts) with ER+, HER2– Locally Advanced/Metastatic Breast Cancer (LA/MBC): Primary Analysis of the Phase II, Randomised, Open-Label AcelERA BC Study. Ann. Oncol. 2022, 33, S633–S634. [Google Scholar] [CrossRef]
- Lawson, M.; Cureton, N.; Ros, S.; Cheraghchi-Bashi-Astaneh, A.; Urosevic, J.; D’Arcy, S.; Delpuech, O.; DuPont, M.; Fisher, D.I.; Gangl, E.T.; et al. The Next-Generation Oral Selective Estrogen Receptor Degrader Camizestrant (AZD9833) Suppresses ER+ Breast Cancer Growth and Overcomes Endocrine and CDK4/6 Inhibitor Resistance. Cancer Res. 2023, 83, 3989–4004. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Pominchuck, D.; Nowecki, Z.; Hamilton, E.; Kulyaba, Y.; Andabekov, T.; Hotko, Y.; Melkadze, T.; Nemsadze, G.; Neven, P.; et al. Abstract GS3-02: GS3-02 Camizestrant, a next Generation Oral SERD vs Fulvestrant in Post-Menopausal Women with Advanced ER-Positive HER2-Negative Breast Cancer: Results of the Randomized, Multi-Dose Phase 2 SERENA-2 Trial. Cancer Res. 2023, 83, GS3-02. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Schott, A.F.; Nanda, R.; Lu, H.; Keung, C.F.; Gedrich, R.; Parameswaran, J.; Han, H.S.; Hurvitz, S.A. ARV-471, an Estrogen Receptor (ER) PROTAC Degrader, Combined with Palbociclib in Advanced ER+/Human Epidermal Growth Factor Receptor 2–Negative (HER2-) Breast Cancer: Phase 1b Cohort (Part C) of a Phase 1/2 Study. J. Clin. Oncol. 2022, 40, TPS1120. [Google Scholar] [CrossRef]
- Johnston, S.R.D.; Pluard, T.J.; Wang, J.S.; Hamilton, E.P.; Juric, D.; Scholz, C.R.; Hnitecki, E.; Gao, L.; Cantagallo, L.; Korpal, M.; et al. Phase 1b Study of H3B-6545 in Combination with Palbociclib in Women with Metastatic Estrogen Receptor–Positive (ER+), Human Epidermal Growth Factor Receptor 2 (HER2)-Negative Breast Cancer. J. Clin. Oncol. 2021, 39, e13025. [Google Scholar] [CrossRef]
- Hamilton, E.; Meisel, J.; Alemany, C.; Virginia, B.; Lin, N.; Wesolowski, R.; Mathauda-Sahota, G.; Makower, D.; Lawrence, J.; Faltaos, D.; et al. Phase 1b Results from OP-1250-001, a Dose Escalation and Dose Expansion Study of OP-1250, an Oral CERAN, in Subjects with Advanced and/or Metastatic Estrogen Receptor (ER)-Positive, HER2-Negative Breast Cancer (NCT04505826). Eur. J. Cancer 2022, 174, S36. [Google Scholar] [CrossRef]
- Turner, N.C.; Jhaveri, K.L.; Bardia, A.; Niikura, N.; Dieras, V.; Barrios, C.H.; Im, S.-A.; Mueller, V.; Bellet, M.; Chang, C.-W.; et al. PersevERA Breast Cancer (BC): Phase III Study Evaluating the Efficacy and Safety of Giredestrant (GDC-9545) + Palbociclib versus Letrozole + Palbociclib in Patients (Pts) with Estrogen-Receptor-Positive, HER2-Negative Locally Advanced or Metastatic BC (ER+/HER2–LA/MBC). J. Clin. Oncol. 2021, 39, TPS1103. [Google Scholar] [CrossRef]
- Im, S.-A.; Hamilton, E.P.; Cussac, A.L.; Baird, R.D.; Ettl, J.; Goetz, M.P.; Iwata, H.; Joy, A.A.; Neven, P.; Haddad, V.; et al. SERENA-4: A Phase 3 Comparison of AZD9833 (Camizestrant) plus Palbociclib, versus Anastrozole plus Palbociclib, for Patients with ER-Positive, HER2-Negative Advanced Breast Cancer Who Have Not Previously Received Systemic Treatment for Advanced Disease. J. Clin. Oncol. 2021, 39, TPS1101. [Google Scholar] [CrossRef]
- Mayer, E.L.; Tolaney, S.; Brufsky, A.M.; Gradishar, W.; Jhaveri, K.; Martín, M.; Moscetti, L.; Vidal, G.; Cortazar, P.; Feldman, M.; et al. Abstract OT2-01-07: EvERA Breast Cancer: A Phase III Study of Giredestrant (GDC-9545) + Everolimus vs Exemestane + Everolimus in Patients with Estrogen Receptor+, HER2– Locally Advanced or Metastatic Breast Cancer. Cancer Res. 2023, 83, OT2-01. [Google Scholar] [CrossRef]
- Oliveira, M.; Sonnenblick, A.; Rugo, H.S.; Jung, K.H.; Yam, E.G.; Hurvitz, S.A.; Hernando, C.; Im, S.-A.; Breton, V.; Collier, A.; et al. Interim Analyses (IA) of the Giredestrant (G), G + Abemaciclib (A), and G + Ribociclib (R) Arms in MORPHEUS Breast Cancer (BC): A Phase I/II Study of G Treatment (Tx) Combinations in Patients (Pts) with Estrogen Receptor-Positive, HER2-Negative Locally Advanced/Metastatic BC (ER+, HER2–LA/MBC). J. Clin. Oncol. 2023, 41, 1061. [Google Scholar] [CrossRef]
- Turner, N.; Huang-Bartlett, C.; Kalinsky, K.; Cristofanilli, M.; Bianchini, G.; Chia, S.; Iwata, H.; Janni, W.; Ma, C.X.; Mayer, E.L.; et al. Design of SERENA-6, a Phase III Switching Trial of Camizestrant in ESR1-Mutant Breast Cancer during First-Line Treatment. Future Oncol. 2023, 19, 559–573. [Google Scholar] [CrossRef]
- Jhaveri, K.; Harbeck, N.; Aftimos, P.; Kim, S.-B.; Pivot, X.; Saura, C.; Curigliano, G.; Casalnuovo, M.; Wang, X.A.; Young, S.R.L.; et al. Abstract OT2-11-01: EMBER-3: A Randomized Phase 3 Study of LY3484356, a Novel, Oral Selective Estrogen Receptor Degrader vs Investigator’s Choice of Endocrine Therapy of Either Fulvestrant or Exemestane, in Patients with Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative, Locally Advanced or Metastatic Breast Cancer Previously Treated with Endocrine-Based Therapy. Cancer Res. 2022, 82, OT2-11. [Google Scholar] [CrossRef]
- Andreano, K.J.; Wardell, S.E.; Baker, J.G.; Desautels, T.K.; Baldi, R.; Chao, C.A.; Heetderks, K.A.; Bae, Y.; Xiong, R.; Tonetti, D.A.; et al. G1T48, an Oral Selective Estrogen Receptor Degrader, and the CDK4/6 Inhibitor Lerociclib Inhibit Tumor Growth in Animal Models of Endocrine-Resistant Breast Cancer. Breast Cancer Res. Treat 2020, 180, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Keogh, G.P.; Papish, S.; Piskorski, W.; Ulanska, M.; Jackson, B.; Suster, M.; Ptaszynski, M.; Mina, L. 564TiP A Phase Ib Dose-Escalation Study of ZN-C5, an Oral Selective Estrogen Receptor Degrader (SERD), in Combination with Abemaciclib in Patients with Advanced Estrogen Receptor (ER)+/HER2- Breast Cancer. Ann. Oncol. 2021, 32, S618–S619. [Google Scholar] [CrossRef]
- Abramson, V.; Linden, H.M.; Crew, K.; Mortimer, J.; Alidzanovic, J.; Nangia, J.; Layman, R.; Vranjes, Z.J.; Andric, Z.; Milovic-Kovacevic, M.; et al. 565TiP A Phase I/II Dose-Escalation and Expansion Study of ZN-C5, an Oral Selective Estrogen Receptor Degrader (SERD), as Monotherapy and in Combination with Palbociclib in Patients with Advanced Estrogen Receptor (ER)+/HER2- Breast Cancer. Ann. Oncol. 2021, 32, S619. [Google Scholar] [CrossRef]
- Osborne, C.; Richards, D.A.; Wilks, S.T.; Diab, S.; Juric, D.; Lathrop, K.; Silber, A.; Edenfield, W.; Aulakh, A.; Cho, B.; et al. Abstract PS11-26: A Phase 1 Study of D-0502, an Orally Bioavailable SERD, for Advanced or Metastatic HR-Positive and HER2-Negative Breast Cancer. Cancer Res. 2021, 81, PS11-26. [Google Scholar] [CrossRef]
- Maenhoudt, N.; Defraye, C.; Boretto, M.; Jan, Z.; Heremans, R.; Boeckx, B.; Hermans, F.; Arijs, I.; Cox, B.; Van Nieuwenhuysen, E.; et al. Developing Organoids from Ovarian Cancer as Experimental and Preclinical Models. Stem Cell Rep. 2020, 14, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Dumont, S.; Jan, Z.; Heremans, R.; Van Gorp, T.; Vergote, I.; Timmerman, D. Organoids of Epithelial Ovarian Cancer as an Emerging Preclinical in Vitro Tool: A Review. J. Ovarian Res. 2019, 12, 105. [Google Scholar] [CrossRef]
| Drug | Mode of Action | Status | Phase | Indication | Biomarkers for Patient Selection | Enrolment | Clinicaltrials.gov |
|---|---|---|---|---|---|---|---|
| Ribociclib Letrozole | CDK4/6 inhibitor Aromatase inhibitor | Active, not recruiting | II | Recurrent LGSOC | NA | 51 (actual) | NCT03673124 |
| Abemaciclib Fulvestrant | CDK4/6 inhibitor SERD | Active, not recruiting | II | Advanced LGSOC, first-line, neo-adjuvant treatment | NA | 18 (actual) | NCT03531645 |
| Abemaciclib Letrozole | CDK4/6 inhibitor Aromatase inhibitor | Recruiting | II | Recurrent LGSOC | ER positivity | 100 (estimated) | NCT05872204 |
| Palbociclib Binimetinib | CDK4/6 inhibitor MEK inhibitor | Recruiting | II | RAS-mutant cancers, including LGSOC | KRAS/NRAS/HRAS or BRAF alterations, RAF mutations or RAF fusions | 199 (estimated) | NCT05554367 |
| Avutometinib (VS-6766) +/− Defactinib (VS-6063) | Dual RAF/MEK inhibitor FAK inhibitor | Recruiting | II | Recurrent LGSOC with or without a KRAS mutation | KRAS wild-type KRAS mutation | 225 (estimated) | NCT04625270 |
| Avutometinib (VS-6766) + Defactinib (VS-6063) | Dual RAF/MEK inhibitor FAK inhibitor | Recruiting | III | Recurrent LGSOC | KRAS wild-type KRAS mutation | 270 (estimated) | NCT06072781 |
| Pimasertib Voxtalisib (SAR245409) | MEK1/2 inhibitor PI3K/mTOR inhibitor | Completed | II | Recurrent borderline/low malignant potential and LGSOC | NA | 65 (actual) | NCT01936363 [46] |
| Regorafenib Fulvestrant | Multikinase inhibitor SERD | Recruiting | II | Recurrent LGSOC | NA | 31 (estimated) | NCT05113368 |
| Letrozole Alpelisib OR Letrozole Ribociclib based on presence of PI3KCA mutation status | Aromatase inhibitor PI3Ka-inhibitor OR Aromatase inhibitor CDK4/6 inhibitor | Recruiting | II | Advanced gynecological cancers that express hormone receptors | ER and/or PR positivity | 100 (actual) | ACTRN12621000639820 |
| Biomarker-driven therapy * | AKT inhibitor MEK inhibitor Chemotherapy CDK4/6 inhibitor PI3K α inhibitor SERD Aromatase inhibitor VEGF-A inhibitor PARP inhibitor PD-L1 inhibitor Anti-HER2 antibody + cytotoxic agent | Prematurely terminated | II | Recurrent rare epithelial ovarian tumors | See footnote of this table | 500 (estimated) | NCT04931342 |
| Drug(s) | Mode of Action | Clinical Trial | Phase | Indication | Treatment Arms | Outcome Primary Endpoints | Reference |
|---|---|---|---|---|---|---|---|
| Lasofoxifene | SERM | ELAINE I NCT03781063 | II | ER+/HER2- advanced or metastatic breast cancer with an ESR1 mutation | Lasofoxifene vs. fulvestrant |
| Goetz et al. [79] |
| Elacestrant | SERD/ SERM | EMERALD NCT03778931 | III | ER+/HER2- advanced or metastatic breast cancer with 1–2 previous lines of endocrine therapy, including a CDK4/6 inhibitor | Elacestrant vs. standard endocrine therapy (fulvestrant/AI) | Median PFS: 2.8 months (elacestrant) vs. 1.9 months (standard endocrine therapy); HR: 0.664; p = 0.0019 | Bidard et al. [80] |
| Amcenestrant | SERD | AMEERA-3 NCT04059484 | II | ER+/HER2- advanced or metastatic breast cancer that progressed after prior treatment with an AI and a CDK4/6 inhibitor | Amcenestrant vs. physician’s choice of endocrine treatment | Median PFS: 3.6 months (amcenestrant) vs. 3.7 months (physician’s choice of endocrine treatment); HR: 1.05, p = 0.64 | Tolaney et al. [81] |
| Giredestrant | SERD | acelERA NCT04576455 | II | ER+/HER2- advanced or metastatic breast cancer that received 1 or 2 prior lines of systemic therapy | Giredestrant vs. physician’s choice of endocrine treatment | Median PFS: 5.6 months (giredestrant) vs. 5.4 months (physician’s choice of endocrine treatment); HR 0.81, p = 0.18 | Martin et al. Martin Jimenez et al. [82,83] |
| Camizestrant | SERD | SERENA-2 NCT04214288 | II | ER+/HER2- advanced or metastatic breast cancer | Camizestrant 75, 150 or 300 mg vs. Fulvestrant | Median PFS: 7.2 months (camizestrant 75 mg) vs. 3.7 (fulvestrant); p = 0.0124 Median PFS: 7.7 months (camizestrant 150 mg) vs. 3.7 (fulvestrant); p = 0.0161 | Lawson et al. Oliveira et al. [84,85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottenbourgs, T.; Van Nieuwenhuysen, E. Novel Endocrine Therapeutic Opportunities for Estrogen Receptor-Positive Ovarian Cancer—What Can We Learn from Breast Cancer? Cancers 2024, 16, 1862. https://doi.org/10.3390/cancers16101862
Ottenbourgs T, Van Nieuwenhuysen E. Novel Endocrine Therapeutic Opportunities for Estrogen Receptor-Positive Ovarian Cancer—What Can We Learn from Breast Cancer? Cancers. 2024; 16(10):1862. https://doi.org/10.3390/cancers16101862
Chicago/Turabian StyleOttenbourgs, Tine, and Els Van Nieuwenhuysen. 2024. "Novel Endocrine Therapeutic Opportunities for Estrogen Receptor-Positive Ovarian Cancer—What Can We Learn from Breast Cancer?" Cancers 16, no. 10: 1862. https://doi.org/10.3390/cancers16101862
APA StyleOttenbourgs, T., & Van Nieuwenhuysen, E. (2024). Novel Endocrine Therapeutic Opportunities for Estrogen Receptor-Positive Ovarian Cancer—What Can We Learn from Breast Cancer? Cancers, 16(10), 1862. https://doi.org/10.3390/cancers16101862