
Exploiting the DNA Damage Response for Prostate Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction

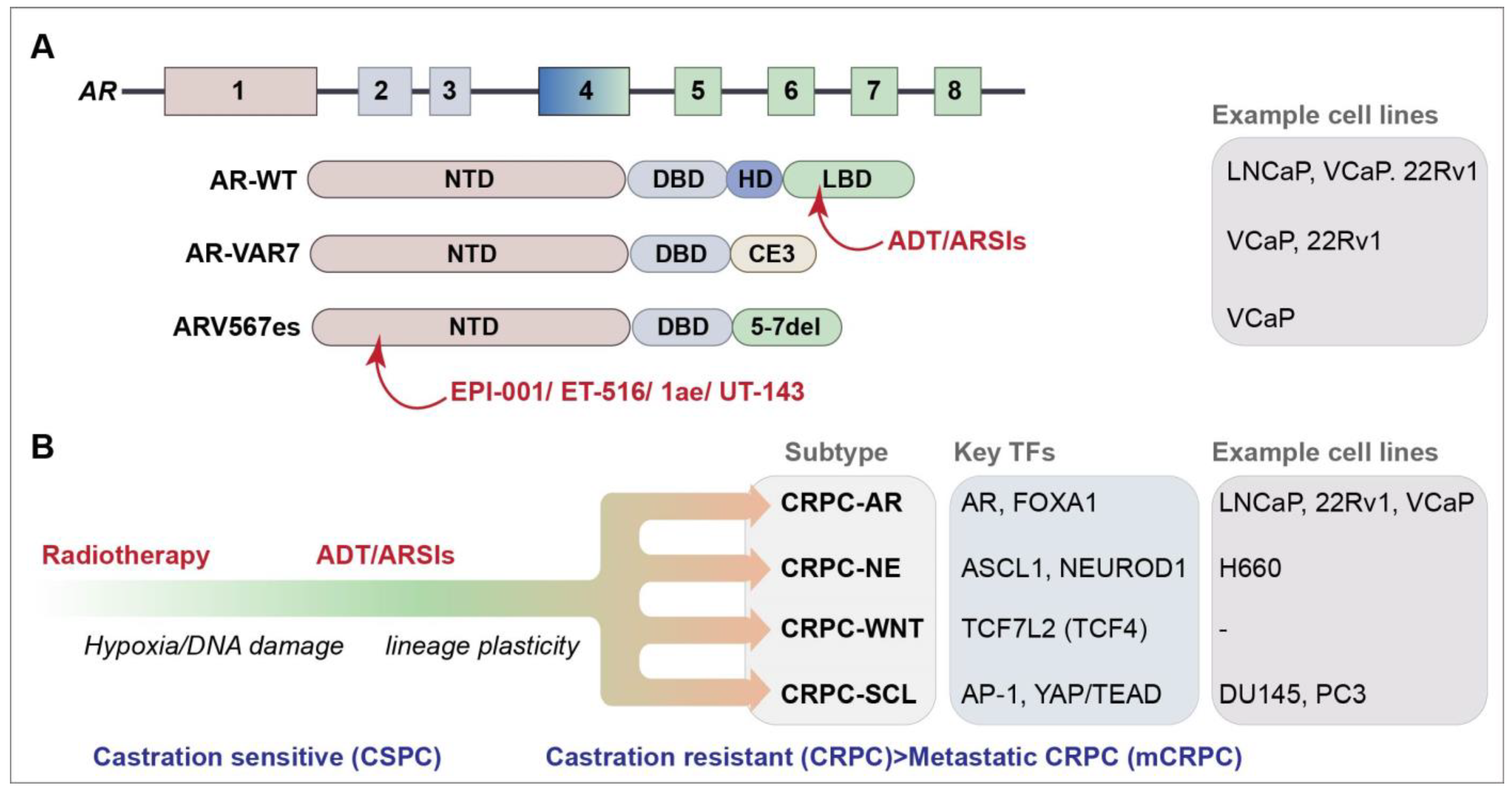
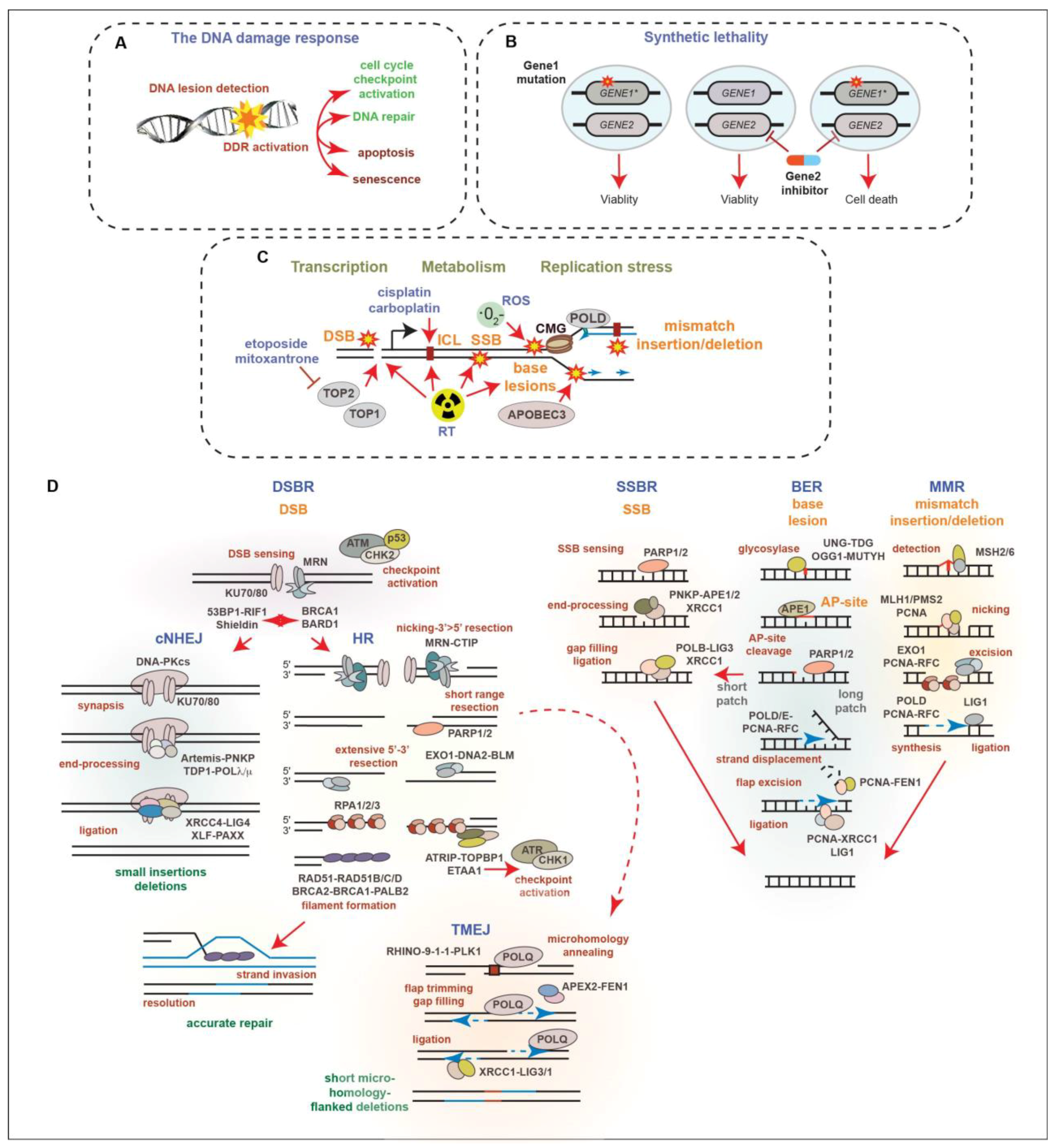
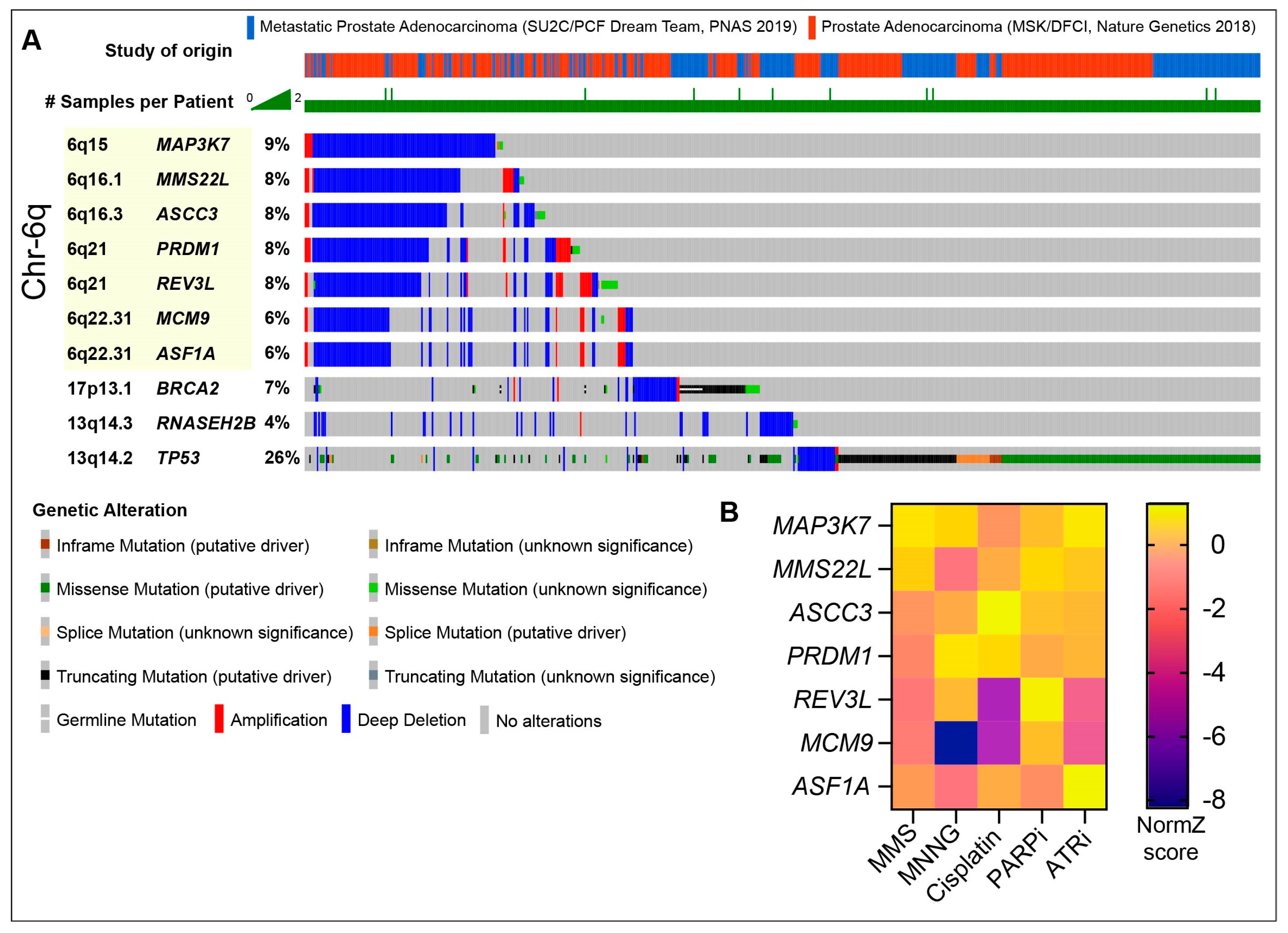
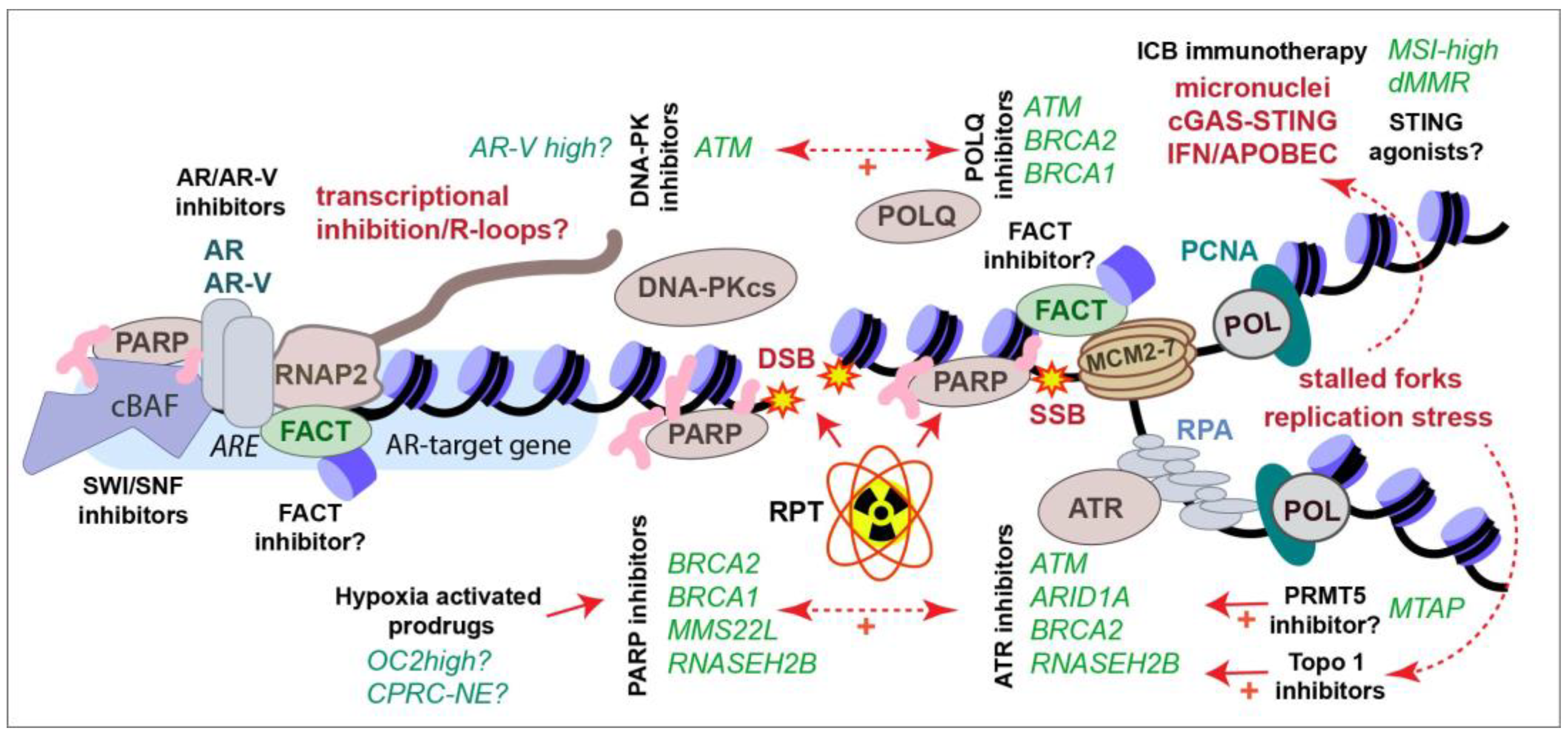
2. The DNA Damage Response in Prostate Cancer
3. New Approaches for Targeting AR-Variants
4. PARP Inhibitors in CRPC
5. Targeting DDR Kinases in CRPC Therapy
6. Generating Targeted DNA Damage with Radiopharmaceutical Therapy
7. Crosstalk between the Immune System and the DDR in CRPC
8. Targeting the Hypoxic TME in CRPC Therapy
9. Emerging Targets in CRPC
9.1. SWI/SNF
9.2. POLQ
9.3. PRMT1
9.4. PRMT5
9.5. FACT
9.6. The Tousled-Like Kinases (TLKs)
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | Androgen deprivation therapy |
| Alt-NHEJ | Alternative non-homologous end joining |
| APOBEC | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide |
| AR | Androgen receptor |
| AR-V | AR-variants |
| ARE | Androgen response element |
| ARSI | Androgen receptor signaling inhibitor |
| BER | Base excision repair |
| BRCA | Breast cancer associated |
| CE3 | Cryptic exon 3 |
| cNHEJ | Classical non-homologous end-joining |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CRPC | Castration resistant prostate cancer |
| CRPC-AR | CRPC-androgen receptor subtype |
| CRPC-NE | CRPC-neuroendocrine subtype |
| CRPC-SCL | CRPC-stem cell like subtype |
| CRPC-WNT | CRPC-WNT dependent subtype |
| DBD | DNA binding domain |
| DDR | DNA damage response |
| dMMR | Mismatch Repair deficiency |
| DSB | Double-strand break |
| DSBR | DSB repair |
| dsDNA | Double-strand DNA |
| EBRT | External beam radiotherapy |
| HAP | Hypoxia activated prodrug |
| HD | Hinge domain |
| HIF | Hypoxia inducible factor |
| HR | Homologous recombination |
| HRD | Homologous recombination deficiency |
| ICB | Immune checkpoint blockade |
| LBD | Ligand binding domain |
| mCRPC | Metastatic CRPC |
| MMR | Mismatch Repair |
| MMS | Methyl methanesulfonate |
| MNNG | Methylnitronitrosoguanidine |
| MRN | MRE11-RAD50-NBS1 |
| MSI | Microsatellite instability |
| NTD | N-Terminal domain |
| PARP | Poly (ADP ribose) polymerase |
| PCa | Prostate cancer |
| PSA | Prostate specific membrane antigen |
| RNASEH2 | Ribonuclease H2 |
| ROS | Reactive oxygen species |
| RPT | Radiopharmaceutical therapy |
| RT | Radiotherapy |
| SBS | Single base substitution |
| SSB | Single-strand break |
| SSBR | SSB Repair |
| ssDNA | Single-strand DNA |
| TF | Transcription factors |
| TLK | Tousled-like kinase |
| TMB | Tumor mutational burden |
| TME | Tumor microenvironment |
| TMEJ | Polymerase theta-mediated end-joining |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- Lavaud, P.; Dumont, C.; Thibault, C.; Albiges, L.; Baciarello, G.; Colomba, E.; Flippot, R.; Fuerea, A.; Loriot, Y.; Fizazi, K. Next-Generation Androgen Receptor Inhibitors in Non-Metastatic Castration-Resistant Prostate Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920978134. [Google Scholar] [CrossRef] [PubMed]
- Estébanez-Perpiñá, E.; Bevan, C.L.; McEwan, I.J. Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes On. Cancers 2021, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Androgen Receptor Splice Variants and Prostate Cancer: From Bench to Bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed]
- Tietz, K.T.; Dehm, S.M. Androgen Receptor Variants: RNA-Based Mechanisms and Therapeutic Targets. Hum. Mol. Genet. 2020, 29, R19–R26. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Brown, L.C.; Antonarakis, E.S.; Armstrong, A.J.; Luo, J. Androgen Receptor Variant-Driven Prostate Cancer II: Advances in Laboratory Investigations. Prostate Cancer Prostatic Dis. 2020, 23, 381–397. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Graf, R.P.; Hullings, M.; Barnett, E.S.; Carbone, E.; Dittamore, R.; Scher, H.I. Clinical Utility of the Nuclear-Localized AR-V7 Biomarker in Circulating Tumor Cells in Improving Physician Treatment Choice in Castration-Resistant Prostate Cancer. Eur. Urol. 2020, 77, 170–177. [Google Scholar] [CrossRef]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Rothwell, C.J.; et al. Prospective Multicenter Validation of Androgen Receptor Splice Variant 7 and Hormone Therapy Resistance in High-Risk Castration-Resistant Prostate Cancer: The PROPHECY Study. J. Clin. Oncol. 2019, 37, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Lu, D.; Louw, J.; Danila, D.C.; Dugan, L.; Johnson, A.; Heller, G.; et al. Nuclear-Specific AR-V7 Protein Localization Is Necessary to Guide Treatment Selection in Metastatic Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker with Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen Receptor Splice Variant-7 Expression Emerges with Castration Resistance in Prostate Cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Modonutti, D.; Majdalany, S.E.; Corsi, N.; Li, P.; Sood, A.; Dalela, D.; Jamil, M.L.; Hwang, C.; Menon, M.; Rogers, C.G.; et al. A Novel Prognostic Model Predicting Overall Survival in Patients with Metastatic Castration-Resistant Prostate Cancer Receiving Standard Chemotherapy: A Multi-Trial Cohort Analysis. Prostate 2022, 82, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Halabi, S.; Lin, C.-Y.; Kelly, W.K.; Fizazi, K.S.; Moul, J.W.; Kaplan, E.B.; Morris, M.J.; Small, E.J. Updated Prognostic Model for Predicting Overall Survival in First-Line Chemotherapy for Patients with Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2014, 32, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.-K.; Watt, K.; Tam, T.; Yang, Y.C.; Bañuelos, C.A.; et al. Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef]
- Basu, S.; Martinez-Cristobal, P.; Pesarrodona, M.; Frigole-Vivas, M.; Szulc, E.; Lewis, M.; Banuelos, A.; Sanchez-Zarzalejo, C.; Bielskute, S.; Zhu, J.; et al. Androgen Receptor Condensates as Drug Targets. BioRxiv 2022. [Google Scholar] [CrossRef]
- Xie, J.; He, H.; Kong, W.; Li, Z.; Gao, Z.; Xie, D.; Sun, L.; Fan, X.; Jiang, X.; Zheng, Q.; et al. Targeting Androgen Receptor Phase Separation to Overcome Antiandrogen Resistance. Nat. Chem. Biol. 2022, 18, 1341–1350. [Google Scholar] [CrossRef]
- Thiyagarajan, T.; Ponnusamy, S.; Hwang, D.-J.; He, Y.; Asemota, S.; Young, K.L.; Johnson, D.L.; Bocharova, V.; Zhou, W.; Jain, A.K.; et al. Inhibiting Androgen Receptor Splice Variants with Cysteine-Selective Irreversible Covalent Inhibitors to Treat Prostate Cancer. Proc. Natl. Acad. Sci. USA 2023, 120, e2211832120. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Xu, D.; Wang, S.; Wong, C.K.; Martinez-Fundichely, A.; Lee, C.J.; Cohen, S.; Park, J.; Hill, C.E.; Eng, K.; et al. Chromatin Profiles Classify Castration-Resistant Prostate Cancers Suggesting Therapeutic Targets. Science 2022, 376, eabe1505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.D.; Deng, S.; Hoover, E.; Chen, C.-C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e11. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Manzar, N.; Ateeq, B. Dynamics of Cellular Plasticity in Prostate Cancer Progression. Front. Mol. Biosci. 2020, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Abd, G.M.; Laird, M.C.; Ku, J.C.; Li, Y. Hypoxia-Induced Cancer Cell Reprogramming: A Review on How Cancer Stem Cells Arise. Front. Oncol. 2023, 13, 1227884. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-Inducible Factors: Cancer Progression and Clinical Translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why Is the Partial Oxygen Pressure of Human Tissues a Crucial Parameter? Small Molecules and Hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours-Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Movsas, B.; Chapman, J.D.; Greenberg, R.E.; Hanlon, A.L.; Horwitz, E.M.; Pinover, W.H.; Stobbe, C.; Hanks, G.E. Increasing Levels of Hypoxia in Prostate Carcinoma Correlate Significantly with Increasing Clinical Stage and Patient Age: An Eppendorf pO(2) Study. Cancer 2000, 89, 2018–2024. [Google Scholar] [CrossRef]
- Cameron, S.; Deblois, G.; Hawley, J.R.; Qamra, A.; Zhou, S.; Tonekaboni, S.A.M.; Murison, A.; Van Vliet, R.; Liu, J.; Locasale, J.W.; et al. Chronic Hypoxia Favours Adoption to a Castration-Resistant Cell State in Prostate Cancer. Oncogene 2023, 42, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Xue, C.; Mendonca, J.; Sun, X.-X.; Liu, Q.; Reardon, P.N.; Chen, Y.; Qian, K.; Hua, V.; Chen, A.; et al. Interplay between Hypoxia and Androgen Controls a Metabolic Switch Conferring Resistance to Androgen/AR-Targeted Therapy. Nat. Commun. 2018, 9, 4972. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, K.T.; McCarthy, H.O.; Devlin, A.; Ming, L.; Robson, T.; McKeown, S.R.; Worthington, J. Hypoxia Selects for Androgen Independent LNCaP Cells with a More Malignant Geno- and Phenotype. Int. J. Cancer 2008, 123, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.; Bristow, R. Bad Neighbours: Hypoxia and Genomic Instability in Prostate Cancer. Br. J. Radiol 2020, 93, 20200087. [Google Scholar] [CrossRef] [PubMed]
- Douglass, M.; Hall, E.J.; Amato, J. Giaccia: Radiobiology for the Radiologist. Australas. Phys. Eng. Sci. Med. 2018, 41, 1129–1130. [Google Scholar] [CrossRef]
- Singleton, D.C.; Macann, A.; Wilson, W.R. Therapeutic Targeting of the Hypoxic Tumour Microenvironment. Nat. Rev. Clin. Oncol. 2021, 18, 751–772. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human Topoisomerases and Their Roles in Genome Stability and Organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef]
- Semlow, D.R.; Walter, J.C. Mechanisms of Vertebrate DNA Interstrand Cross-Link Repair. Annu. Rev. Biochem. 2021, 90, 107–135. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA Single-Strand Break Repair and Human Genetic Disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef]
- Ramsden, D.A.; Carvajal-Garcia, J.; Gupta, G.P. Mechanism, Cellular Functions and Cancer Roles of Polymerase-Theta-Mediated DNA End Joining. Nat. Rev. Mol. Cell Biol. 2022, 23, 125–140. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Mertz, T.M.; Collins, C.D.; Dennis, M.; Coxon, M.; Roberts, S.A. APOBEC-Induced Mutagenesis in Cancer. Annu. Rev. Genet. 2022, 56, 229–252. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.A.; Lara, P.N.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and Estramustine Compared with Mitoxantrone and Prednisone for Advanced Refractory Prostate Cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Teply, B.A.; Hauke, R.J. Chemotherapy Options in Castration-Resistant Prostate Cancer. Indian J. Urol. 2016, 32, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. JCO Oncol. Pract. 2022, 18, 45–55. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- De Marco Zompit, M.; Esteban, M.T.; Mooser, C.; Adam, S.; Rossi, S.E.; Jeanrenaud, A.; Leimbacher, P.-A.; Fink, D.; Shorrocks, A.-M.K.; Blackford, A.N.; et al. The CIP2A-TOPBP1 Complex Safeguards Chromosomal Stability during Mitosis. Nat. Commun. 2022, 13, 4143. [Google Scholar] [CrossRef]
- Trivedi, P.; Steele, C.D.; Au, F.K.C.; Alexandrov, L.B.; Cleveland, D.W. Mitotic Tethering Enables Inheritance of Shattered Micronuclear Chromosomes. Nature 2023, 618, 1049–1056. [Google Scholar] [CrossRef]
- Lin, Y.-F.; Hu, Q.; Mazzagatti, A.; Valle-Inclán, J.E.; Maurais, E.G.; Dahiya, R.; Guyer, A.; Sanders, J.T.; Engel, J.L.; Nguyen, G.; et al. Mitotic Clustering of Pulverized Chromosomes from Micronuclei. Nature 2023, 618, 1041–1048. [Google Scholar] [CrossRef]
- Brambati, A.; Sacco, O.; Porcella, S.; Heyza, J.; Kareh, M.; Schmidt, J.C.; Sfeir, A. RHINO Directs MMEJ to Repair DNA Breaks in Mitosis. Science 2023, 381, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Gelot, C.; Kovacs, M.T.; Miron, S.; Mylne, E.; Haan, A.; Boeffard-Dosierre, L.; Ghouil, R.; Popova, T.; Dingli, F.; Loew, D.; et al. Polθ Is Phosphorylated by PLK1 to Repair Double-Strand Breaks in Mitosis. Nature 2023, 621, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Olave, M.C.; Graham, R.P. Mismatch Repair Deficiency: The What, How and Why It Is Important. Genes Chromosomes Cancer 2022, 61, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.-C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA Damage Response in Immuno-Oncology: Developments and Opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Kanu, N.; Cerone, M.A.; Goh, G.; Zalmas, L.-P.; Bartkova, J.; Dietzen, M.; McGranahan, N.; Rogers, R.; Law, E.K.; Gromova, I.; et al. DNA Replication Stress Mediates APOBEC3 Family Mutagenesis in Breast Cancer. Genome Biol. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Dananberg, A.; Chu, K.; Bergstrom, E.N.; Striepen, J.; von Morgen, P.; Chen, Y.; Shah, H.; Sale, J.E.; Alexandrov, L.B.; et al. Mechanisms of APOBEC3 Mutagenesis in Human Cancer Cells. Nature 2022, 607, 799–807. [Google Scholar] [CrossRef]
- DeWeerd, R.A.; Németh, E.; Póti, Á.; Petryk, N.; Chen, C.-L.; Hyrien, O.; Szüts, D.; Green, A.M. Prospectively Defined Patterns of APOBEC3A Mutagenesis Are Prevalent in Human Cancers. Cell Rep. 2022, 38, 110555. [Google Scholar] [CrossRef]
- Zou, X.; Koh, G.C.C.; Nanda, A.S.; Degasperi, A.; Urgo, K.; Roumeliotis, T.I.; Agu, C.A.; Badja, C.; Momen, S.; Young, J.; et al. A Systematic CRISPR Screen Defines Mutational Mechanisms Underpinning Signatures Caused by Replication Errors and Endogenous DNA Damage. Nat. Cancer 2021, 2, 643–657. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wedge, D.C.; Gundem, G.; Mitchell, T.; Woodcock, D.J.; Martincorena, I.; Ghori, M.; Zamora, J.; Butler, A.; Whitaker, H.; Kote-Jarai, Z.; et al. Sequencing of Prostate Cancers Identifies New Cancer Genes, Routes of Progression and Drug Targets. Nat. Genet. 2018, 50, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Ali, S.; Genega, E.; Reed, D.; Sokol, E.; Mathew, P. Aggressive-Variant Microsatellite-Stable POLE Mutant Prostate Cancer with High Mutation Burden and Durable Response to Immune Checkpoint Inhibitor Therapy. JCO Precis. Oncol. 2018, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Wang, H.; He, K.; Peng, M.; Wang, Y.; Li, Y.; Jiang, S.; Li, J.; Yi, L.; Cui, R. Clinical Characterization of Mismatch Repair Gene-Deficient Metastatic Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 533282. [Google Scholar] [CrossRef] [PubMed]
- Ritch, E.; Fu, S.Y.F.; Herberts, C.; Wang, G.; Warner, E.W.; Schönlau, E.; Taavitsainen, S.; Murtha, A.J.; Vandekerkhove, G.; Beja, K.; et al. Identification of Hypermutation and Defective Mismatch Repair in ctDNA from Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Movasati, A.; Brunner, S.R.; Nguyen, L.; Priestley, P.; Cuppen, E.; Van Hoeck, A. Pan-Cancer Whole-Genome Comparison of Primary and Metastatic Solid Tumours. Nature 2023, 618, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Deng, S.; Zhu, G.; Wang, C.; Johnson, N.A.; Zhang, Z.; Tirado, C.R.; Xu, Y.; Metang, L.A.; et al. Loss of SYNCRIP Unleashes APOBEC-Driven Mutagenesis, Tumor Heterogeneity, and AR-Targeted Therapy Resistance in Prostate Cancer. Cancer Cell 2023, 41, 1427–1449. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef]
- Kohaar, I.; Zhang, X.; Tan, S.-H.; Nousome, D.; Babcock, K.; Ravindranath, L.; Sukumar, G.; Mcgrath-Martinez, E.; Rosenberger, J.; Alba, C.; et al. Germline Mutation Landscape of DNA Damage Repair Genes in African Americans with Prostate Cancer Highlights Potentially Targetable RAD Genes. Nat. Commun. 2022, 13, 1361. [Google Scholar] [CrossRef]
- Zhang, W.; van Gent, D.C.; Incrocci, L.; van Weerden, W.M.; Nonnekens, J. Role of the DNA Damage Response in Prostate Cancer Formation, Progression and Treatment. Prostate Cancer Prostatic Dis. 2020, 23, 24–37. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.-Y.; Gleave, M.E.; Beltran, H. Towards Precision Oncology in Advanced Prostate Cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.T.; Huitema, A.D.R.; Chau, C.H.; Figg, W.D. Resistance to Second-Generation Androgen Receptor Antagonists in Prostate Cancer. Nat. Rev. Urol. 2021, 18, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, R.; Xu, K.; Ding, S.; Li, J.; Baek, G.; Ramanand, S.G.; Ding, S.; Liu, Z.; Gao, Y.; et al. Androgen Receptor Variants Mediate DNA Repair after Prostate Cancer Irradiation. Cancer Res. 2017, 77, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Tolkach, Y.; Kremer, A.; Lotz, G.; Schmid, M.; Mayr, T.; Förster, S.; Garbe, S.; Hosni, S.; Cronauer, M.V.; Kocsmár, I.; et al. Androgen Receptor Splice Variants Contribute to the Upregulation of DNA Repair in Prostate Cancer. Cancers 2022, 14, 4441. [Google Scholar] [CrossRef]
- Basu, S.; Martínez-Cristóbal, P.; Frigolé-Vivas, M.; Pesarrodona, M.; Lewis, M.; Szulc, E.; Bañuelos, C.A.; Sánchez-Zarzalejo, C.; Bielskutė, S.; Zhu, J.; et al. Rational Optimization of a Transcription Factor Activation Domain Inhibitor. Nat. Struct. Mol. Biol. 2023, 30, 1958–1969. [Google Scholar] [CrossRef]
- Kounatidou, E.; Nakjang, S.; McCracken, S.R.C.; Dehm, S.M.; Robson, C.N.; Jones, D.; Gaughan, L. A Novel CRISPR-Engineered Prostate Cancer Cell Line Defines the AR-V Transcriptome and Identifies PARP Inhibitor Sensitivities. Nucleic Acids Res. 2019, 47, 5634–5647. [Google Scholar] [CrossRef]
- Gui, B.; Gui, F.; Takai, T.; Feng, C.; Bai, X.; Fazli, L.; Dong, X.; Liu, S.; Zhang, X.; Zhang, W.; et al. Selective Targeting of PARP-2 Inhibits Androgen Receptor Signaling and Prostate Cancer Growth through Disruption of FOXA1 Function. Proc. Natl. Acad. Sci. USA 2019, 116, 14573–14582. [Google Scholar] [CrossRef]
- Alghoul, E.; Basbous, J.; Constantinou, A. Compartmentalization of the DNA Damage Response: Mechanisms and Functions. DNA Repair 2023, 128, 103524. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers 2022, 14, 1420. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Ryan, C.J.; Lord, C.J. Exploiting Cancer Synthetic Lethality in Cancer—Lessons Learnt from PARP Inhibitors. In Targeting the DNA Damage Response for Cancer Therapy; Yap, T.A., Shapiro, G.I., Eds.; Cancer Treatment and Research; Springer International Publishing: Cham, Switzerland, 2023; pp. 13–23. ISBN 978-3-031-30065-3. [Google Scholar]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate Cancer and PARP Inhibitors: Progress and Challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Iannantuono, G.M.; Chandran, E.; Floudas, C.S.; Choo-Wosoba, H.; Butera, G.; Roselli, M.; Gulley, J.L.; Karzai, F. Efficacy and Safety of PARP Inhibitors in Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis of Clinical Trials. Cancer Treat. Rev. 2023, 120, 102623. [Google Scholar] [CrossRef]
- Beatson, E.L.; Chau, C.H.; Price, D.K.; Figg, W.D. PARP Inhibitors on the Move in Prostate Cancer: Spotlight on Niraparib & Update on PARP Inhibitor Combination Trials. Am. J. Clin. Exp. Urol. 2022, 10, 252–257. [Google Scholar]
- Mateo, J.; de Bono, J.S.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Agarwal, N.; Olmos, D.; Thiery-Vuillemin, A.; et al. Olaparib for the Treatment of Patients with Metastatic Castration-Resistant Prostate Cancer and Alterations in BRCA1 and/or BRCA2 in the PROfound Trial. JCO 2023. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Bryce, A.H.; Shapiro, J.; Bambury, R.M.; Zhang, J.; Burke, J.M.; Castellano, D.; Font, A.; et al. Rucaparib for the Treatment of Metastatic Castration-Resistant Prostate Cancer Associated with a DNA Damage Repair Gene Alteration: Final Results from the Phase 2 TRITON2 Study. Eur. Urol. 2023, 84, 321–330. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis from the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in Patients with Metastatic Castration-Resistant Prostate Cancer and DNA Repair Gene Defects (GALAHAD): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib Monotherapy in Metastatic Castration-Resistant Prostate Cancer with DNA Repair Alterations (TALAPRO-1): An Open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Hwang, J.; Shi, X.; Elliott, A.; Arnoff, T.E.; McGrath, J.; Xiu, J.; Walker, P.; Bergom, H.E.; Day, A.; Ahmed, S.; et al. Metastatic Prostate Cancers with BRCA2 versus ATM Mutations Exhibit Divergent Molecular Features and Clinical Outcomes. Clin. Cancer Res. 2023, 29, 2702–2713. [Google Scholar] [CrossRef]
- Su, C.T.; Nizialek, E.; Berchuck, J.E.; Vlachostergios, P.J.; Ashkar, R.; Sokolova, A.; Barata, P.C.; Aggarwal, R.R.; McKay, R.R.; Agarwal, N.; et al. Differential Responses to Taxanes and PARP Inhibitors in ATM-versus BRCA2-Mutated Metastatic Castrate-Resistant Prostate Cancer. Prostate 2023, 83, 227–236. [Google Scholar] [CrossRef]
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential Response to Olaparib Treatment among Men with Metastatic Castration-Resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 2019, 76, 452–458. [Google Scholar] [CrossRef]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM Orchestrates the DNA-Damage Response to Counter Toxic Non-Homologous End-Joining at Broken Replication Forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef]
- Williamson, C.T.; Muzik, H.; Turhan, A.G.; Zamò, A.; O’Connor, M.J.; Bebb, D.G.; Lees-Miller, S.P. ATM Deficiency Sensitizes Mantle Cell Lymphoma Cells to Poly(ADP-Ribose) Polymerase-1 Inhibitors. Mol. Cancer Ther. 2010, 9, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Prodosmo, A.; Stagni, V.; Merli, D.; Monteonofrio, L.; Gatti, V.; Gentileschi, M.P.; Barilà, D.; Soddu, S. ATM-Depletion in Breast Cancer Cells Confers Sensitivity to PARP Inhibition. J. Exp. Clin. Cancer Res. 2013, 32, 95. [Google Scholar] [CrossRef] [PubMed]
- Taza, F.; Holler, A.E.; Fu, W.; Wang, H.; Adra, N.; Albany, C.; Ashkar, R.; Cheng, H.H.; Sokolova, A.O.; Agarwal, N.; et al. Differential Activity of PARP Inhibitors in BRCA1- Versus BRCA2-Altered Metastatic Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2021, 5, 1200–1220. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Duro, E.; Lundin, C.; Ask, K.; Sanchez-Pulido, L.; MacArtney, T.J.; Toth, R.; Ponting, C.P.; Groth, A.; Helleday, T.; Rouse, J. Identification of the MMS22L-TONSL Complex That Promotes Homologous Recombination. Mol. Cell 2010, 40, 632–644. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Adamson, B.; Lydeard, J.R.; Sowa, M.E.; Ciccia, A.; Bredemeyer, A.L.; Schlabach, M.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. A Genome-Wide Camptothecin Sensitivity Screen Identifies a Mammalian MMS22L-NFKBIL2 Complex Required for Genomic Stability. Mol. Cell 2010, 40, 645–657. [Google Scholar] [CrossRef]
- O’Donnell, L.; Panier, S.; Wildenhain, J.; Tkach, J.M.; Al-Hakim, A.; Landry, M.-C.; Escribano-Diaz, C.; Szilard, R.K.; Young, J.T.F.; Munro, M.; et al. The MMS22L-TONSL Complex Mediates Recovery from Replication Stress and Homologous Recombination. Mol. Cell 2010, 40, 619–631. [Google Scholar] [CrossRef]
- Tsujino, T.; Takai, T.; Hinohara, K.; Gui, F.; Tsutsumi, T.; Bai, X.; Miao, C.; Feng, C.; Gui, B.; Sztupinszki, Z.; et al. CRISPR Screens Reveal Genetic Determinants of PARP Inhibitor Sensitivity and Resistance in Prostate Cancer. Nat. Commun. 2023, 14, 252. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A Compendium of Mutational Cancer Driver Genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Bennett, C.B.; Lewis, L.K.; Karthikeyan, G.; Lobachev, K.S.; Jin, Y.H.; Sterling, J.F.; Snipe, J.R.; Resnick, M.A. Genes Required for Ionizing Radiation Resistance in Yeast. Nat. Genet. 2001, 29, 426–434. [Google Scholar] [CrossRef]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Tsao, N.; Schärer, O.D.; Mosammaparast, N. The Complexity and Regulation of Repair of Alkylation Damage to Nucleic Acids. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 125–136. [Google Scholar] [CrossRef]
- Martin, S.K.; Wood, R.D. DNA Polymerase ζ in DNA Replication and Repair. Nucleic Acids Res. 2019, 47, 8348–8361. [Google Scholar] [CrossRef]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.-W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ Maintains the Viability of Homologous Recombination-Deficient Cancer Cells through Mutagenic Repair of PRIMPOL-Dependent ssDNA Gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef]
- Park, J.; Long, D.T.; Lee, K.Y.; Abbas, T.; Shibata, E.; Negishi, M.; Luo, Y.; Schimenti, J.C.; Gambus, A.; Walter, J.C.; et al. The MCM8-MCM9 Complex Promotes RAD51 Recruitment at DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell Biol. 2013, 33, 1632–1644. [Google Scholar] [CrossRef]
- Lee, K.Y.; Im, J.-S.; Shibata, E.; Park, J.; Handa, N.; Kowalczykowski, S.C.; Dutta, A. MCM8-9 Complex Promotes Resection of Double-Strand Break Ends by MRE11-RAD50-NBS1 Complex. Nat. Commun. 2015, 6, 7744. [Google Scholar] [CrossRef]
- Hustedt, N.; Saito, Y.; Zimmermann, M.; Álvarez-Quilón, A.; Setiaputra, D.; Adam, S.; McEwan, A.; Yuan, J.Y.; Olivieri, M.; Zhao, Y.; et al. Control of Homologous Recombination by the HROB-MCM8-MCM9 Pathway. Genes Dev. 2019, 33, 1397–1415. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Z.; Su, D.; Tang, M.; Nie, L.; Zhang, H.; Feng, X.; Wang, R.; Shen, X.; Srivastava, M.; et al. C17orf53 Is Identified as a Novel Gene Involved in Inter-Strand Crosslink Repair. DNA Repair 2020, 95, 102946. [Google Scholar] [CrossRef]
- Huang, J.-W.; Acharya, A.; Taglialatela, A.; Nambiar, T.S.; Cuella-Martin, R.; Leuzzi, G.; Hayward, S.B.; Joseph, S.A.; Brunette, G.J.; Anand, R.; et al. MCM8IP Activates the MCM8-9 Helicase to Promote DNA Synthesis and Homologous Recombination upon DNA Damage. Nat. Commun. 2020, 11, 2948. [Google Scholar] [CrossRef]
- Lee, K.Y.; Im, J.-S.; Shibata, E.; Dutta, A. ASF1a Promotes Non-Homologous End Joining Repair by Facilitating Phosphorylation of MDC1 by ATM at Double-Strand Breaks. Mol. Cell 2017, 68, 61–75.e5. [Google Scholar] [CrossRef]
- Lee, K.Y.; Dutta, A. Chk1 Promotes Non-Homologous End Joining in G1 through Direct Phosphorylation of ASF1A. Cell Rep. 2021, 34, 108680. [Google Scholar] [CrossRef]
- Huang, T.-H.; Fowler, F.; Chen, C.-C.; Shen, Z.-J.; Sleckman, B.; Tyler, J.K. The Histone Chaperones ASF1 and CAF-1 Promote MMS22L-TONSL-Mediated Rad51 Loading onto ssDNA during Homologous Recombination in Human Cells. Mol. Cell 2018, 69, 879–892.e5. [Google Scholar] [CrossRef]
- Feng, S.; Ma, S.; Li, K.; Gao, S.; Ning, S.; Shang, J.; Guo, R.; Chen, Y.; Blumenfeld, B.; Simon, I.; et al. RIF1-ASF1-Mediated High-Order Chromatin Structure Safeguards Genome Integrity. Nat. Commun. 2022, 13, 957. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Z.; Wang, C.; Feng, X.; Lee, N.; Huang, M.; Zhang, H.; Li, S.; Xiong, Y.; Chen, J. Histone Chaperone ASF1 Acts with RIF1 to Promote DNA End-Joining in BRCA1-Deficient Cells. J. Biol. Chem. 2022, 298, 101979. [Google Scholar] [CrossRef]
- Washino, S.; Rider, L.C.; Romero, L.; Jillson, L.K.; Affandi, T.; Ohm, A.M.; Lam, E.T.; Reyland, M.E.; Costello, J.C.; Cramer, S.D. Loss of MAP3K7 Sensitizes Prostate Cancer Cells to CDK1/2 Inhibition and DNA Damage by Disrupting Homologous Recombination. Mol. Cancer Res. 2019, 17, 1985–1998. [Google Scholar] [CrossRef]
- Huang, Z.; Tang, B.; Yang, Y.; Yang, Z.; Shi, L.; Bai, Y.; Yan, B.; Karnes, R.J.; Zhang, J.; Jimenez, R.; et al. MAP3K7-IKK Inflammatory Signaling Modulates AR Protein Degradation and Prostate Cancer Progression. Cancer Res. 2021, 81, 4471–4484. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The Long Tail of Oncogenic Drivers in Prostate Cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Reijns, M.A.M.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic Removal of Ribonucleotides from DNA Is Essential for Mammalian Genome Integrity and Development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR Screens Identify Genomic Ribonucleotides as a Source of PARP-Trapping Lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Wang, C.; Wang, G.; Feng, X.; Shepherd, P.; Zhang, J.; Tang, M.; Chen, Z.; Srivastava, M.; McLaughlin, M.E.; Navone, N.M.; et al. Genome-Wide CRISPR Screens Reveal Synthetic Lethality of RNASEH2 Deficiency and ATR Inhibition. Oncogene 2019, 38, 2451–2463. [Google Scholar] [CrossRef]
- Miao, C.; Tsujino, T.; Takai, T.; Gui, F.; Tsutsumi, T.; Sztupinszki, Z.; Wang, Z.; Azuma, H.; Szallasi, Z.; Mouw, K.W.; et al. RB1 Loss Overrides PARP Inhibitor Sensitivity Driven by RNASEH2B Loss in Prostate Cancer. Sci. Adv. 2022, 8, eabl9794. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 175, 889. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of Lethal Prostate Cancer at Diagnosis and Castration Resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Måseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic Hypoxia Decreases Synthesis of Homologous Recombination Proteins to Offset Chemoresistance and Radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T.; et al. Contextual Synthetic Lethality of Cancer Cell Kill Based on the Tumor Microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef]
- Mehibel, M.; Xu, Y.; Li, C.G.; Moon, E.J.; Thakkar, K.N.; Diep, A.N.; Kim, R.K.; Bloomstein, J.D.; Xiao, Y.; Bacal, J.; et al. Eliminating Hypoxic Tumor Cells Improves Response to PARP Inhibitors in Homologous Recombination–Deficient Cancer Models. J. Clin. Investig. 2021, 131, e146256. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef]
- Hustedt, N.; Álvarez-Quilón, A.; McEwan, A.; Yuan, J.Y.; Cho, T.; Koob, L.; Hart, T.; Durocher, D. A Consensus Set of Genetic Vulnerabilities to ATR Inhibition. Open Biol. 2019, 9, 190156. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A Cell-Based Screen Identifies ATR Inhibitors with Synthetic Lethal Properties for Cancer-Associated Mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef]
- Qiu, Z.; Fa, P.; Liu, T.; Prasad, C.B.; Ma, S.; Hong, Z.; Chan, E.R.; Wang, H.; Li, Z.; He, K.; et al. A Genome-Wide Pooled shRNA Screen Identifies PPP2R2A as a Predictive Biomarker for the Response to ATR and CHK1 Inhibitors. Cancer Res. 2020, 80, 3305–3318. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic Stress Sensitizes Murine Cancers to Hypomorphic Suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.D.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting Oncogene-Induced Replicative Stress for the Selective Killing of Myc-Driven Tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef]
- Mohni, K.N.; Thompson, P.S.; Luzwick, J.W.; Glick, G.G.; Pendleton, C.S.; Lehmann, B.D.; Pietenpol, J.A.; Cortez, D. A Synthetic Lethal Screen Identifies DNA Repair Pathways That Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS ONE 2015, 10, e0125482. [Google Scholar] [CrossRef]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-Wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting Replication Stress in Cancer Therapy. Nat. Rev. Drug Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef]
- Roulston, A.; Zimmermann, M.; Papp, R.; Skeldon, A.; Pellerin, C.; Dumas-Bérube, É.; Dumais, V.; Dorich, S.; Fader, L.D.; Fournier, S.; et al. RP-3500: A Novel, Potent, and Selective ATR Inhibitor That Is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 245–256. [Google Scholar] [CrossRef]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a Clinically-Successful ATR Inhibitor for Cancer Therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef]
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR Inhibitors VE-821 and VX-970 Sensitize Cancer Cells to Topoisomerase i Inhibitors by Disabling DNA Replication Initiation and Fork Elongation Responses. Cancer Res. 2014, 74, 6968–6979. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR Inhibition Potentiates Genome Instability and Cell Death in ATM-Deficient Cancer Cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Jette, N.R.; Radhamani, S.; Arthur, G.; Ye, R.; Goutam, S.; Bolyos, A.; Petersen, L.F.; Bose, P.; Bebb, D.G.; Lees-Miller, S.P. Combined Poly-ADP Ribose Polymerase and Ataxia-Telangiectasia Mutated/Rad3-Related Inhibition Targets Ataxia-Telangiectasia Mutated-Deficient Lung Cancer Cells. Br. J. Cancer 2019, 121, 600–610. [Google Scholar] [CrossRef]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.-Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Abel, M.L.; Takahashi, N.; Peer, C.; Redon, C.E.; Nichols, S.; Vilimas, R.; Lee, M.-J.; Lee, S.; Shelat, M.; Kattappuram, R.; et al. Targeting Replication Stress and Chemotherapy Resistance with a Combination of Sacituzumab Govitecan and Berzosertib: A Phase I Clinical Trial. Clin. Cancer Res. 2023, 29, 3603–3611. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Bernier, C.; Kaiser, B.; Fournier, S.; Li, L.; Desjardins, J.; Skeldon, A.; Rimkunas, V.; Veloso, A.; Young, J.T.F.; et al. Guiding ATR and PARP Inhibitor Combinationswith Chemogenomic Screens. Cell Rep. 2022, 40, 111081. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Kothari, V.; Drake, J.M.; Zhao, S.; Dylgjeri, E.; Dean, J.L.; Schiewer, M.J.; McNair, C.; Jones, J.K.; Aytes, A.; et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015, 28, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Livingstone, J.; Wrana, J.L.; Finelli, A.; He, H.H.; van der Kwast, T.; Zlotta, A.R.; Bristow, R.G.; Boutros, P.C. Somatic Driver Mutation Prevalence in 1844 Prostate Cancers Identifies ZNRF3 Loss as a Predictor of Metastatic Relapse. Nat. Commun. 2021, 12, 6248. [Google Scholar] [CrossRef]
- Adamson, B.; Brittain, N.; Walker, L.; Duncan, R.; Luzzi, S.; Rescigno, P.; Smith, G.R.; McGill, S.; Burchmore, R.J.; Willmore, E.; et al. The Catalytic Subunit of DNA-PK Regulates Transcription and Splicing of AR in Advanced Prostate Cancer. J. Clin. Investig. 2023, 133, e169200. [Google Scholar] [CrossRef]
- Puc, J.; Kozbial, P.; Li, W.; Tan, Y.; Liu, Z.; Suter, T.; Ohgi, K.A.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Ligand-Dependent Enhancer Activation Regulated by Topoisomerase-I Activity. Cell 2015, 160, 367–380. [Google Scholar] [CrossRef]
- Tan, Y.; Yao, L.; Gamliel, A.; Nair, S.J.; Taylor, H.; Ohgi, K.; Aggarwal, A.K.; Rosenfeld, M.G. Signal-Induced Enhancer Activation Requires Ku70 to Read Topoisomerase1-DNA Covalent Complexes. Nat. Struct. Mol. Biol. 2023, 30, 148–158. [Google Scholar] [CrossRef]
- Shao, Z.; Flynn, R.A.; Crowe, J.L.; Zhu, Y.; Liang, J.; Jiang, W.; Aryan, F.; Aoude, P.; Bertozzi, C.R.; Estes, V.M.; et al. DNA-PKcs Has KU-Dependent Function in rRNA Processing and Haematopoiesis. Nature 2020, 579, 291–296. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Kothari, V.; Shafi, A.A.; Semenova, G.; Gallagher, P.T.; Guan, Y.F.; Pang, A.; Goodwin, J.F.; Irani, S.; McCann, J.J.; et al. A Novel Role for DNA-PK in Metabolism by Regulating Glycolysis in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2022, 28, 1446–1459. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Zhao, J.L.; Antonarakis, E.S.; Cheng, H.H.; George, D.J.; Aggarwal, R.; Riedel, E.; Sumiyoshi, T.; Schonhoft, J.D.; Anderson, A.; Mao, N.; et al. Phase 1b Study of Enzalutamide plus CC-115, a Dual mTORC1/2 and DNA-PK Inhibitor, in Men with Metastatic Castration-Resistant Prostate Cancer (mCRPC). Br. J. Cancer 2023, 1–10. [Google Scholar] [CrossRef]
- Chao, O.S.; Goodman, O.B. DNA-PKc Inhibition Overcomes Taxane Resistance by Promoting Taxane-Induced DNA Damage in Prostate Cancer Cells. Prostate 2021, 81, 1032–1048. [Google Scholar] [CrossRef]
- Salerno, K.E.; Roy, S.; Ribaudo, C.; Fisher, T.; Patel, R.B.; Mena, E.; Escorcia, F.E. A Primer on Radiopharmaceutical Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2023, 115, 48–59. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- Miyahira, A.K.; Soule, H.R. The History of Prostate-Specific Membrane Antigen as a Theranostic Target in Prostate Cancer: The Cornerstone Role of the Prostate Cancer Foundation. J. Nucl. Med. 2022, 63, 331–338. [Google Scholar] [CrossRef]
- Feuerecker, B.; Tauber, R.; Knorr, K.; Heck, M.; Beheshti, A.; Seidl, C.; Bruchertseifer, F.; Pickhard, A.; Gafita, A.; Kratochwil, C.; et al. Activity and Adverse Events of Actinium-225-PSMA-617 in Advanced Metastatic Castration-Resistant Prostate Cancer After Failure of Lutetium-177-PSMA. Eur. Urol. 2021, 79, 343–350. [Google Scholar] [CrossRef]
- Jani, A.B.; Ravizzini, G.C.; Gartrell, B.A.; Siegel, B.A.; Twardowski, P.; Saltzstein, D.; Fleming, M.T.; Chau, A.; Davis, P.; Chapin, B.F.; et al. Diagnostic Performance and Safety of 18F-rhPSMA-7.3 Positron Emission Tomography in Men With Suspected Prostate Cancer Recurrence: Results From a Phase 3, Prospective, Multicenter Study (SPOTLIGHT). J. Urol. 2023, 210, 299–311. [Google Scholar] [CrossRef]
- Bundschuh, R.A.; Pfob, C.H.; Wienand, G.; Dierks, A.; Kircher, M.; Lapa, C. 177 Lu-rhPSMA-10.1 Induces Tumor Response in a Patient with mCRPC After PSMA-Directed Radioligand Therapy with 177 Lu-PSMA-I&T. Clin. Nucl. Med. 2023, 48, 337–338. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Graf, R.P.; Fisher, V.; Weberpals, J.; Gjoerup, O.; Tierno, M.B.; Huang, R.S.P.; Sayegh, N.; Lin, D.I.; Raskina, K.; Schrock, A.B.; et al. Comparative Effectiveness of Immune Checkpoint Inhibitors vs. Chemotherapy by Tumor Mutational Burden in Metastatic Castration-Resistant Prostate Cancer. JAMA Netw. Open 2022, 5, e225394. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The Impact of Nonsense-Mediated mRNA Decay on Genetic Disease, Gene Editing and Cancer Immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Lim, E.L.; Xu, H.; Liu, P.; Al-Bakir, M.; Wong, Y.N.S.; Rowan, A.; Funt, S.A.; Merghoub, T.; et al. Escape from Nonsense-Mediated Decay Associates with Anti-Tumor Immunogenicity. Nat. Commun. 2020, 11, 3800. [Google Scholar] [CrossRef]
- Reading, J.L.; Caswell, D.R.; Swanton, C. Tumor Heterogeneity Impairs Immunogenicity in Mismatch Repair Deficient Tumors. Nat. Genet. 2023, 55, 1610–1612. [Google Scholar] [CrossRef]
- Jaratlerdsiri, W.; Jiang, J.; Gong, T.; Patrick, S.M.; Willet, C.; Chew, T.; Lyons, R.J.; Haynes, A.-M.; Pasqualim, G.; Louw, M.; et al. African-Specific Molecular Taxonomy of Prostate Cancer. Nature 2022, 609, 552–559. [Google Scholar] [CrossRef]
- Serrano, J.C.; von Trentini, D.; Berríos, K.N.; Barka, A.; Dmochowski, I.J.; Kohli, R.M. Structure-Guided Design of a Potent and Specific Inhibitor against the Genomic Mutator APOBEC3A. ACS Chem. Biol. 2022, 17, 3379–3388. [Google Scholar] [CrossRef]
- Petljak, M.; Green, A.M.; Maciejowski, J.; Weitzman, M.D. Addressing the Benefits of Inhibiting APOBEC3-Dependent Mutagenesis in Cancer. Nat. Genet. 2022, 54, 1599–1608. [Google Scholar] [CrossRef]
- Kvach, M.V.; Barzak, F.M.; Harjes, S.; Schares, H.A.M.; Jameson, G.B.; Ayoub, A.M.; Moorthy, R.; Aihara, H.; Harris, R.S.; Filichev, V.V.; et al. Inhibiting APOBEC3 Activity with Single-Stranded DNA Containing 2′-Deoxyzebularine Analogues. Biochemistry 2019, 58, 391–400. [Google Scholar] [CrossRef]
- Barzak, F.M.; Harjes, S.; Kvach, M.V.; Kurup, H.M.; Jameson, G.B.; Filichev, V.V.; Harjes, E. Selective Inhibition of APOBEC3 Enzymes by Single-Stranded DNAs Containing 2′-Deoxyzebularine. Org. Biomol. Chem. 2019, 17, 9435–9441. [Google Scholar] [CrossRef]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef]
- Isozaki, H.; Sakhtemani, R.; Abbasi, A.; Nikpour, N.; Stanzione, M.; Oh, S.; Langenbucher, A.; Monroe, S.; Su, W.; Cabanos, H.F.; et al. Therapy-Induced APOBEC3A Drives Evolution of Persistent Cancer Cells. Nature 2023, 620, 393–401. [Google Scholar] [CrossRef]
- Calagua, C.; Ficial, M.; Jansen, C.S.; Hirz, T.; Del Balzo, L.; Wilkinson, S.; Lake, R.; Ku, A.T.; Voznesensky, O.; Sykes, D.B.; et al. A Subset of Localized Prostate Cancer Displays an Immunogenic Phenotype Associated with Losses of Key Tumor Suppressor Genes. Clin. Cancer Res. 2021, 27, 4836–4847. [Google Scholar] [CrossRef]
- Plas, S.; Pircher, A.; Heidegger, I. Pembrolizumab in mCRPC-Combination Therapies as Breakthrough to Success? Curr. Opin. Urol. 2023, 33, 458–471. [Google Scholar] [CrossRef]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-Dose Targeted Radionuclide Therapy Renders Immunologically Cold Tumors Responsive to Immune Checkpoint Blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-Induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.-Y. ATM Inhibition Enhances Cancer Immunotherapy by Promoting mtDNA Leakage and cGAS/STING Activation. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 Abrogation Triggers Innate Immune Responses Potentiated by Treatment with PARP Inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef]
- Wang, W.; McMillan, M.T.; Zhao, X.; Wang, Z.; Jiang, L.; Karnak, D.; Lima, F.; Parsels, J.D.; Parsels, L.A.; Lawrence, T.S.; et al. DNA-PK Inhibition and Radiation Promote Antitumoral Immunity through RNA Polymerase III in Pancreatic Cancer. Mol. Cancer Res. 2022, 20, 1137–1150. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib Activates Interferon Signaling and Potentiates Anti-PD-1 Antibody Efficacy in Tumor Models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef]
- Carr, M.I.; Chiu, L.-Y.; Guo, Y.; Xu, C.; Lazorchak, A.S.; Yu, H.; Qin, G.; Qi, J.; Marelli, B.; Lan, Y.; et al. DNA-PK Inhibitor Peposertib Amplifies Radiation-Induced Inflammatory Micronucleation and Enhances TGFβ/PD-L1 Targeted Cancer Immunotherapy. Mol. Cancer Res. 2022, 20, 568–582. [Google Scholar] [CrossRef]
- Luo, L.Y.; O’Hara, M.H.; Mitchell, T.C.; Vonderheide, R.H.; Wherry, E.J.; Minn, A.J.; Maity, A. Combining Radiation with Immunotherapy: The University of Pennsylvania Experience. Semin. Radiat. Oncol. 2020, 30, 173–180. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of cGAS-STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Beyer, U.; Brand, F.; Martens, H.; Weder, J.; Christians, A.; Elyan, N.; Hentschel, B.; Westphal, M.; Schackert, G.; Pietsch, T.; et al. Rare ADAR and RNASEH2B Variants and a Type I Interferon Signature in Glioma and Prostate Carcinoma Risk and Tumorigenesis. Acta Neuropathol. 2017, 134, 905–922. [Google Scholar] [CrossRef]
- Liu, A.; Ying, S. Aicardi-Goutières Syndrome: A Monogenic Type I Interferonopathy. Scand. J. Immunol. 2023, 98, e13314. [Google Scholar] [CrossRef]
- Samson, N.; Ablasser, A. The cGAS-STING Pathway and Cancer. Nat. Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef]
- Curran, E.; Chen, X.; Corrales, L.; Kline, D.E.; Dubensky, T.W.; Duttagupta, P.; Kortylewski, M.; Kline, J. STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep. 2016, 15, 2357–2366. [Google Scholar] [CrossRef]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial Watch: STING Agonists in Cancer Therapy. Oncoimmunology 2020, 9, 1777624. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Lamb, T.J.; Pawelec, G. Here, There, and Everywhere: Myeloid-Derived Suppressor Cells in Immunology. J. Immunol. 2023, 210, 1183–1197. [Google Scholar] [CrossRef]
- Siemińska, I.; Baran, J. Myeloid-Derived Suppressor Cells as Key Players and Promising Therapy Targets in Prostate Cancer. Front. Oncol. 2022, 12, 862416. [Google Scholar] [CrossRef]
- Roth, T.J.; Sheinin, Y.; Lohse, C.M.; Kuntz, S.M.; Frigola, X.; Inman, B.A.; Krambeck, A.E.; McKenney, M.E.; Karnes, R.J.; Blute, M.L.; et al. B7-H3 Ligand Expression by Prostate Cancer: A Novel Marker of Prognosis and Potential Target for Therapy. Cancer Res. 2007, 67, 7893–7900. [Google Scholar] [CrossRef]
- Guo, C.; Figueiredo, I.; Gurel, B.; Neeb, A.; Seed, G.; Crespo, M.; Carreira, S.; Rekowski, J.; Buroni, L.; Welti, J.; et al. B7-H3 as a Therapeutic Target in Advanced Prostate Cancer. Eur. Urol. 2023, 83, 224–238. [Google Scholar] [CrossRef]
- Shi, W.; Wang, Y.; Zhao, Y.; Kim, J.J.; Li, H.; Meng, C.; Chen, F.; Zhang, J.; Mak, D.H.; Van, V.; et al. Immune Checkpoint B7-H3 Is a Therapeutic Vulnerability in Prostate Cancer Harboring PTEN and TP53 Deficiencies. Sci. Transl. Med. 2023, 15, eadf6724. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective Combinatorial Immunotherapy for Castration-Resistant Prostate Cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef]
- Guo, C.; Sharp, A.; Gurel, B.; Crespo, M.; Figueiredo, I.; Jain, S.; Vogl, U.; Rekowski, J.; Rouhifard, M.; Gallagher, L.; et al. Targeting Myeloid Chemotaxis to Reverse Prostate Cancer Therapy Resistance. Nature 2023, 623, 1053–1061. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular Landmarks of Tumor Hypoxia across Cancer Types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Duan, J.-X.; Jiao, H.; Kaizerman, J.; Stanton, T.; Evans, J.W.; Lan, L.; Lorente, G.; Banica, M.; Jung, D.; Wang, J.; et al. Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs. J. Med. Chem. 2008, 51, 2412–2420. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, Q.; Zhang, Y.; Liu, Z.; Zheng, Z.; Liu, S.; Meng, L.; Xin, Y.; Jiang, X. Targeting Hypoxia in the Tumor Microenvironment: A Potential Strategy to Improve Cancer Immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 24. [Google Scholar] [CrossRef]
- van der Wiel, A.M.A.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.M.; Lieuwes, N.G.; Biemans, R.; Lin, X.; et al. Selectively Targeting Tumor Hypoxia with the Hypoxia-Activated Prodrug CP-506. Mol. Cancer Ther. 2021, 20, 2372–2383. [Google Scholar] [CrossRef]
- Yaromina, A.; Koi, L.; Schuitmaker, L.; van der Wiel, A.M.-M.A.; Dubois, L.J.; Krause, M.; Lambin, P. Overcoming Radioresistance with the Hypoxia-Activated Prodrug CP-506: A Pre-Clinical Study of Local Tumour Control Probability. Radiother. Oncol. 2023, 186, 109738. [Google Scholar] [CrossRef]
- Puca, L.; Vlachostergios, P.J.; Beltran, H. Neuroendocrine Differentiation in Prostate Cancer: Emerging Biology, Models, and Therapies. Cold Spring Harb. Perspect. Med. 2019, 9, a030593. [Google Scholar] [CrossRef]
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, H.; Li, E.; et al. ONECUT2 Is a Driver of Neuroendocrine Prostate Cancer. Nat. Commun. 2019, 10, 278. [Google Scholar] [CrossRef]
- Qian, C.; Yang, Q.; Freedland, S.J.; Di Vizio, D.; Ellis, L.; You, S.; Freeman, M.R. Activation of ONECUT2 by RB1 Loss in Castration-Resistant Prostate Cancer. Am. J. Clin. Exp. Urol. 2022, 10, 397–407. [Google Scholar]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Ribeiro-Silva, C.; Vermeulen, W.; Lans, H. SWI/SNF: Complex Complexes in Genome Stability and Cancer. DNA Repair 2019, 77, 87–95. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A Deficiency Promotes Mutability and Potentiates Therapeutic Antitumor Immunity Unleashed by Immune Checkpoint Blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef]
- Bayona-Feliu, A.; Barroso, S.; Muñoz, S.; Aguilera, A. The SWI/SNF Chromatin Remodeling Complex Helps Resolve R-Loop-Mediated Transcription-Replication Conflicts. Nat. Genet. 2021, 53, 1050–1063. [Google Scholar] [CrossRef]
- Iurlaro, M.; Stadler, M.B.; Masoni, F.; Jagani, Z.; Galli, G.G.; Schübeler, D. Mammalian SWI/SNF Continuously Restores Local Accessibility to Chromatin. Nat. Genet. 2021, 53, 279–287. [Google Scholar] [CrossRef]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in Enhancer-Addicted Prostate Cancer. Nature 2022, 601, 434–439. [Google Scholar] [CrossRef]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin Remodeling ATPase BRG1 and PTEN Are Synthetic Lethal in Prostate Cancer. J. Clin. Investig. 2019, 129, 759–773. [Google Scholar] [CrossRef]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ Inhibitors Elicit BRCA-Gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Bubenik, M.; Mader, P.; Mochirian, P.; Vallée, F.; Clark, J.; Truchon, J.-F.; Perryman, A.L.; Pau, V.; Kurinov, I.; Zahn, K.E.; et al. Identification of RP-6685, an Orally Bioavailable Compound That Inhibits the DNA Polymerase Activity of Polθ. J. Med. Chem. 2022, 65, 13198–13215. [Google Scholar] [CrossRef]
- Llorens-Agost, M.; Ensminger, M.; Le, H.P.; Gawai, A.; Liu, J.; Cruz-García, A.; Bhetawal, S.; Wood, R.D.; Heyer, W.-D.; Löbrich, M. POLθ-Mediated End Joining Is Restricted by RAD52 and BRCA2 until the Onset of Mitosis. Nat. Cell Biol. 2021, 23, 1095–1104. [Google Scholar] [CrossRef]
- Oh, G.; Wang, A.; Wang, L.; Li, J.; Werba, G.; Weissinger, D.; Zhao, E.; Dhara, S.; Hernandez, R.E.; Ackermann, A.; et al. POLQ Inhibition Elicits an Immune Response in Homologous Recombination–Deficient Pancreatic Adenocarcinoma via cGAS/STING Signaling. J. Clin. Investig. 2023, 133, e165934. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian Polymerase θ Promotes Alternative NHEJ and Suppresses Recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef]
- Wang, Z.; Song, Y.; Li, S.; Kurian, S.; Xiang, R.; Chiba, T.; Wu, X. DNA Polymerase θ (POLQ) Is Important for Repair of DNA Double-Strand Breaks Caused by Fork Collapse. J. Biol. Chem. 2019, 294, 3909–3919. [Google Scholar] [CrossRef]
- Patterson-Fortin, J.; Bose, A.; Tsai, W.-C.; Grochala, C.; Nguyen, H.; Zhou, J.; Parmar, K.; Lazaro, J.-B.; Liu, J.; McQueen, K.; et al. Targeting DNA Repair with Combined Inhibition of NHEJ and MMEJ Induces Synthetic Lethality in TP53-Mutant Cancers. Cancer Res. 2022, 82, 3815–3829. [Google Scholar] [CrossRef]
- Kumar, R.J.; Chao, H.X.; Simpson, D.A.; Feng, W.; Cho, M.-G.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Wozny, A.-S.; Fagan-Solis, K.; et al. Dual Inhibition of DNA-PK and DNA Polymerase Theta Overcomes Radiation Resistance Induced by P53 Deficiency. NAR Cancer 2020, 2, zcaa038. [Google Scholar] [CrossRef]
- Hwang, J.W.; Cho, Y.; Bae, G.-U.; Kim, S.-N.; Kim, Y.K. Protein Arginine Methyltransferases: Promising Targets for Cancer Therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein Arginine Methylation: From Enigmatic Functions to Therapeutic Targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Liu, J.; Bu, X.; Chu, C.; Dai, X.; Asara, J.M.; Sicinski, P.; Freeman, G.J.; Wei, W. PRMT1 Mediated Methylation of cGAS Suppresses Anti-Tumor Immunity. Nat. Commun. 2023, 14, 2806. [Google Scholar] [CrossRef]
- Boisvert, F.-M.; Rhie, A.; Richard, S.; Doherty, A.J. The GAR Motif of 53BP1 Is Arginine Methylated by PRMT1 and Is Necessary for 53BP1 DNA Binding Activity. Cell Cycle 2005, 4, 1834–1841. [Google Scholar] [CrossRef]
- He, L.; Hu, Z.; Sun, Y.; Zhang, M.; Zhu, H.; Jiang, L.; Zhang, Q.; Mu, D.; Zhang, J.; Gu, L.; et al. PRMT1 Is Critical to FEN1 Expression and Drug Resistance in Lung Cancer Cells. DNA Repair 2020, 95, 102953. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, M.F.; González-Guerrero, R.; Sánchez-Del-Campo, L.; Piñero-Madrona, A.; Cabezas-Herrera, J.; Rodríguez-López, J.N. PRMT1-Dependent Methylation of BRCA1 Contributes to the Epigenetic Defense of Breast Cancer Cells against Ionizing Radiation. Sci. Rep. 2020, 10, 13275. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Bailon, M.P.; Choi, S.-Y.; Dufficy, E.R.; Sharma, K.; McNee, G.S.; Gunnell, E.; Chiang, K.; Sahay, D.; Maslen, S.; Stewart, G.S.; et al. Arginine Methylation and Ubiquitylation Crosstalk Controls DNA End-Resection and Homologous Recombination Repair. Nat. Commun. 2021, 12, 6313. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Vogel, G.; Coulombe, Y.; Dubeau, D.; Spehalski, E.; Hébert, J.; Ferguson, D.O.; Masson, J.Y.; Richard, S. The MRE11 GAR Motif Regulates DNA Double-Strand Break Processing and ATR Activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Vadnais, C.; Chen, R.; Fraszczak, J.; Yu, Z.; Boulais, J.; Pinder, J.; Frank, D.; Khandanpour, C.; Hébert, J.; Dellaire, G.; et al. GFI1 Facilitates Efficient DNA Repair by Regulating PRMT1 Dependent Methylation of MRE11 and 53BP1. Nat. Commun. 2018, 9, 1418. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, T.; Hébert, J.; Li, E.; Richard, S. A Mouse PRMT1 Null Allele Defines an Essential Role for Arginine Methylation in Genome Maintenance and Cell Proliferation. Mol. Cell Biol. 2009, 29, 2982–2996. [Google Scholar] [CrossRef] [PubMed]
- Grypari, I.M.; Logotheti, S.; Zolota, V.; Troncoso, P.; Efstathiou, E.; Bravou, V.; Melachrinou, M.; Logothetis, C.; Tzelepi, V. The Protein Arginine Methyltransferases (PRMTs) PRMT1 and CARM1 as Candidate Epigenetic Drivers in Prostate Cancer Progression. Medicine 2021, 100, e27094. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Sethunath, V.; Metaferia, N.Y.; Nogueira, M.F.; Gallant, D.S.; Garner, E.R.; Lairson, L.A.; Penney, C.M.; Li, J.; Gelbard, M.K.; et al. A Genome-Scale CRISPR Screen Reveals PRMT1 as a Critical Regulator of Androgen Receptor Signaling in Prostate Cancer. Cell Rep. 2022, 38, 110417. [Google Scholar] [CrossRef]
- Li, X.; Li, F.; Ye, F.; Guo, H.; Chen, W.; Jin, J.; Wang, Y.; Dai, P.; Shi, H.; Tao, H.; et al. Spermine Is a Natural Suppressor of AR Signaling in Castration-Resistant Prostate Cancer. Cell Rep. 2023, 42, 112798. [Google Scholar] [CrossRef]
- Beketova, E.; Owens, J.L.; Asberry, A.M.; Hu, C.-D. PRMT5: A Putative Oncogene and Therapeutic Target in Prostate Cancer. Cancer Gene Ther. 2021, 29, 264–276. [Google Scholar] [CrossRef]
- Kim, H.; Ronai, Z.A. PRMT5 Function and Targeting in Cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916.e7. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP Deletion Confers Enhanced Dependency on the PRMT5 Arginine Methyltransferase in Cancer Cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Huang, M.; Yao, F.; Nie, L.; Wang, C.; Su, D.; Zhang, H.; Li, S.; Tang, M.; Feng, X.; Yu, B.; et al. FACS-Based Genome-Wide CRISPR Screens Define Key Regulators of DNA Damage Signaling Pathways. Mol. Cell 2023, 83, 2810–2828.e6. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 Control of cGAS/STING and NLRC5 Pathways Defines Melanoma Response to Antitumor Immunity. Sci. Transl. Med. 2020, 12, eaaz5683. [Google Scholar] [CrossRef]
- Hu, R.; Zhou, B.; Chen, Z.; Chen, S.; Chen, N.; Shen, L.; Xiao, H.; Zheng, Y. PRMT5 Inhibition Promotes PD-L1 Expression and Immuno-Resistance in Lung Cancer. Front. Immunol. 2021, 12, 722188. [Google Scholar] [CrossRef]
- Owens, J.L.; Beketova, E.; Liu, S.; Shen, Q.; Pawar, J.S.; Asberry, A.M.; Yang, J.; Deng, X.; Elzey, B.D.; Ratliff, T.L.; et al. Targeting Protein Arginine Methyltransferase 5 Suppresses Radiation-Induced Neuroendocrine Differentiation and Sensitizes Prostate Cancer Cells to Radiation. Mol. Cancer Ther. 2022, 21, 448–459. [Google Scholar] [CrossRef]
- Pawar, J.S.; Al-Amin, M.Y.; Hu, C.-D. JNJ-64619178 Radiosensitizes and Suppresses Fractionated Ionizing Radiation-Induced Neuroendocrine Differentiation (NED) in Prostate Cancer. Front. Oncol. 2023, 13, 1126482. [Google Scholar] [CrossRef]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR Screening Identifies PRMT5 as Synthetic Lethality Combinatorial Target with Gemcitabine in Pancreatic Cancer Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28068–28079. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Ray, S. The FAcilitates Chromatin Transcription (FACT) Complex: Its Roles in DNA Repair and Implications for Cancer Therapy. DNA Repair 2022, 109, 103246. [Google Scholar] [CrossRef]
- Gurova, K.V.; Garcia, H.; Miecznikowski, J.; Omilian, A.R.; Morrison, C. Level of SSRP1 in Cancer as a Prognostic Marker of Aggressive Disease. Am. J. Clin. Pathol. 2013, 140, A152. [Google Scholar] [CrossRef]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer Compounds That Simultaneously Suppress NF-κB and Activate P53 by Targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef]
- De, S.; Lindner, D.J.; Coleman, C.J.; Wildey, G.; Dowlati, A.; Stark, G.R. The FACT Inhibitor CBL0137 Synergizes with Cisplatin in Small-Cell Lung Cancer by Increasing NOTCH1 Expression and Targeting Tumor-Initiating Cells. Cancer Res. 2018, 78, 2396–2406. [Google Scholar] [CrossRef]
- Song, H.; Xi, S.; Chen, Y.; Pramanik, S.; Zeng, J.; Roychoudhury, S.; Harris, H.; Akbar, A.; Elhag, S.S.; Coulter, D.W.; et al. Histone Chaperone FACT Complex Inhibitor CBL0137 Interferes with DNA Damage Repair and Enhances Sensitivity of Medulloblastoma to Chemotherapy and Radiation. Cancer Lett. 2021, 520, 201–212. [Google Scholar] [CrossRef]
- Tallman, M.M.; Zalenski, A.A.; Deighen, A.M.; Schrock, M.S.; Mortach, S.; Grubb, T.M.; Kastury, P.S.; Huntoon, K.; Summers, M.K.; Venere, M. The Small Molecule Drug CBL0137 Increases the Level of DNA Damage and the Efficacy of Radiotherapy for Glioblastoma. Cancer Lett. 2021, 499, 232–242. [Google Scholar] [CrossRef]
- Somers, K.; Kosciolek, A.; Bongers, A.; El-Ayoubi, A.; Karsa, M.; Mayoh, C.; Wadham, C.; Middlemiss, S.; Neznanov, N.; Kees, U.R.; et al. Potent Antileukemic Activity of Curaxin CBL0137 against MLL-Rearranged Leukemia. Int. J. Cancer 2020, 146, 1902–1916. [Google Scholar] [CrossRef]
- Xiao, L.; Somers, K.; Murray, J.; Pandher, R.; Karsa, M.; Ronca, E.; Bongers, A.; Terry, R.; Ehteda, A.; Gamble, L.D.; et al. Dual Targeting of Chromatin Stability by the Curaxin CBL0137 and Histone Deacetylase Inhibitor Panobinostat Shows Significant Preclinical Efficacy in Neuroblastoma. Clin. Cancer Res. 2021, 27, 4338–4352. [Google Scholar] [CrossRef]
- Ehteda, A.; Simon, S.; Franshaw, L.; Giorgi, F.M.; Liu, J.; Joshi, S.; Rouaen, J.R.C.; Pang, C.N.I.; Pandher, R.; Mayoh, C.; et al. Dual Targeting of the Epigenome via FACT Complex and Histone Deacetylase Is a Potent Treatment Strategy for DIPG. Cell Rep. 2021, 35, 108994. [Google Scholar] [CrossRef]
- Forgione, M.O.; McClure, B.J.; Page, E.C.; Yeung, D.T.; Eadie, L.N.; White, D.L. TP53 Loss-of-function Mutations Reduce Sensitivity of Acute Leukaemia to the Curaxin CBL0137. Oncol. Rep. 2022, 47, 99. [Google Scholar] [CrossRef]
- Lu, X.; He, Y.; Johnston, R.L.; Nanayakarra, D.; Sankarasubramanian, S.; Lopez, J.A.; Friedlander, M.; Kalimutho, M.; Hooper, J.D.; Raninga, P.V.; et al. CBL0137 Impairs Homologous Recombination Repair and Sensitizes High-Grade Serous Ovarian Carcinoma to PARP Inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 355. [Google Scholar] [CrossRef]
- Zhou, D.; Wu, Z.; Park, J.-G.; Fiches, G.N.; Li, T.-W.; Ma, Q.; Huang, H.; Biswas, A.; Martinez-Sobrido, L.; Santoso, N.G.; et al. FACT Subunit SUPT16H Associates with BRD4 and Contributes to Silencing of Interferon Signaling. Nucleic Acids Res. 2022, 50, 8700–8718. [Google Scholar] [CrossRef]
- Huang, H.; Santoso, N.; Power, D.; Simpson, S.; Dieringer, M.; Miao, H.; Gurova, K.; Giam, C.-Z.; Elledge, S.J.; Zhu, J. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J. Biol. Chem. 2015, 290, 27297–27310. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, W.; Xie, D.; Gao, T.; Dong, Z.; Lu, X. Histone Chaperone FACT Represses Retrotransposon MERVL and MERVL-Derived Cryptic Promoters. Nucleic Acids Res. 2020, 48, 10211–10225. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C.; et al. ADAR1 Masks the Cancer Immunotherapeutic Promise of ZBP1-Driven Necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or Opponent in the Tumour Microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef]
- Shen, J.; Yang, C.; Zhang, M.S.; Chin, D.W.-C.; Chan, F.-F.; Law, C.-T.; Wang, G.; Cheng, C.L.-H.; Chen, M.; Wan, R.T.-C.; et al. Histone Chaperone FACT Complex Coordinates with HIF to Mediate an Expeditious Transcription Program to Adapt to Poorly Oxygenated Cancers. Cell Rep. 2022, 38, 110304. [Google Scholar] [CrossRef]
- Krause, D.R.; Jonnalagadda, J.C.; Gatei, M.H.; Sillje, H.H.W.; Zhou, B.-B.; Nigg, E.A.; Khanna, K. Suppression of Tousled-like Kinase Activity after DNA Damage or Replication Block Requires ATM, NBS1 and Chk1. Oncogene 2003, 22, 5927–5937. [Google Scholar] [CrossRef]
- Groth, A.; Lukas, J.; Nigg, E.A.; Silljé, H.H.W.; Wernstedt, C.; Bartek, J.; Hansen, K. Human Tousled like Kinases Are Targeted by an ATM- and Chk1-Dependent DNA Damage Checkpoint. EMBO J. 2003, 22, 1676–1687. [Google Scholar] [CrossRef]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A Phospho-Proteomic Screen Identifies Substrates of the Checkpoint Kinase Chk1. Genome Biol. 2011, 12, R78. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Stracker, T.H. The Tousled-like Kinases Regulate Genome and Epigenome Stability: Implications in Development and Disease. Cell. Mol. Life Sci. 2019, 76, 3827–3841. [Google Scholar] [CrossRef]
- Ghosh, I.; De Benedetti, A. Untousling the Role of Tousled-like Kinase 1 in DNA Damage Repair. Int. J. Mol. Sci. 2023, 24, 13369. [Google Scholar] [CrossRef] [PubMed]
- Canfield, C.; Rains, J.; De Benedetti, A. TLK1B Promotes Repair of DSBs via Its Interaction with Rad9 and Asf1. BMC Mol. Biol. 2009, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Sunavala-Dossabhoy, G.; De Benedetti, A. Tousled Homolog, TLK1, Binds and Phosphorylates Rad9; TLK1 Acts as a Molecular Chaperone in DNA Repair. DNA Repair 2009, 8, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.; Davey, S.K. Tousled-like Kinase-Dependent Phosphorylation of Rad9 Plays a Role in Cell Cycle Progression and G2/M Checkpoint Exit. PLoS ONE 2013, 8, e85859. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Kwon, Y.; Shabestari, A.B.; Chikhale, R.; Chen, J.; Wiese, C.; Sung, P.; De Benedetti, A. TLK1-Mediated RAD54 Phosphorylation Spatio-Temporally Regulates Homologous Recombination Repair. Nucleic Acids Res. 2023, 51, 8643–8662. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-B.; Segura-Bayona, S.; Villamor-Payà, M.; Saredi, G.; Todd, M.A.M.; Attolini, C.S.-O.; Chang, T.-Y.; Stracker, T.H.; Groth, A. Tousled-like Kinases Stabilize Replication Forks and Show Synthetic Lethality with Checkpoint and PARP Inhibitors. Sci. Adv. 2018, 4, eaat4985. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Villamor-Payà, M.; Attolini, C.S.-O.; Koenig, L.M.; Sanchiz-Calvo, M.; Boulton, S.J.; Stracker, T.H. Tousled-Like Kinases Suppress Innate Immune Signaling Triggered by Alternative Lengthening of Telomeres. Cell Rep. 2020, 32, 107983. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Luster, T.A.; Hu, H.; Zhang, H.; Ng, W.-L.; Khodadadi-Jamayran, A.; Wang, W.; Chen, T.; Deng, J.; et al. In Vivo Epigenetic CRISPR Screen Identifies Asf1a as an Immunotherapeutic Target in Kras-Mutant Lung Adenocarcinoma. Cancer Discov. 2020, 10, 270–287. [Google Scholar] [CrossRef]
- Kim, J.-A.; Tan, Y.; Wang, X.; Cao, X.; Veeraraghavan, J.; Liang, Y.; Edwards, D.P.; Huang, S.; Pan, X.; Li, K.; et al. Comprehensive Functional Analysis of the Tousled-like Kinase 2 Frequently Amplified in Aggressive Luminal Breast Cancers. Nat. Commun. 2016, 7, 12991. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR Axis with Thioridazine Suppresses Outgrowth of Androgen Independent Prostate Tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23, 101474. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-B.; Chang, T.-Y.; Lee, N.-Z.; Yu, Z.-Y.; Liu, C.-Y.; Lee, H.-Y. Design, Synthesis and Biological Evaluation of Bisindole Derivatives as Anticancer Agents against Tousled-like Kinases. Eur. J. Med. Chem. 2022, 227, 113904. [Google Scholar] [CrossRef] [PubMed]
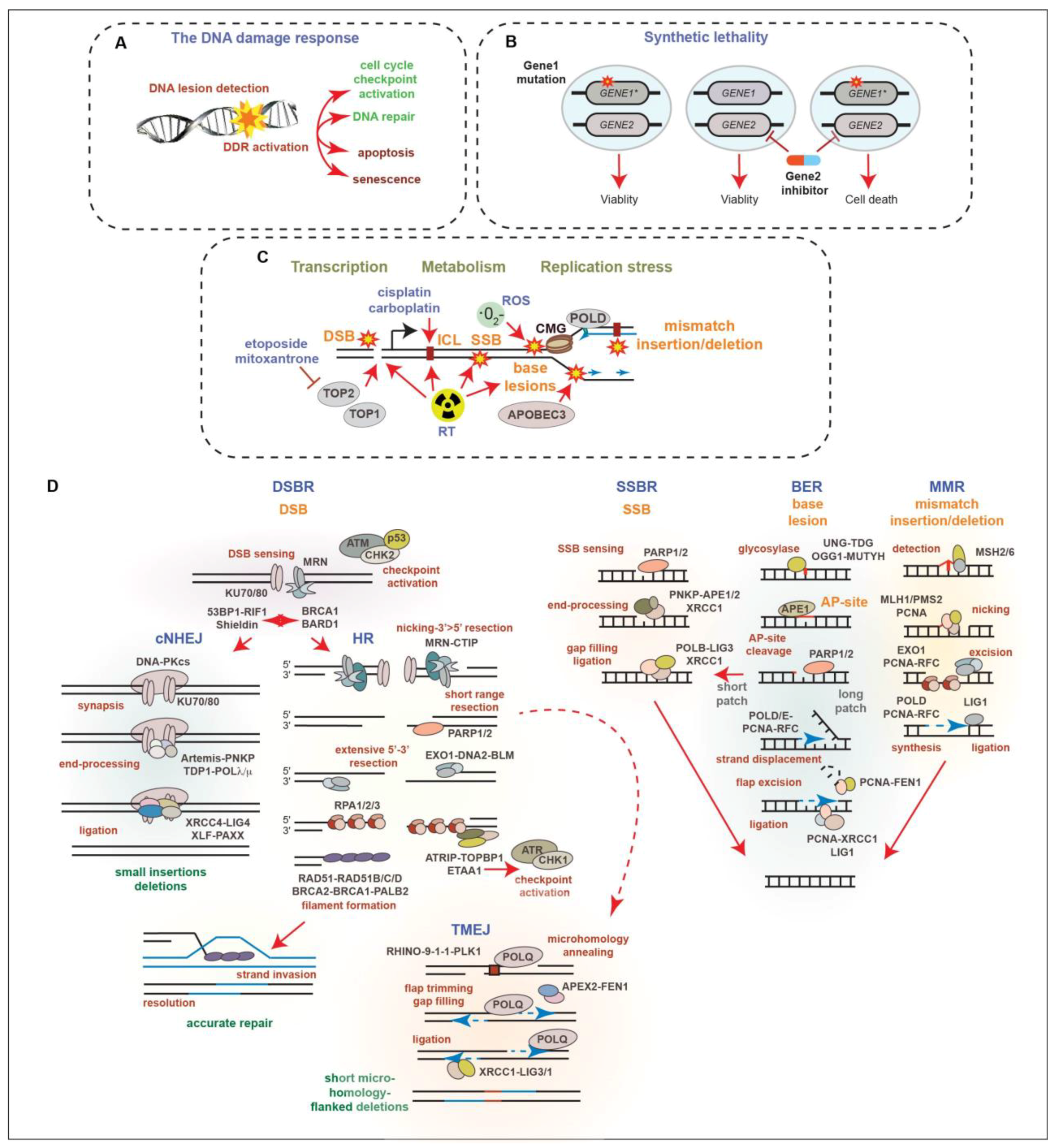
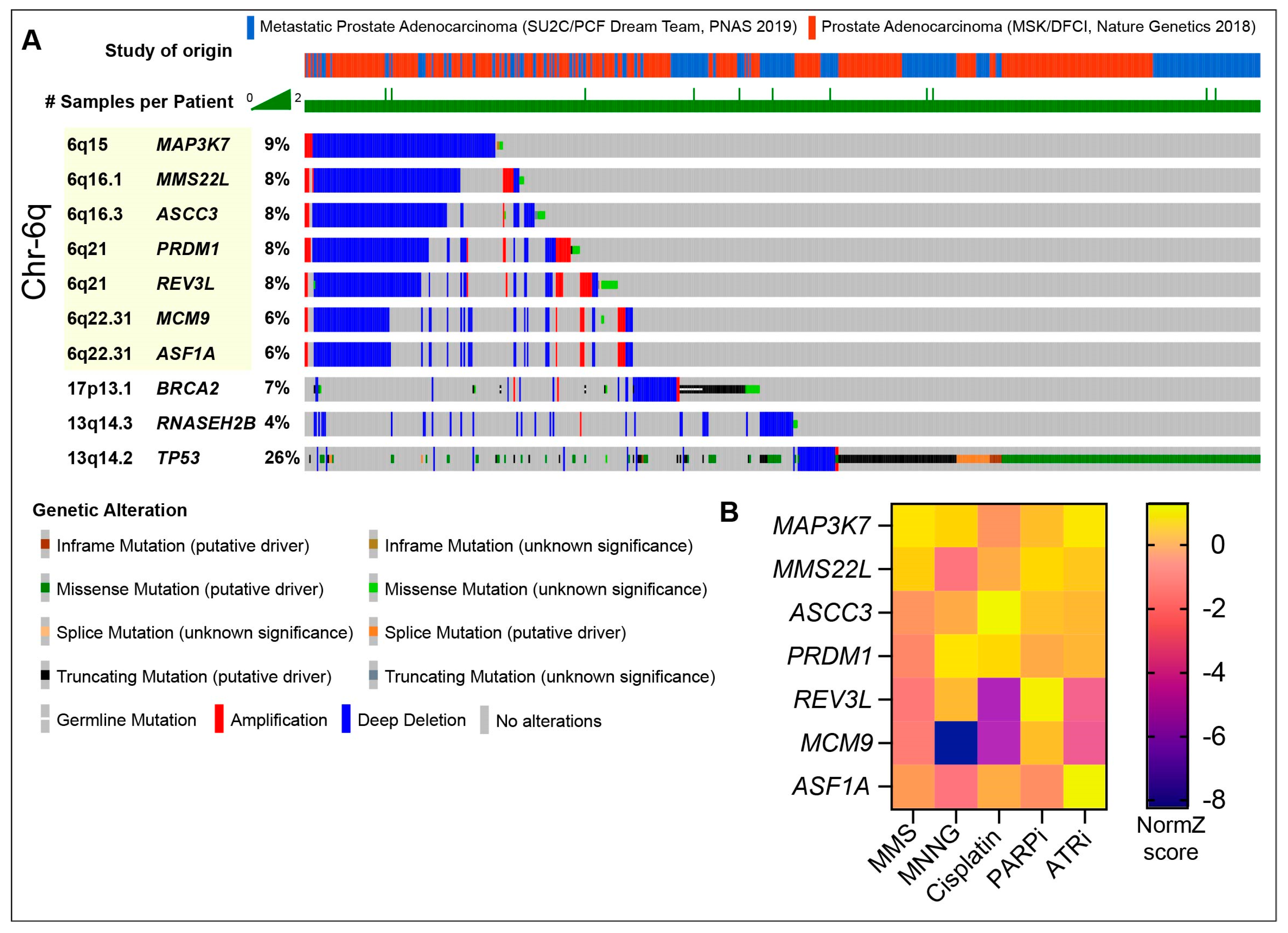
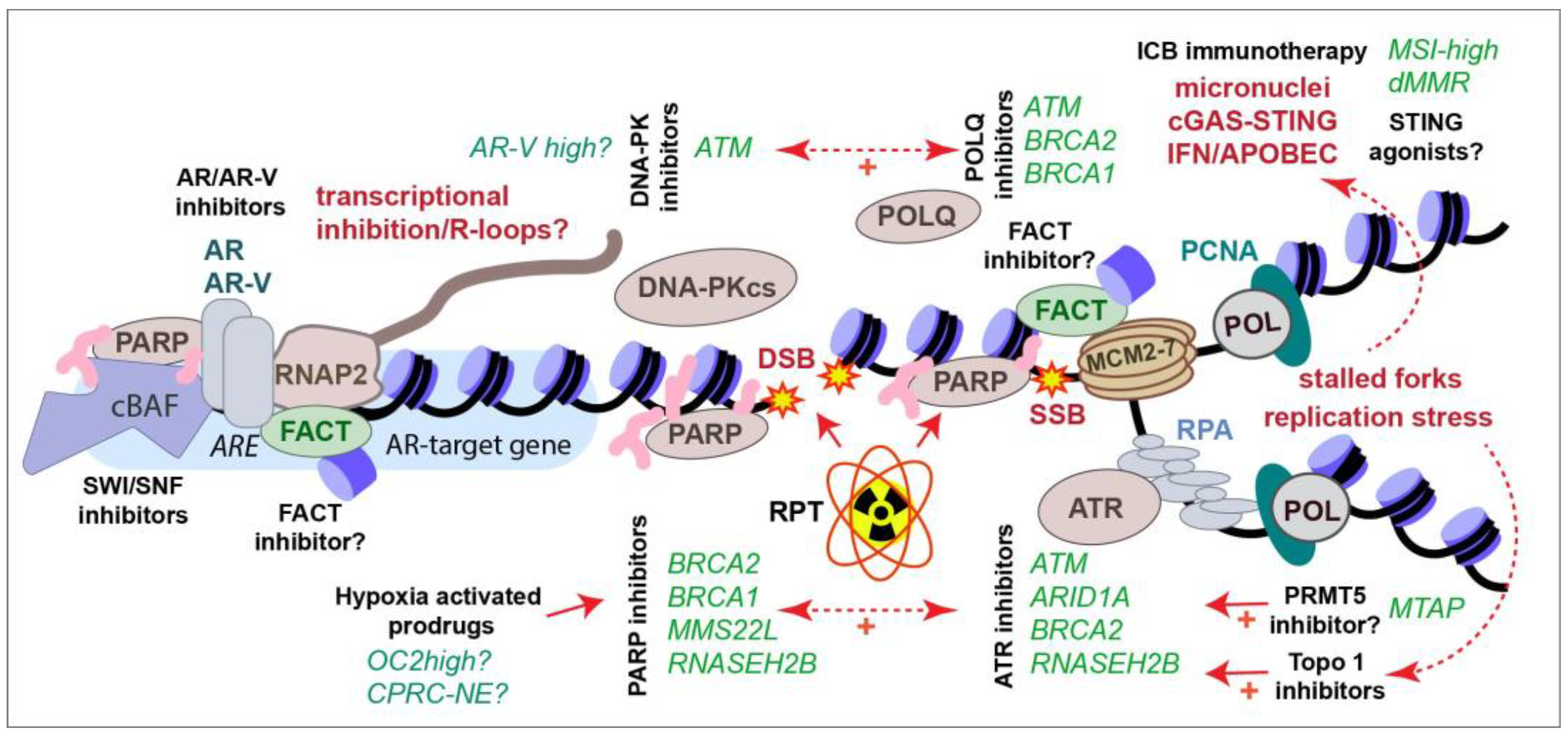
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stracker, T.H.; Osagie, O.I.; Escorcia, F.E.; Citrin, D.E. Exploiting the DNA Damage Response for Prostate Cancer Therapy. Cancers 2024, 16, 83. https://doi.org/10.3390/cancers16010083
Stracker TH, Osagie OI, Escorcia FE, Citrin DE. Exploiting the DNA Damage Response for Prostate Cancer Therapy. Cancers. 2024; 16(1):83. https://doi.org/10.3390/cancers16010083
Chicago/Turabian StyleStracker, Travis H., Oloruntoba I. Osagie, Freddy E. Escorcia, and Deborah E. Citrin. 2024. "Exploiting the DNA Damage Response for Prostate Cancer Therapy" Cancers 16, no. 1: 83. https://doi.org/10.3390/cancers16010083
APA StyleStracker, T. H., Osagie, O. I., Escorcia, F. E., & Citrin, D. E. (2024). Exploiting the DNA Damage Response for Prostate Cancer Therapy. Cancers, 16(1), 83. https://doi.org/10.3390/cancers16010083