
A Novel Tissue-Free Method to Estimate Tumor-Derived Cell-Free DNA Quantity Using Tumor Methylation Patterns

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Origin and Prior Processing
2.2. Data Processing and Statistical Analysis
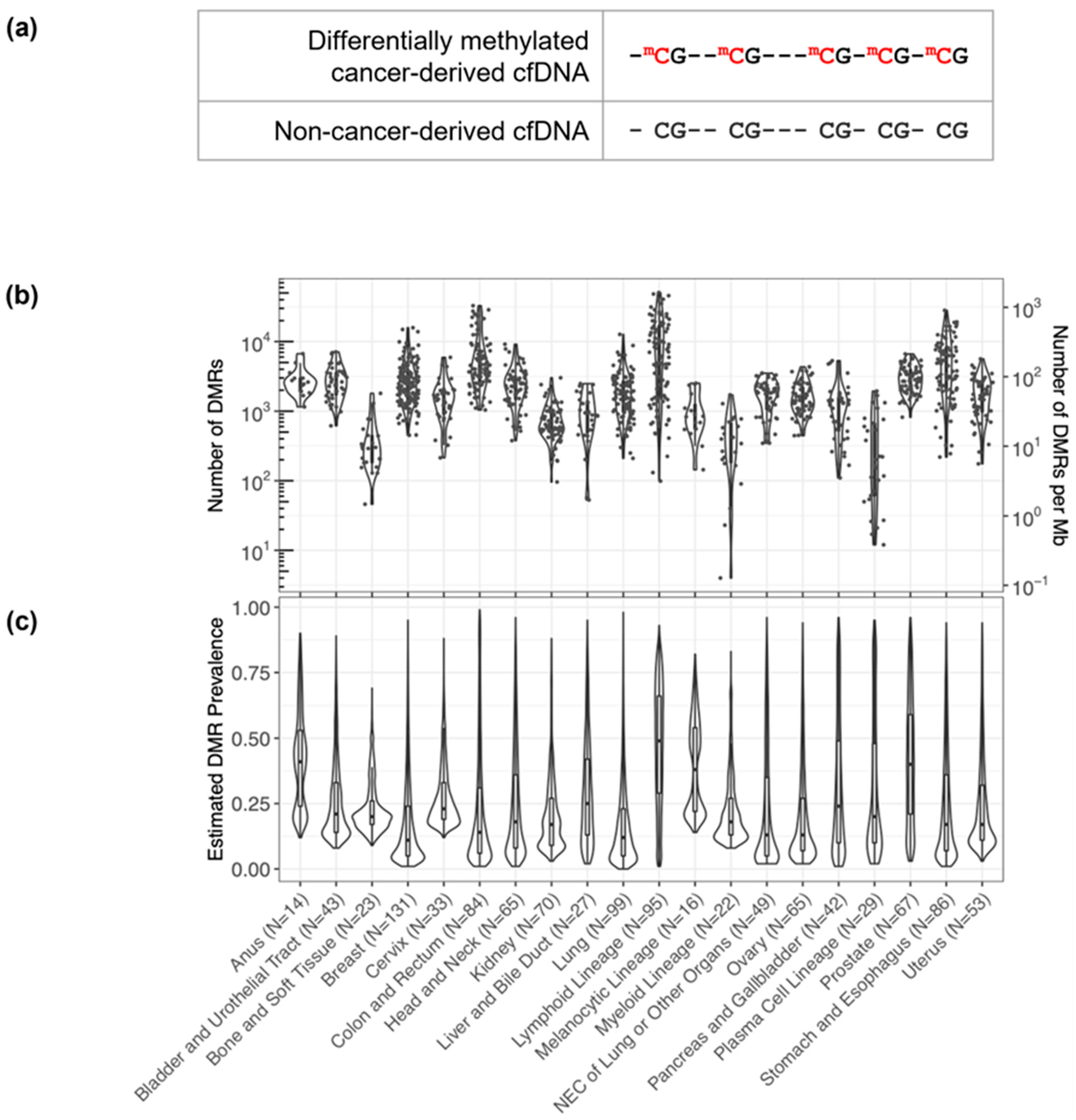
2.3. Data Processing and Statistical Analysis—Differentially Methylated Region Calling
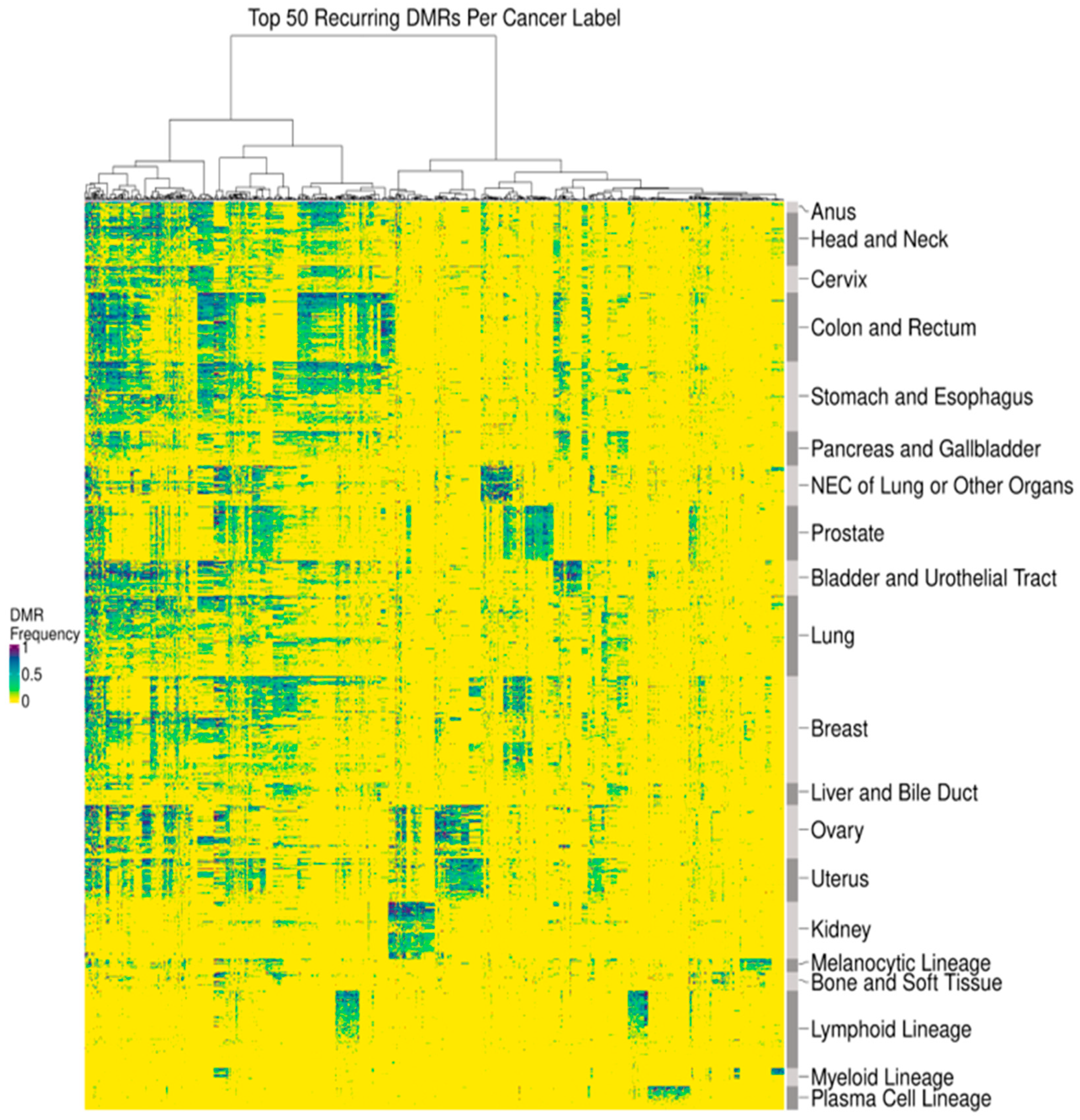
2.4. Data Processing and Statistical Analysis—DMR Clustering
2.5. Data Processing and Statistical Analysis—DMR Heme Filtering
2.6. Data Processing and Statistical Analysis—DMR Prevalence Estimation
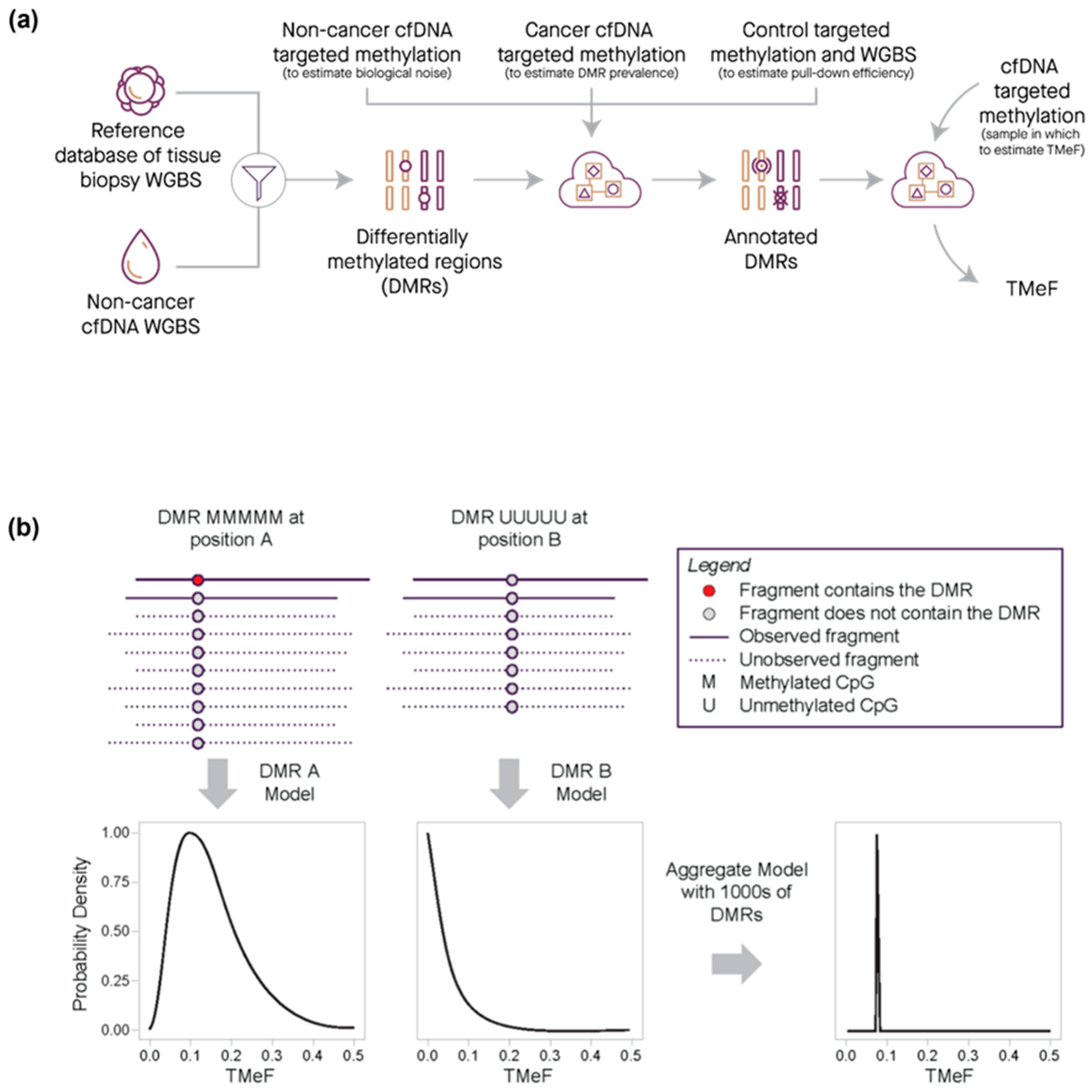
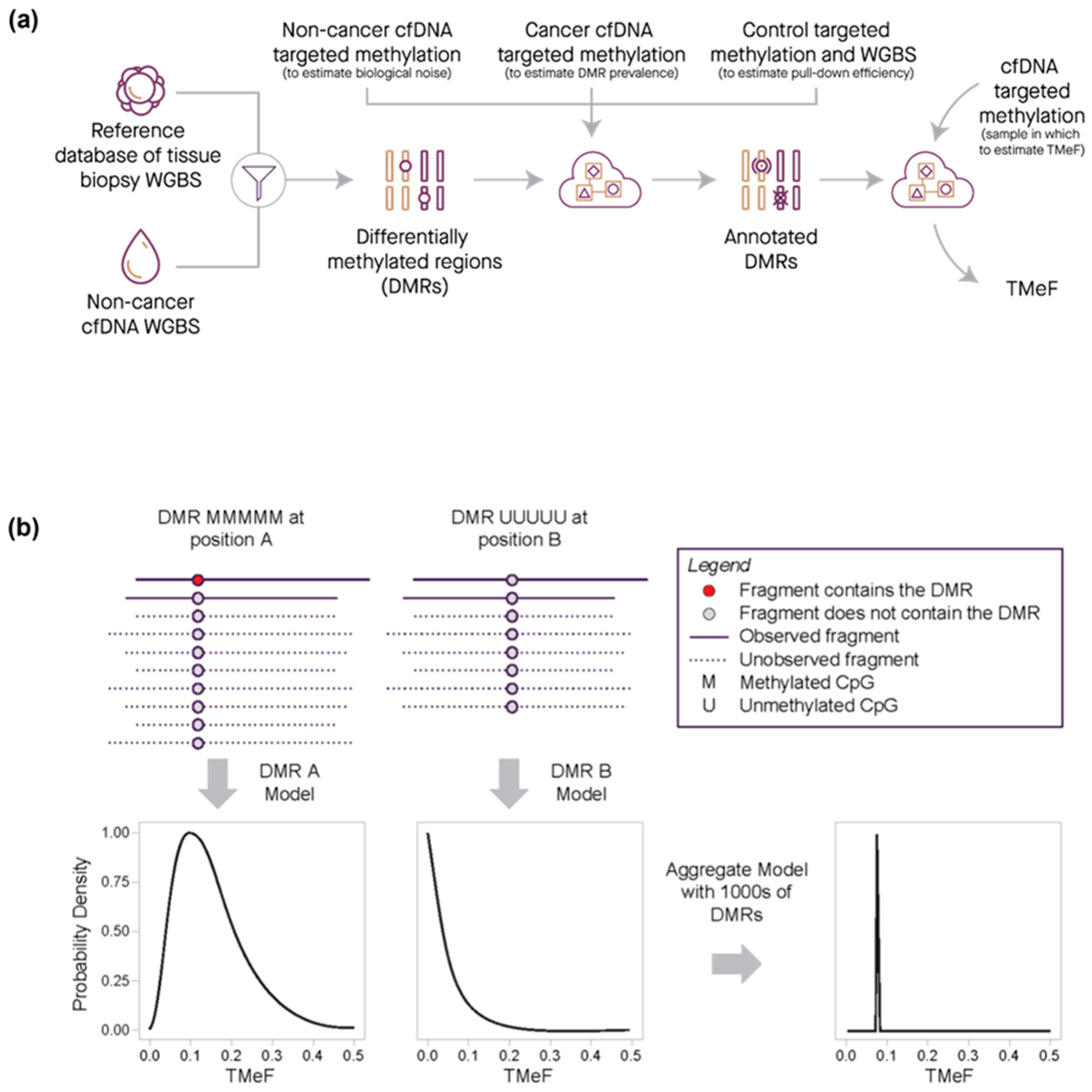
2.7. Data Processing and Statistical Analysis—Tumor Methylated Fraction Estimation
2.8. Synthetic Dilutions
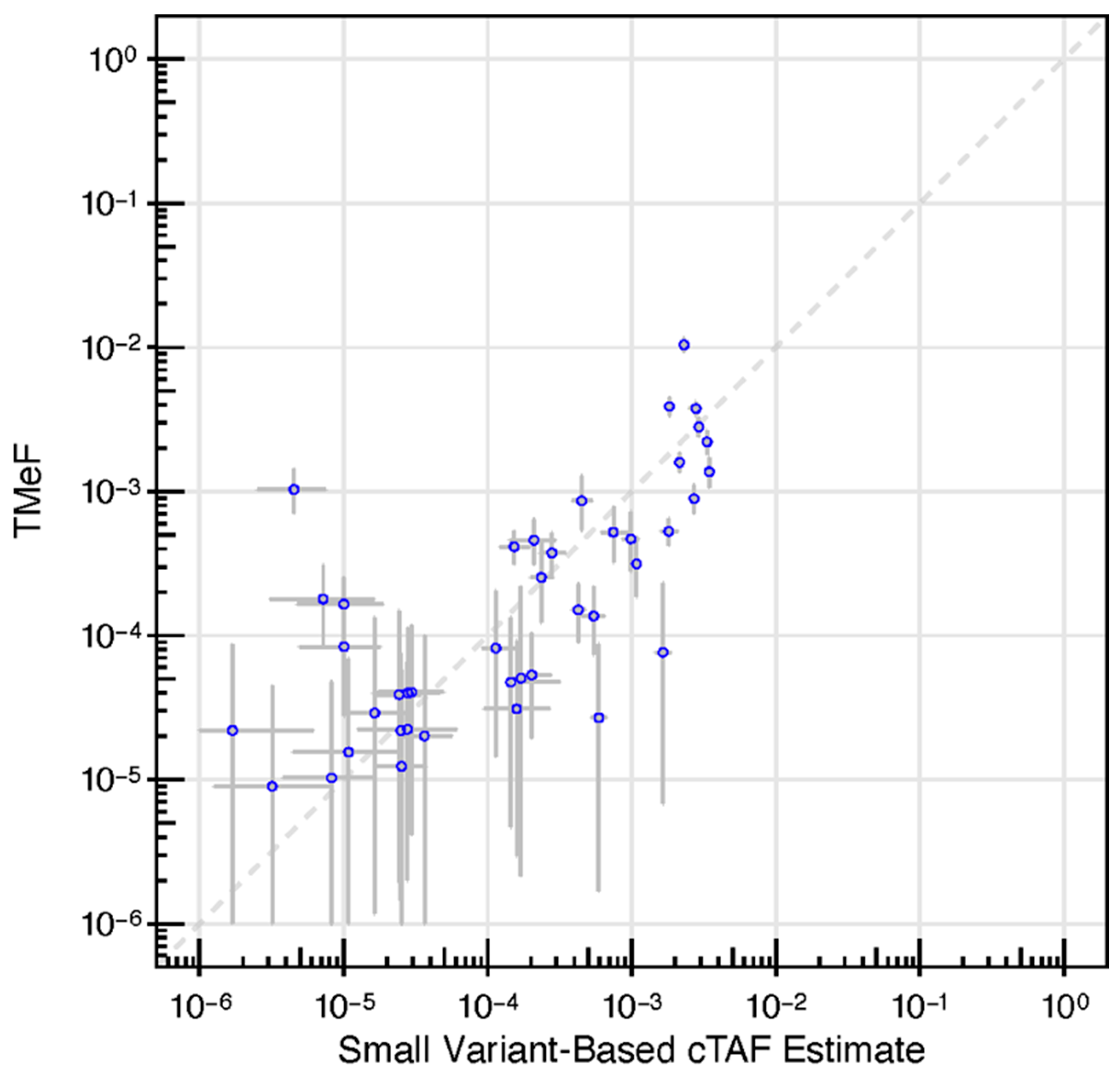
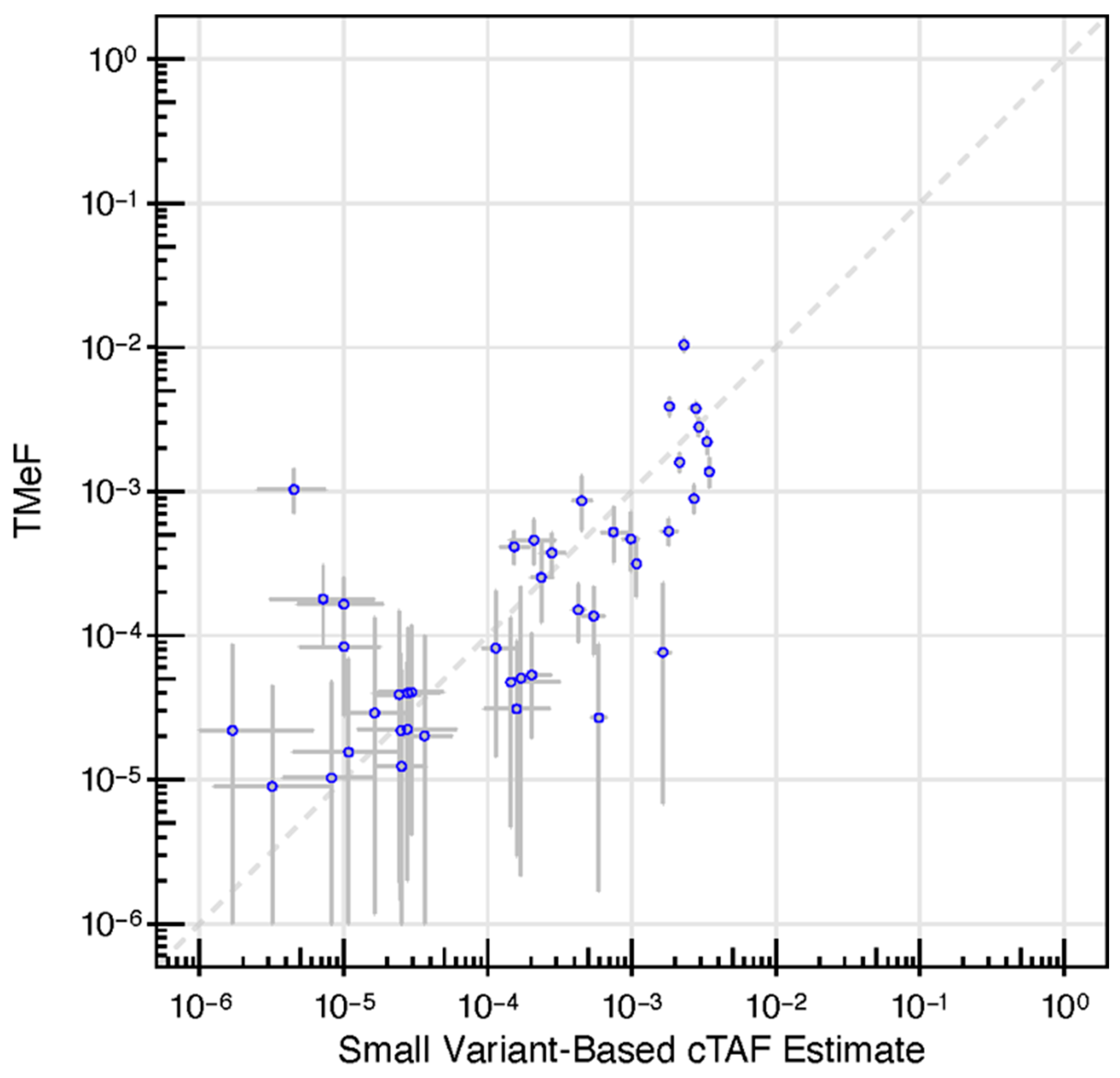
2.9. Small Variant Allele Fraction Estimates
2.10. Biophysical Modeling of ctDNA Shedding
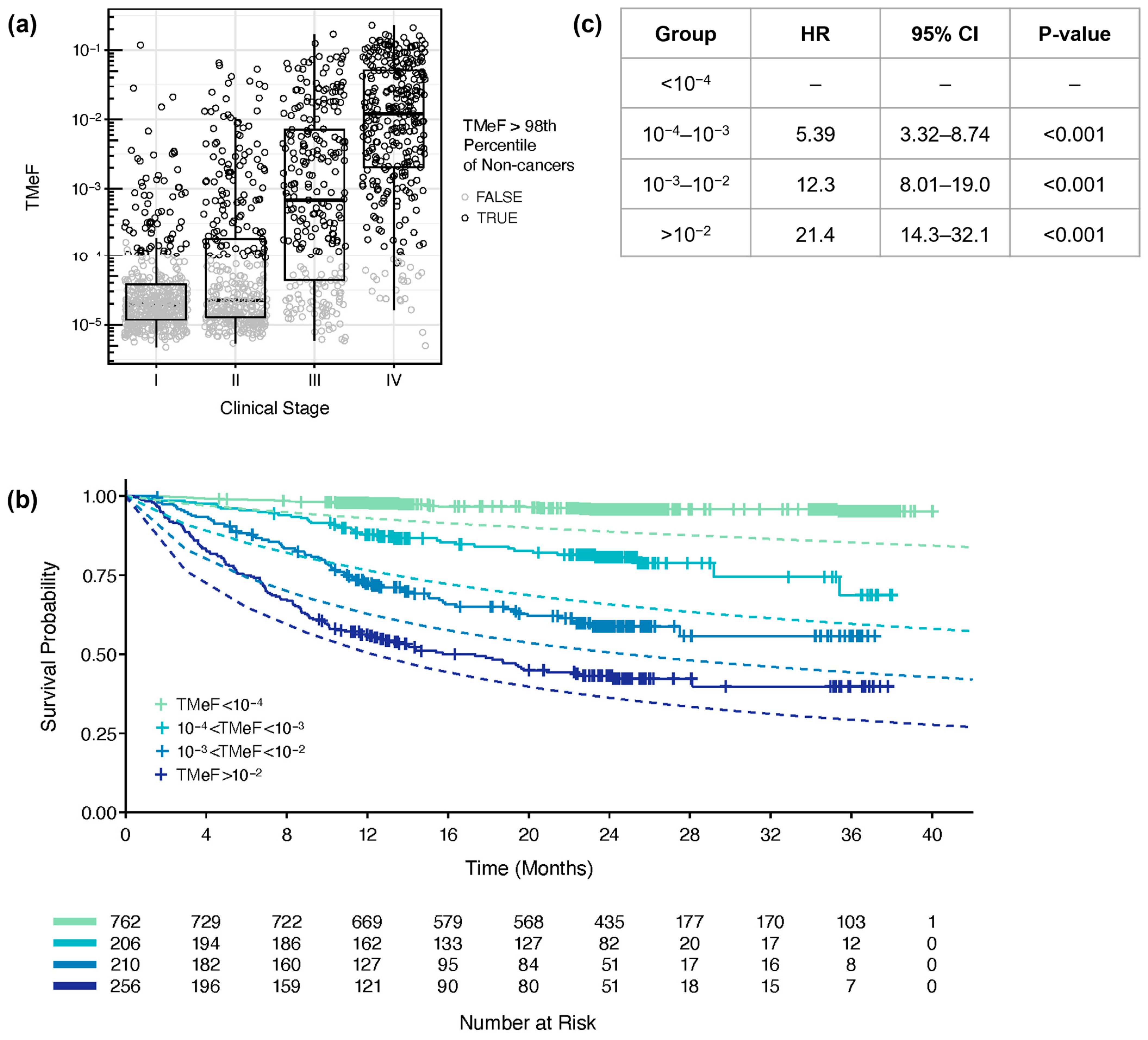
2.11. Survival Modeling
3. Results
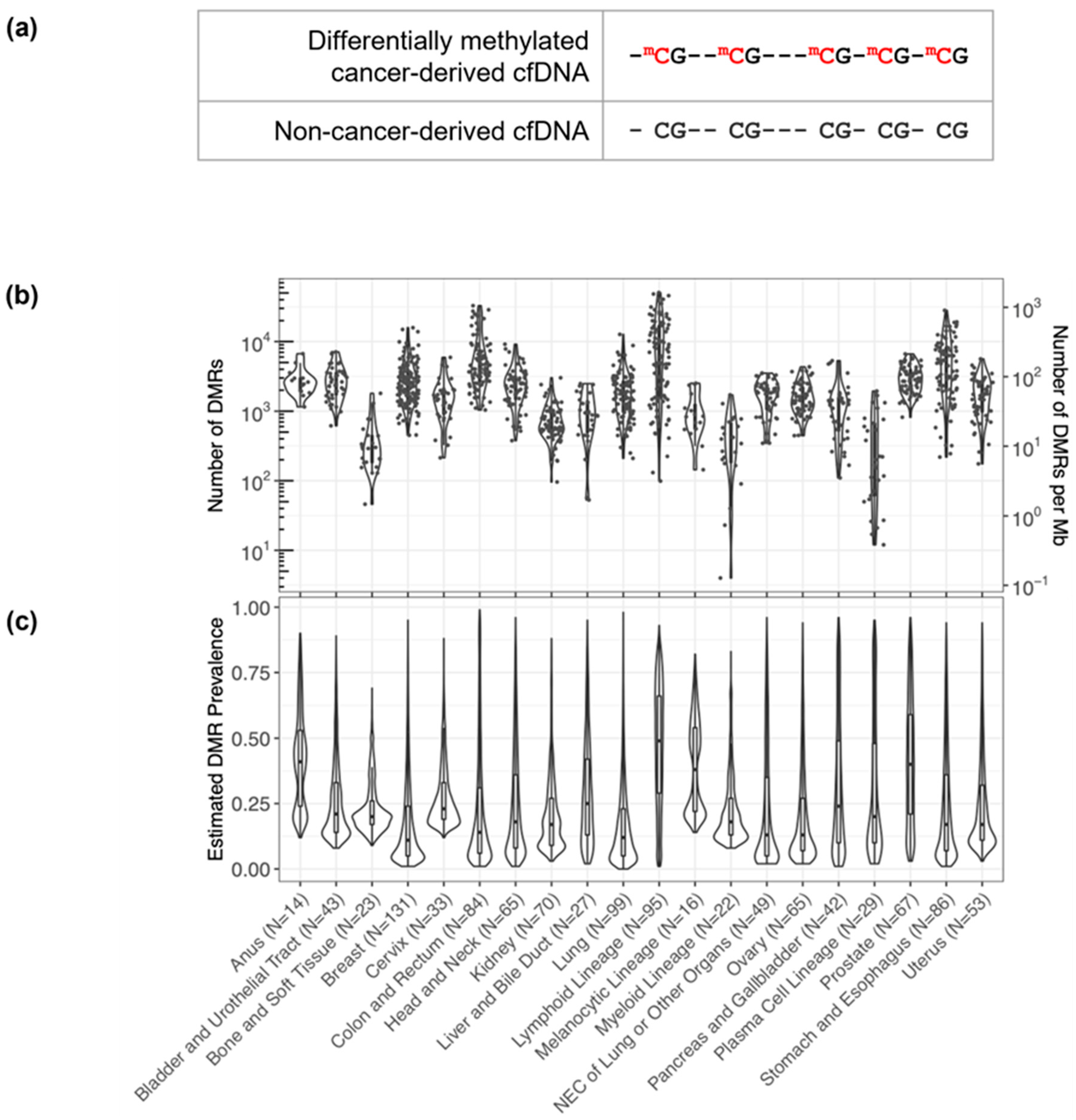
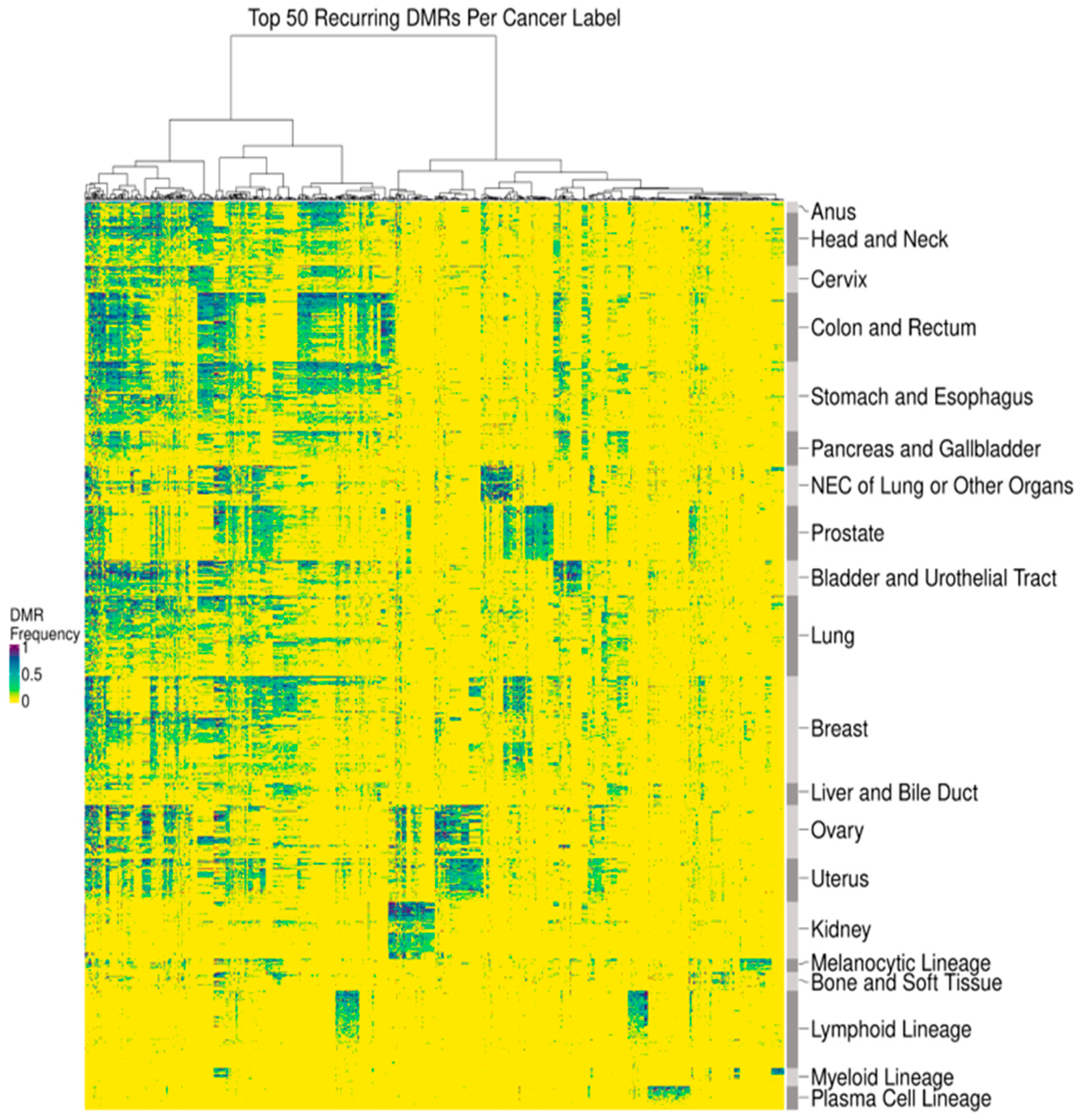
3.1. DMRs Are Diverse and Cluster by Cancer Type
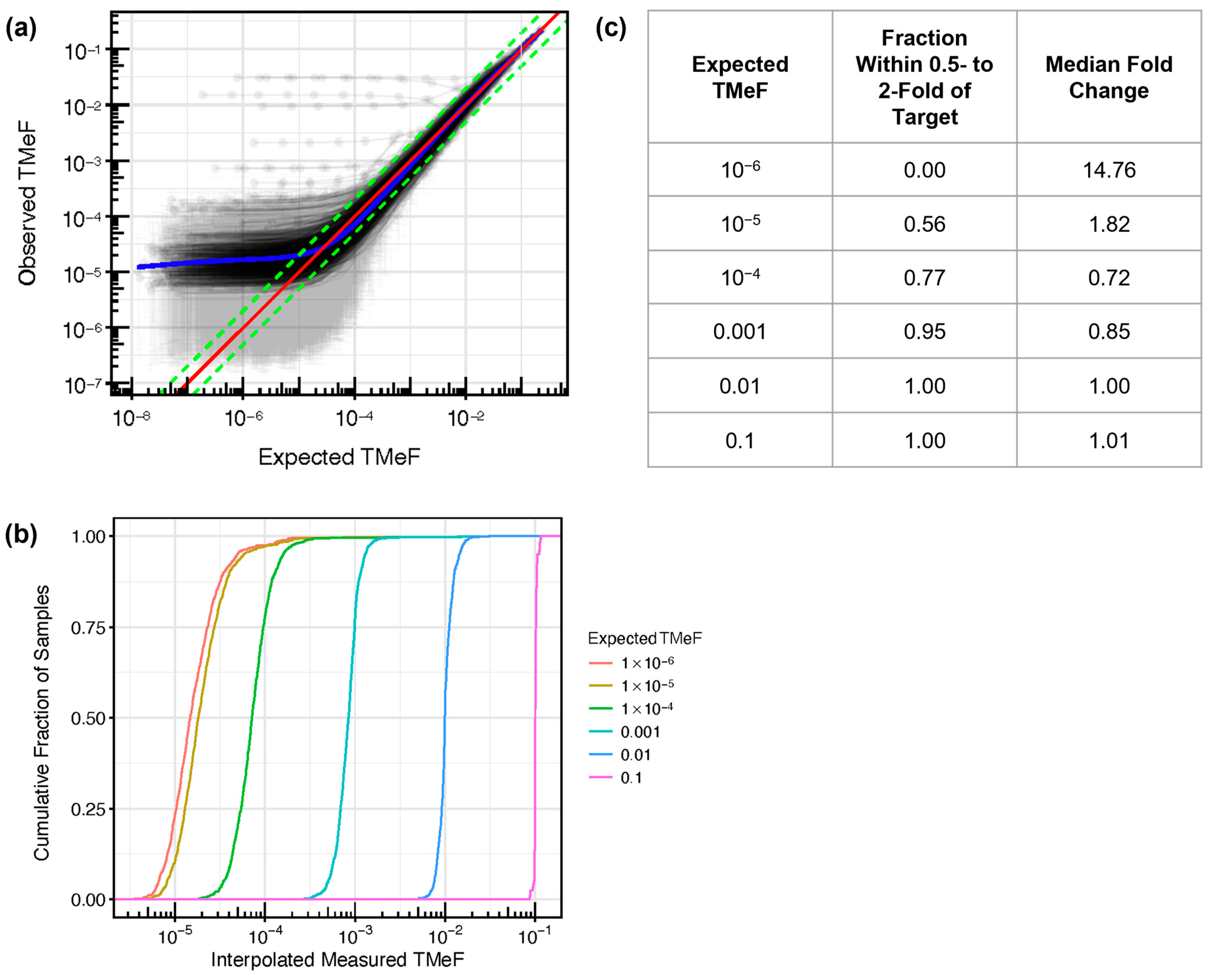
3.2. TMeF Can Accurately Quantify ctDNA Abundance
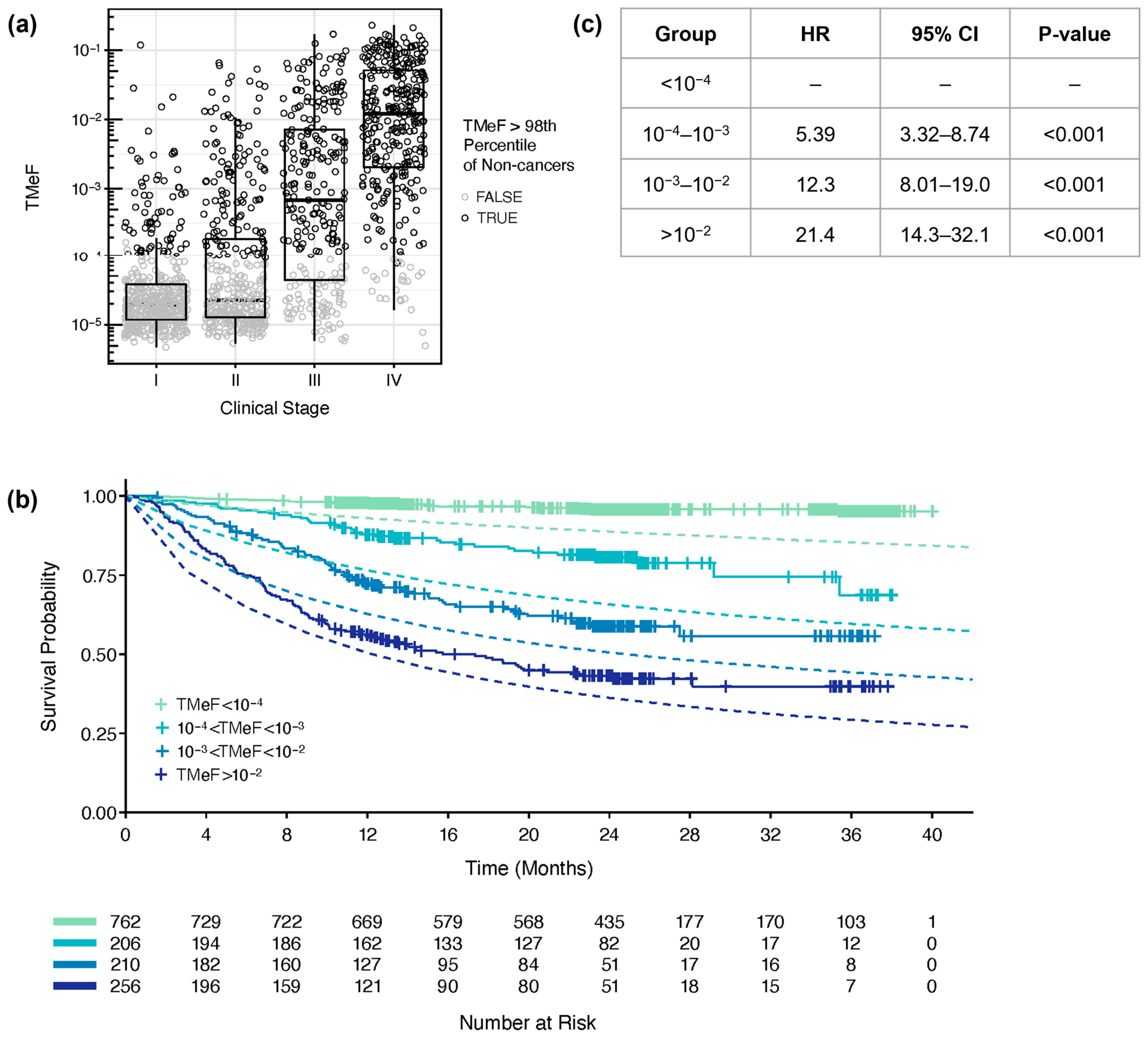
3.3. TMeF Is Associated with Clinical Stage and Tumor Size
3.4. TMeF Is Associated with Overall Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Jia, B.; Zhang, X.; Mo, Y.; Chen, B.; Long, H.; Rong, T.; Su, X. The Study of Tumor Volume as a Prognostic Factor in T Staging System for Non-Small Cell Lung Cancer: An Exploratory Study. Technol. Cancer Res. Treat. 2020, 19, 1533033820980106. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A. Tumour Size Predicts Long-Term Survival among Women with Lymph Node-Positive Breast Cancer. Curr. Oncol. 2012, 19, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Dall’Olio, F.G.; Marabelle, A.; Caramella, C.; Garcia, C.; Aldea, M.; Chaput, N.; Robert, C.; Besse, B. Tumour Burden and Efficacy of Immune-Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2022, 19, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.-J.; Tsui, D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer Oxf. Engl. 1990 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.C.; Schwartz, L.H.; Zhao, B. The Imaging Viewpoint: How Imaging Affects Determination of Progression-Free Survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2621–2628. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Heuvelmans, M.A.; Oudkerk, M. Volume versus Diameter Assessment of Small Pulmonary Nodules in CT Lung Cancer Screening. Transl. Lung Cancer Res. 2017, 6, 52–61. [Google Scholar] [CrossRef] [PubMed]
- James, K.; Eisenhauer, E.; Christian, M.; Terenziani, M.; Vena, D.; Muldal, A.; Therasse, P. Measuring Response in Solid Tumors: Unidimensional versus Bidimensional Measurement. J. Natl. Cancer Inst. 1999, 91, 523–528. [Google Scholar] [CrossRef]
- Jaffe, C.C. Measures of Response: RECIST, WHO, and New Alternatives. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3245–3251. [Google Scholar] [CrossRef]
- Sargent, D.J.; Rubinstein, L.; Schwartz, L.; Dancey, J.E.; Gatsonis, C.; Dodd, L.E.; Shankar, L.K. Validation of Novel Imaging Methodologies for Use as Cancer Clinical Trial End-Points. Eur. J. Cancer Oxf. Engl. 1990 2009, 45, 290–299. [Google Scholar] [CrossRef]
- Boellaard, R.; Delgado-Bolton, R.; Oyen, W.J.G.; Giammarile, F.; Tatsch, K.; Eschner, W.; Verzijlbergen, F.J.; Barrington, S.F.; Pike, L.C.; Weber, W.A.; et al. FDG PET/CT: EANM Procedure Guidelines for Tumour Imaging: Version 2.0. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 328–354. [Google Scholar] [CrossRef]
- Hofman, M.S.; Lawrentschuk, N.; Francis, R.J.; Tang, C.; Vela, I.; Thomas, P.; Rutherford, N.; Martin, J.M.; Frydenberg, M.; Shakher, R.; et al. Prostate-Specific Membrane Antigen PET-CT in Patients with High-Risk Prostate Cancer before Curative-Intent Surgery or Radiotherapy (proPSMA): A Prospective, Randomised, Multicentre Study. Lancet Lond. Engl. 2020, 395, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Petrou, M.; Quint, L.E.; Nan, B.; Baker, L.H. Pulmonary Nodule Volumetric Measurement Variability as a Function of CT Slice Thickness and Nodule Morphology. Am. J. Roentgenol. 2007, 188, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Zhao, B.; Sima, C.S.; Ginsberg, M.S.; James, L.P.; Lefkowitz, R.A.; Guo, P.; Kris, M.G.; Schwartz, L.H.; Riely, G.J. Variability of Lung Tumor Measurements on Repeat Computed Tomography Scans Taken within 15 Minutes. J. Clin. Oncol. 2011, 29, 3114–3119. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Guo, P.; Mann, H.; Marley, S.E.; Juanita Scott, M.L.; Schwartz, L.H.; Ghiorghiu, D.C.; Zhao, B. Assessing the Effect of CT Slice Interval on Unidimensional, Bidimensional and Volumetric Measurements of Solid Tumours. Cancer Imaging 2012, 12, 497–505. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration Computed Tomography (CT). Available online: https://www.fda.gov/radiation-emitting-products/medical-x-ray-imaging/computed-tomography-ct#3 (accessed on 6 October 2023).
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A Mathematical Model of ctDNA Shedding Predicts Tumor Detection Size. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yu, Y.; Shen, M.; Liu, M.; Wu, S.; Liang, L.; Huang, F.; Zhang, C.; Guo, W.; Liu, T. Role of Circulating Free DNA in Evaluating Clinical Tumor Burden and Predicting Survival in Chinese Metastatic Colorectal Cancer Patients. BMC Cancer 2020, 20, 1006. [Google Scholar] [CrossRef]
- Symonds, E.L.; Pedersen, S.K.; Yeo, B.; Al Naji, H.; Byrne, S.E.; Roy, A.; Young, G.P. Assessment of Tumor Burden and Response to Therapy in Patients with Colorectal Cancer Using a Quantitative ctDNA Test for Methylated BCAT1/IKZF1. Mol. Oncol. 2022, 16, 2031–2041. [Google Scholar] [CrossRef]
- Bredno, J.; Lipson, J.; Venn, O.; Aravanis, A.M.; Jamshidi, A. Clinical Correlates of Circulating Cell-Free DNA Tumor Fraction. PLoS ONE 2021, 16, e0256436. [Google Scholar] [CrossRef]
- Bredno, J.; Venn, O.; Chen, X.; Freese, P.; Ofman, J.J. Circulating Tumor DNA Allele Fraction: A Candidate Biological Signal for Multicancer Early Detection Tests to Assess the Clinical Significance of Cancers. Am. J. Pathol. 2022, 192, 1368–1378. [Google Scholar] [CrossRef]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating Genomic Features for Non-Invasive Early Lung Cancer Detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e13. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Dong, Z.; Hubbell, E.; Kurtzman, K.N.; Oxnard, G.R.; Venn, O.; Melton, C.; Clarke, C.A.; Shaknovich, R.; Ma, T.; et al. Prognostic Significance of Blood-Based Multi-Cancer Detection in Plasma Cell-Free DNA. Clin. Cancer Res. 2021, 27, 4221–4229. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.-I.; Chen, K.; Usmani, A.; Chua, C.; Harris, P.K.; Binkley, M.S.; Azad, T.D.; Dudley, J.C.; Chaudhuri, A.A. Detection of Solid Tumor Molecular Residual Disease (MRD) Using Circulating Tumor DNA (ctDNA). Mol. Diagn. Ther. 2019, 23, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Soo, J.; Co Ting Keh, L.; Alig, S.; Chabon, J.J.; Sworder, B.J.; Schultz, A.; Jin, M.C.; Scherer, F.; Garofalo, A.; et al. Enhanced Detection of Minimal Residual Disease by Targeted Sequencing of Phased Variants in Circulating Tumor DNA. Nat. Biotechnol. 2021, 39, 1537–1547. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Heider, K.; Gale, D.; Murphy, S.; Fisher, E.; Mouliere, F.; Ruiz-Valdepenas, A.; Santonja, A.; Morris, J.; Chandrananda, D.; et al. ctDNA Monitoring Using Patient-Specific Sequencing and Integration of Variant Reads. Sci. Transl. Med. 2020, 12, eaaz8084. [Google Scholar] [CrossRef] [PubMed]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.K.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J.; et al. Genome-Wide Cell-Free DNA Mutational Integration Enables Ultra-Sensitive Cancer Monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef]
- Yadav, D.K.; Bai, X.; Yadav, R.K.; Singh, A.; Li, G.; Ma, T.; Chen, W.; Liang, T. Liquid Biopsy in Pancreatic Cancer: The Beginning of a New Era. Oncotarget 2018, 9, 26900–26933. [Google Scholar] [CrossRef]
- McLean, A.E.B.; Barnes, D.J.; Troy, L.K. Diagnosing Lung Cancer: The Complexities of Obtaining a Tissue Diagnosis in the Era of Minimally Invasive and Personalised Medicine. J. Clin. Med. 2018, 7, 163. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Fairchild, L.; Whalen, J.; D’Aco, K.; Wu, J.; Gustafson, C.B.; Solovieff, N.; Su, F.; Leary, R.J.; Campbell, C.D.; Balbin, O.A. Clonal Hematopoiesis Detection in Patients with Cancer Using Cell-Free DNA Sequencing. Sci. Transl. Med. 2023, 15, eabm8729. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Frankell, A.M.; Harrison, T.; Kisistok, J.; Garnett, A.; Johnson, L.; Veeriah, S.; Moreau, M.; Chesh, A.; Chaunzwa, T.L.; et al. Tracking Early Lung Cancer Metastatic Dissemination in TRACERx Using ctDNA. Nature 2023, 616, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Laird, P.W. Interplay between the Cancer Genome and Epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A Human DNA Methylation Atlas Reveals Principles of Cell Type-Specific Methylation and Identifies Thousands of Cell Type-Specific Regulatory Elements. bioRxiv 2022. bioRxiv:2022.01.24.477547. [Google Scholar]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Cummings, S.R.; Absalan, F.; Alexander, G.; Allen, B.; Amini, H.; et al. Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-Free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N. Clinical Validation of a Targeted Methylation-Based Multi-Cancer Early Detection Test Using an Independent Validation Set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive Human Cell-Type Methylation Atlas Reveals Origins of Circulating Cell-Free DNA in Health and Disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.-C.; Matsuoka, L.; Sher, L.; Wong, W.H.; et al. CancerDetector: Ultrasensitive and Non-Invasive Cancer Detection at the Resolution of Individual Reads Using Cell-Free DNA Methylation Sequencing Data. Nucleic Acids Res. 2018, 46, e89. [Google Scholar] [CrossRef]
- Liang, N.; Li, B.; Jia, Z.; Wang, C.; Wu, P.; Zheng, T.; Wang, Y.; Qiu, F.; Wu, Y.; Su, J.; et al. Ultrasensitive Detection of Circulating Tumour DNA via Deep Methylation Sequencing Aided by Machine Learning. Nat. Biomed. Eng. 2021, 5, 586–599. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.-L.; Zhang, K.; Zhang, K. Identification of Methylation Haplotype Blocks Aids in Deconvolution of Heterogeneous Tissue Samples and Tumor Tissue-of-Origin Mapping from Plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive Tumour Detection and Classification Using Plasma Cell-Free DNA Methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef]
- Zhou, X.; Cheng, Z.; Dong, M.; Liu, Q.; Yang, W.; Liu, M.; Tian, J.; Cheng, W. Tumor Fractions Deciphered from Circulating Cell-Free DNA Methylation for Cancer Early Diagnosis. Nat. Commun. 2022, 13, 7694. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jiang, P.; Chan, K.C.A.; Wong, J.; Cheng, Y.K.Y.; Liang, R.H.S.; Chan, W.; Ma, E.S.K.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA Tissue Mapping by Genome-Wide Methylation Sequencing for Noninvasive Prenatal, Cancer, and Transplantation Assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [PubMed]
- Keukeleire, P.; Makrodimitris, S.; Reinders, M. Cell Type Deconvolution of Methylated Cell-Free DNA at the Resolution of Individual Reads. NAR Genom. Bioinform. 2023, 5, lqad048. [Google Scholar] [CrossRef]
- Li, S.; Zeng, W.; Ni, X.; Liu, Q.; Li, W.; Stackpole, M.L.; Zhou, Y.; Gower, A.; Krysan, K.; Ahuja, P.; et al. Comprehensive Tissue Deconvolution of Cell-Free DNA by Deep Learning for Disease Diagnosis and Monitoring. Proc. Natl. Acad. Sci. USA 2023, 120, e2305236120. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, A.; Liu, M.C.; Klein, E.A.; Venn, O.; Hubbell, E.; Beausang, J.F.; Gross, S.; Melton, C.; Fields, A.P.; Liu, Q.; et al. Evaluation of Cell-Free DNA Approaches for Multi-Cancer Early Detection. Cancer Cell 2022, 40, 1537–1549.e12. [Google Scholar] [CrossRef] [PubMed]
- Calef, R.; Gross, S.; Fields, A.; Zhao, M.; Betts, C.; Burkhardt, D.; Venn, O. Multi-Cancer Measurement of Low Tumor Fraction in Cell-Free DNA Using Personalized Targeted Sequencing. In preparation.
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement from the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef]
- Ye, X.; Li, W.; Zhang, L.; Yu, J. Clinical Significance of Circulating Cell-Free DNA Detection in Multiple Myeloma: A Meta-Analysis. Front. Oncol. 2022, 12, 852573. [Google Scholar] [CrossRef]
- Smith, J.T.; Balar, A.; Lakhani, D.A.; Kluwe, C.; Zhao, Z.; Kopparapu, P.; Almodovar, K.; Muterspaugh, A.; Yan, Y.; York, S.; et al. Circulating Tumor DNA as a Potential Biomarker of Radiographic Tumor Burden in Small Cell Lung Cancer. Cancer Res. 2020, 80, 715. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.; Wu, H.-T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients with Urothelial Bladder Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Thankamony, A.P.; Subbalakshmi, A.R.; Jolly, M.K.; Nair, R. Lineage Plasticity in Cancer: The Tale of a Skin-Walker. Cancers 2021, 13, 3602. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Turner, R.; Chen, Y.-W.; Rigas, J.R.; Fernandes, A.W.; Karve, S. Complications and Economic Burden Associated with Obtaining Tissue for Diagnosis and Molecular Analysis in Patients with Non-Small-Cell Lung Cancer in the United States. J. Oncol. Pract. 2019, 15, e717–e727. [Google Scholar] [CrossRef] [PubMed]
- Nørgaard, M.; Bjerre, M.T.; Fredsøe, J.; Vang, S.; Jensen, J.B.; De Laere, B.; Grönberg, H.; Borre, M.; Lindberg, J.; Sørensen, K.D. Prognostic Value of Low-Pass Whole Genome Sequencing of Circulating Tumor DNA in Metastatic Castration-Resistant Prostate Cancer. Clin. Chem. 2023, 69, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable Whole-Exome Sequencing of Cell-Free DNA Reveals High Concordance with Metastatic Tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Shchegrov, S.R.; Chai, S.; Zhou, Y.; Nguyen, T.; Cho, Y.; Melton, C.; Scott, E.; Roychowdhury-Saha, M.; Chang, P.-Y.; et al. Abstract LB297: Analytical Validation of a Tissue-Free, Multi-Cancer, Post-Diagnosis Cancer Research Test That Uses Cell-Free DNA Methylation Profiling. Cancer Res. 2023, 83, LB297. [Google Scholar] [CrossRef]
- Venn, O.; Bredno, J.; Thornton, A.; Chang, C.; Hubbell, E.; Kurtzman, K.; Beausang, J.; Swanton, C. Robustness of a Targeted Methylation-Based Multi-Cancer Early Detection (MCED) Test to Population Differences in Self-Reported Ethnicity. In Proceedings of the AACR Conference on the Science of Cancer Health Disparities in Racial/Ethnic Minorities and the Medically Underserved, Orlando, FL, USA, 29 September–2 October 2023. [Google Scholar]
- Chen, Q.; Zhang, Z.-H.; Wang, S.; Lang, J.-H. Circulating Cell-Free DNA or Circulating Tumor DNA in the Management of Ovarian and Endometrial Cancer. OncoTargets Ther. 2019, 12, 11517–11530. [Google Scholar] [CrossRef]
- Ponti, G.; Manfredini, M.; Tomasi, A. Non-Blood Sources of Cell-Free DNA for Cancer Molecular Profiling in Clinical Pathology and Oncology. Crit. Rev. Oncol. Hematol. 2019, 141, 36–42. [Google Scholar] [CrossRef]
- Roychowdhury-Saha, M.; Adhikari, S.; Aiello, A.C.; L’Hernault, A.; Pathak, A.; Chai, S.; Zhao, M.; Sharma, M.; Shchegrov, S.R.; Nguyen, T.; et al. Analytical Performance of a Cell-Free DNA Targeted Methylation Test for Early Lung Adenocarcinoma (LUAD) Recurrence Prediction. In Proceedings of the North American Conference on Lung Cancer, Chicago, IL, USA, 1–3 December 2023. [Google Scholar]
- Hong, T.H.; Hwang, S.; Abbosh, C.; Dasgupta, A.; Guthrie, V.; Hung, T.; Bredno, J.; Walker, J.; Shi, X.; Milenkova, T.; et al. Tumor-Naïve Pre-Surgical ctDNA Detection Is Prognostic in Stage I Lung Adenocarcinoma, Associating with PD-L1 Positivity and High-Grade Histological Subtype. In Proceedings of the North American Conference on Lung Cancer, Chicago, IL, USA, 1–3 December 2023. [Google Scholar]
- Moding, E.J.; Nabet, B.Y.; Alizadeh, A.A.; Diehn, M. Detecting Liquid Remnants of Solid Tumors: Circulating Tumor DNA Minimal Residual Disease. Cancer Discov. 2021, 11, 2968–2986. [Google Scholar] [CrossRef]
- Corradetti, M.N.; Torok, J.A.; Hatch, A.J.; Xanthopoulos, E.P.; Lafata, K.; Jacobs, C.; Rushing, C.; Calaway, J.; Jones, G.; Kelsey, C.R.; et al. Dynamic Changes in Circulating Tumor DNA During Chemoradiation for Locally Advanced Lung Cancer. Adv. Radiat. Oncol. 2019, 4, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Wald, I. On fast Construction of SAH-based Bounding Volume Hierarchies. In Proceedings of the 2007 IEEE Symposium on Interactive Ray Tracing, Ulm, Germany, 10–12 September 2007; pp. 33–40. [Google Scholar] [CrossRef]
- Alexander, G.E.; Lin, W.; Ortega, F.E.; Ramaiah, M.; Jung, B.; Ji, L.; Revenkova, E.; Shah, P.; Croisetiere, C.; Berman, J.R.; et al. Analytical Validation of a Multi-Cancer Early Detection Test with Cancer Signal Origin Using a Cell-Free DNA-Based Targeted Methylation Assay. PLoS ONE 2023, 18, e0283001. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of DMRs in Each Cancer Label | |||
|---|---|---|---|
| Cancer Label | Unfiltered DMRs | Heme-Filtered DMRs | Fraction Removed |
| Anus | 27,804 | 10,176 | 0.63 |
| Bladder and Urothelial Tract | 39,587 | 16,278 | 0.59 |
| Bone and Soft Tissue | 7389 | 3875 | 0.48 |
| Breast | 76,882 | 32,916 | 0.57 |
| Cervix | 30,740 | 10,397 | 0.66 |
| Colon and Rectum | 111,576 | 47,363 | 0.58 |
| Head and Neck | 51,231 | 21,965 | 0.57 |
| Kidney | 15,173 | 7542 | 0.5 |
| Liver and Bile Duct | 21,186 | 7856 | 0.63 |
| Lung | 66,613 | 28,583 | 0.57 |
| Lymphoid Lineage | 80,078 | 80,078 | 0 |
| Melanocytic Lineage | 11,559 | 5111 | 0.56 |
| Myeloid Lineage | 4876 | 4876 | 0 |
| NEC of Lung or Other Organs | 28,289 | 13,716 | 0.52 |
| Ovary | 30,070 | 14,800 | 0.51 |
| Pancreas and Gallbladder | 33,631 | 10,262 | 0.69 |
| Plasma Cell Lineage | 3558 | 3558 | 0 |
| Prostate | 25,525 | 11,696 | 0.54 |
| Stomach and Esophagus | 99,424 | 41,219 | 0.59 |
| Uterus | 30,335 | 11,671 | 0.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melton, C.A.; Freese, P.; Zhou, Y.; Shenoy, A.; Bagaria, S.; Chang, C.; Kuo, C.-C.; Scott, E.; Srinivasan, S.; Cann, G.; et al. A Novel Tissue-Free Method to Estimate Tumor-Derived Cell-Free DNA Quantity Using Tumor Methylation Patterns. Cancers 2024, 16, 82. https://doi.org/10.3390/cancers16010082
Melton CA, Freese P, Zhou Y, Shenoy A, Bagaria S, Chang C, Kuo C-C, Scott E, Srinivasan S, Cann G, et al. A Novel Tissue-Free Method to Estimate Tumor-Derived Cell-Free DNA Quantity Using Tumor Methylation Patterns. Cancers. 2024; 16(1):82. https://doi.org/10.3390/cancers16010082
Chicago/Turabian StyleMelton, Collin A., Peter Freese, Yifan Zhou, Archana Shenoy, Siddhartha Bagaria, Christopher Chang, Chih-Chung Kuo, Eric Scott, Subashini Srinivasan, Gordon Cann, and et al. 2024. "A Novel Tissue-Free Method to Estimate Tumor-Derived Cell-Free DNA Quantity Using Tumor Methylation Patterns" Cancers 16, no. 1: 82. https://doi.org/10.3390/cancers16010082
APA StyleMelton, C. A., Freese, P., Zhou, Y., Shenoy, A., Bagaria, S., Chang, C., Kuo, C.-C., Scott, E., Srinivasan, S., Cann, G., Roychowdhury-Saha, M., Chang, P.-Y., & Singh, A. H. (2024). A Novel Tissue-Free Method to Estimate Tumor-Derived Cell-Free DNA Quantity Using Tumor Methylation Patterns. Cancers, 16(1), 82. https://doi.org/10.3390/cancers16010082