Initial Phase of Anthracycline Cardiotoxicity Involves Cardiac Fibroblasts Activation and Metabolic Switch

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. In Vivo Treatment with DOX and Animal Protocol
2.2. Heart Function
2.3. Tissue Preparation
2.4. Isolation of Cardiac Primary Fibroblasts
2.5. Protein Extraction and Western Blot Analysis
2.6. Immunofluorescence
2.7. In Vitro Scratch Wound-Healing Assay
2.8. Mitochondrial Cellular Energetics
2.9. Real-Time PCR
| PKM2 Fwd: 5′-ATTACCAGCGACCCCACAGAA-3′ |
| Rev: 5′-ACGGCATCCTTACACAGCACA-3′ |
| LDHA Fwd: 5′-GCACTAAGCGGTCCCAAAAG-3′ |
| Rev: 5′-ACAGCACCAACCCCAACAAC-3′ |
| HPRT Fwd: 5′-TTGTTGGATATGCCCTTGACT-3′ |
| Rev: 5′-CCGCTGTCTTTTAGGCTTTG-3′ |
2.10. Glucose Analog Uptake Assay
2.11. Data Analysis
3. Results
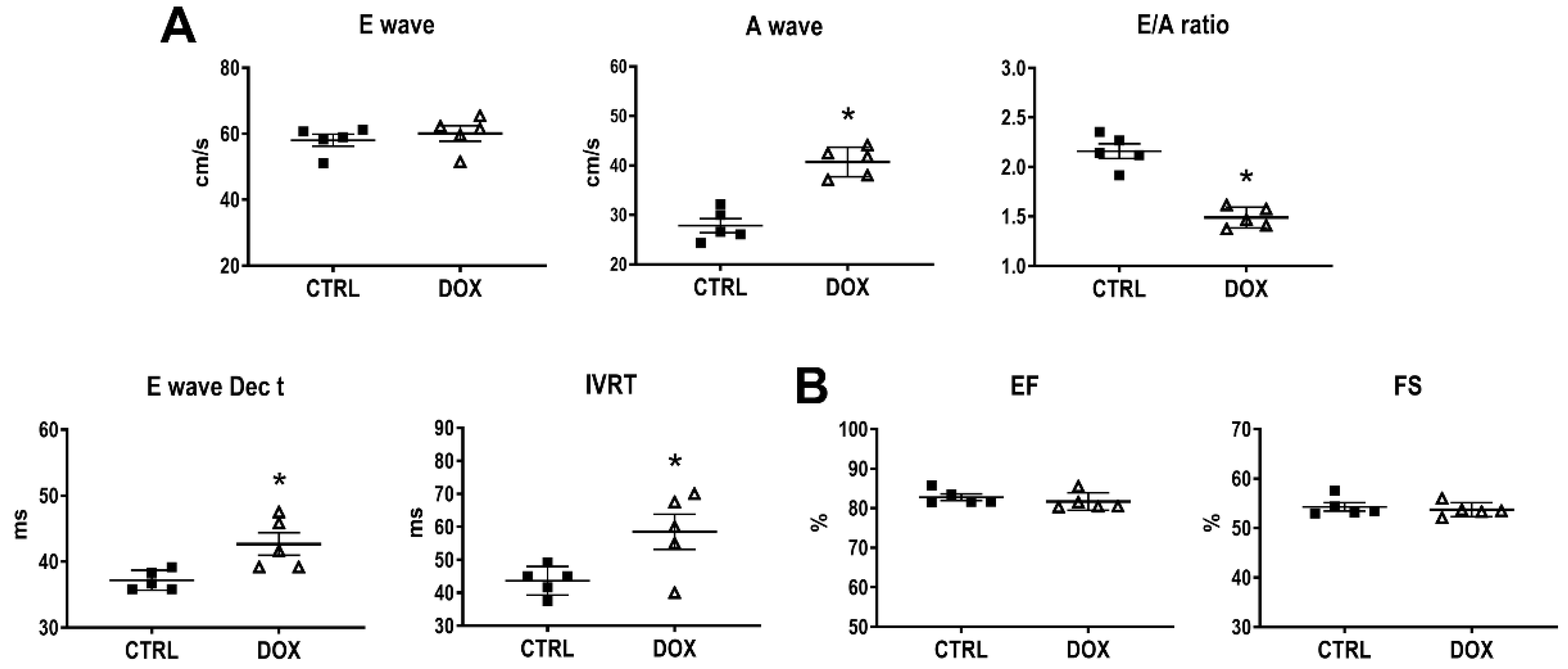
3.1. Cardiac Function
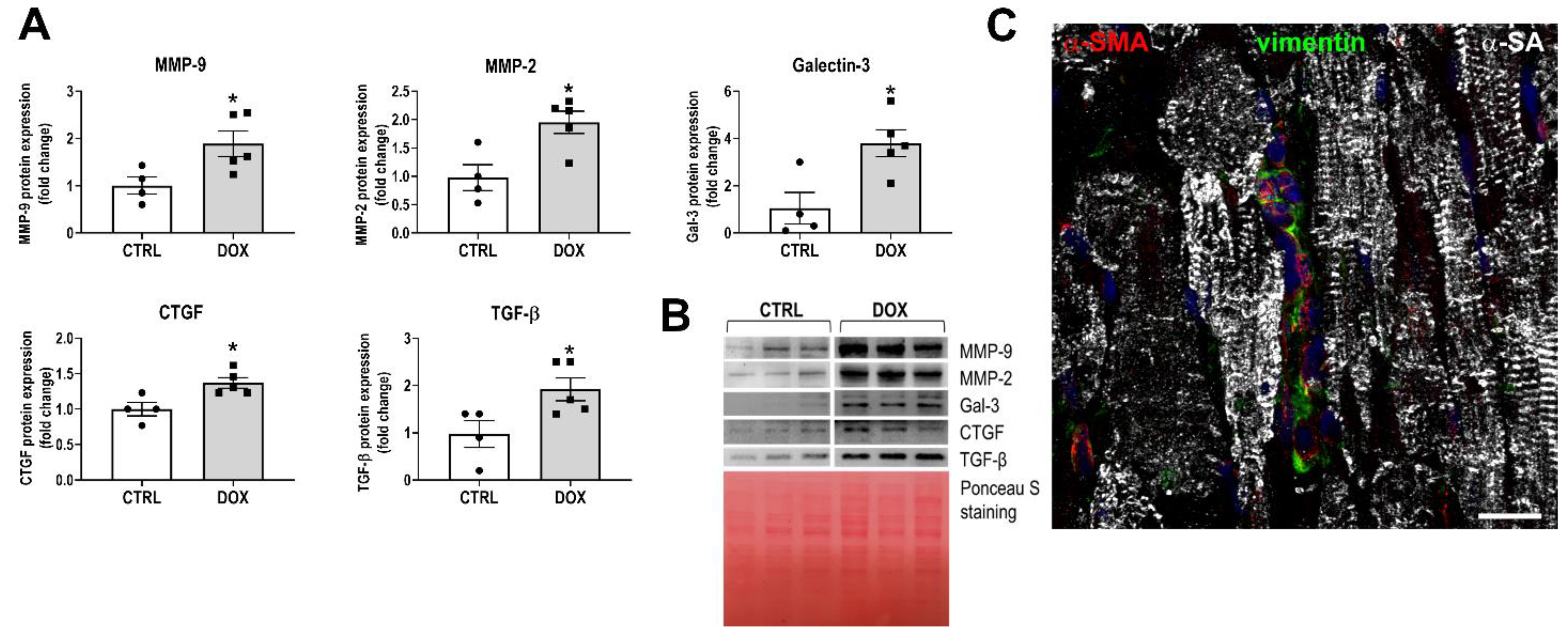
3.2. Development of the Myocardial Fibrotic Phenotype
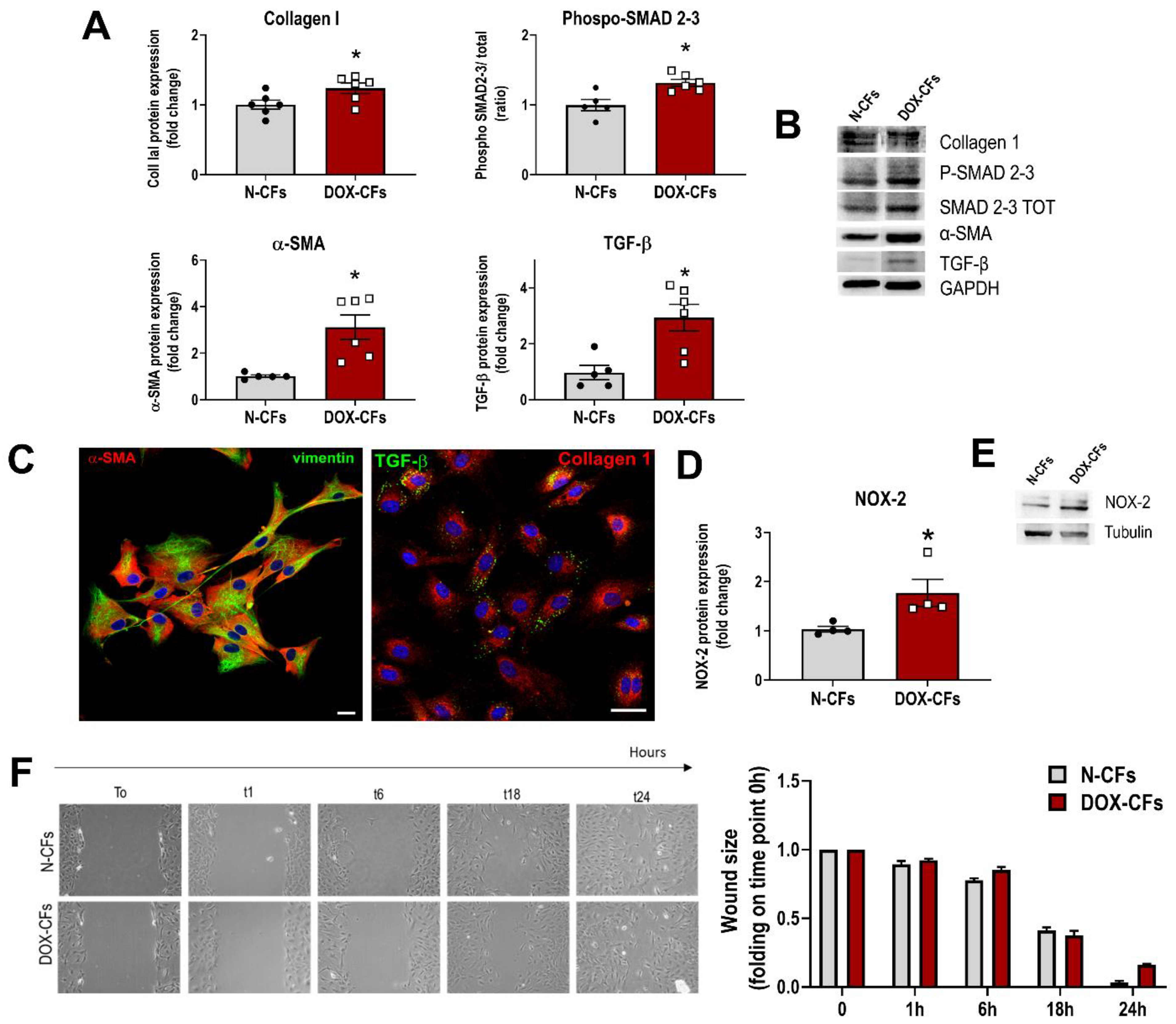
3.3. Cardiac Fibroblasts from DOX-Injected Animals at the Beginning of Treatment
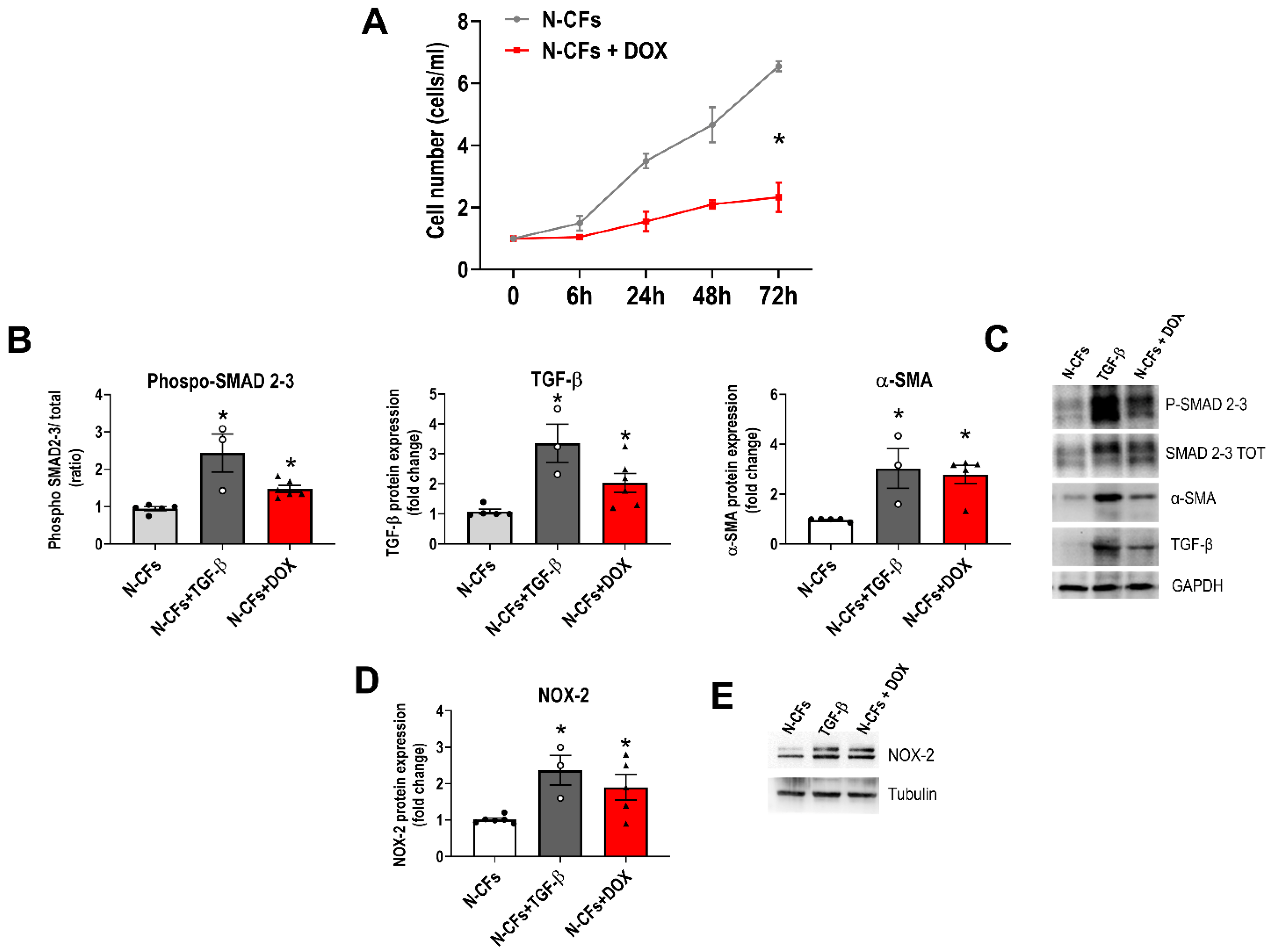
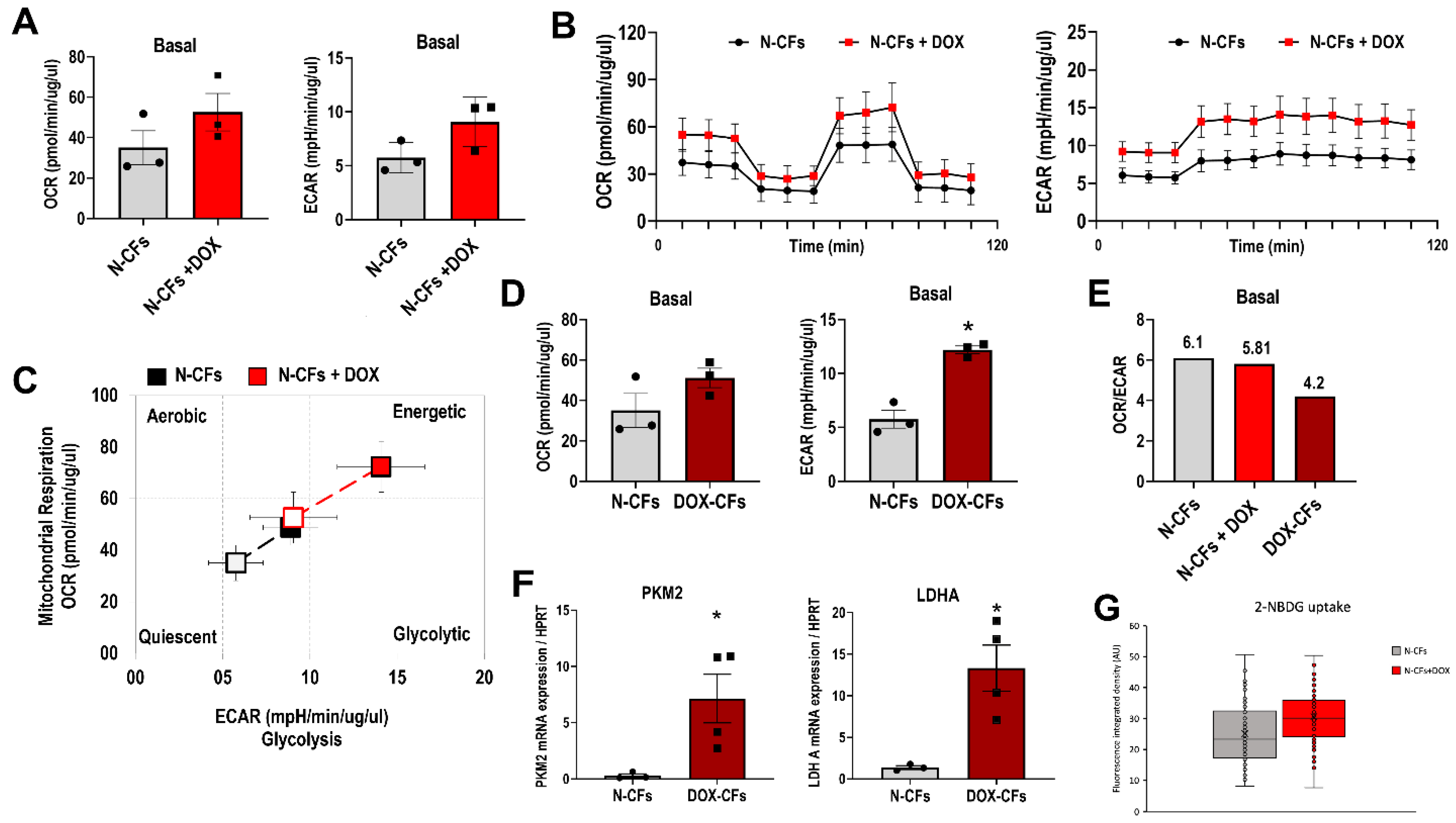
3.4. Naïve Cardiac Fibroblasts Exposed to DOX In Vitro
3.5. DOX and Metabolic Profile of Cardiac Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lyon, A.R.; Lyon, A.R.; López-Fernández, T.; López-Fernández, T.; Couch, L.S.; Couch, L.S.; Asteggiano, R.; Asteggiano, R.; Aznar, M.C.; Aznar, M.C.; et al. 2022 ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Adams, M.J.; Ganatra, S.; Colan, S.D.; Aggarwal, S.; Steiner, R.; Amdani, S.; Lipshultz, E.R.; Lipshultz, S.E. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Burlew, B.S.; Weber, K.T. Cardiac fibrosis as a cause of diastolic dysfunction. Herz 2002, 27, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Gyöngyösi, M.; Winkler, J.; Ramos, I.; Do, Q.; Firat, H.; McDonald, K.; González, A.; Thum, T.; Díez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef]
- Díaz-Araya, G.; Vivar, R.; Humeres, C.; Boza, P.; Bolivar, S.; Muñoz, C. Cardiac fibroblasts as sentinel cells in cardiac tissue: Receptors, signaling pathways and cellular functions. Pharmacol. Res. 2015, 101, 30–40. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Zaugg, M.; Zuppinger, C.; Wallimann, T.; Schlattner, U. New insights into doxorubicin-induced cardiotoxicity: The critical role of cellular energetics. J. Mol. Cell. Cardiol. 2006, 41, 389–405. [Google Scholar] [CrossRef]
- De Angelis, A.; Cappetta, D.; Piegari, E.; Rinaldi, B.; Ciuffreda, L.P.; Esposito, G.; Ferraiolo, F.A.V.; Rivellino, A.; Russo, R.; Donniacuo, M.; et al. Long-term administration of ranolazine attenuates diastolic dysfunction and adverse myocardial remodeling in a model of heart failure with preserved ejection fraction. Int. J. Cardiol. 2016, 217, 69–79. [Google Scholar] [CrossRef]
- Cappetta, D.; Ciuffreda, L.P.; Cozzolino, A.; Esposito, G.; Scavone, C.; Sapio, L.; Naviglio, S.; D’amario, D.; Crea, F.; Rossi, F.; et al. Dipeptidyl Peptidase 4 Inhibition Ameliorates Chronic Kidney Disease in a Model of Salt-Dependent Hypertension. Oxid. Med. Cell. Longev. 2019, 2019, 8912768. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, K.; Cappetta, D.; Bellocchio, G.; Coppola, M.A.; Imbrici, P.; Telesca, M.; Donniacuo, M.; Riemma, M.A.; Mele, E.; Cianflone, E.; et al. Dapagliflozin protects the kidney in a non-diabetic model of cardiorenal syndrome. Pharmacol. Res. 2023, 188, 106659. [Google Scholar] [CrossRef] [PubMed]
- Martinotti, S.; Ranzato, E. Scratch Wound Healing Assay. Methods Mol. Biol. 2020, 2109, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Russo, R.; Della Torre, L.; Cuomo, F.; Dell’Aversana, C.; Sarno, F.; Sgueglia, G.; Di Donato, M.; Rotili, D.; Mai, A.; et al. The Pan-Sirtuin Inhibitor MC2494 Regulates Mitochondrial Function in a Leukemia Cell Line. Front. Oncol. 2020, 10, 820. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Menna, P.; Annibali, O.; Armento, G.; Carpino, A.; Cerchiara, E.; Greco, C.; Marchesi, F.; Spallarossa, P.; Toglia, G.; et al. Early Diastolic Dysfunction after Cancer Chemotherapy: Primary Endpoint Results of a Multicenter Cardio-Oncology Study. Chemotherapy 2018, 63, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Reggiardo, G.; Camilli, M.; Salvatorelli, E.; Menna, P. From Cardiac Anthracycline Accumulation to Real-Life Risk for Early Diastolic Dysfunction: A Translational Approach. JACC CardioOncology 2022, 4, 139–140. [Google Scholar] [CrossRef]
- Narikawa, M.; Umemura, M.; Tanaka, R.; Hikichi, M.; Nagasako, A.; Fujita, T.; Yokoyama, U.; Ishigami, T.; Kimura, K.; Tamura, K.; et al. Doxorubicin induces trans-differentiation and MMP1 expression in cardiac fibroblasts via cell death-independent pathways. PLoS ONE 2019, 14, e0221940. [Google Scholar] [CrossRef]
- De Angelis, A.; Urbanek, K.; Cappetta, D.; Piegari, E.; Ciuffreda, L.P.; Rivellino, A.; Russo, R.; Esposito, G.; Rossi, F.; Berrino, L. Doxorubicin cardiotoxicity and target cells: A broader perspective. Cardiooncology 2016, 2, 2. [Google Scholar] [CrossRef]
- Zhan, H.; Aizawa, K.; Sun, J.; Tomida, S.; Otsu, K.; Conway, S.J.; Mckinnon, P.J.; Manabe, I.; Komuro, I.; Miyagawa, K.; et al. Ataxia telangiectasia mutated in cardiac fibroblasts regulates doxorubicin-induced cardiotoxicity. Cardiovasc. Res. 2016, 110, 85–95. [Google Scholar] [CrossRef]
- Lendahl, U.; Muhl, L.; Betsholtz, C. Identification, discrimination and heterogeneity of fibroblasts. Nat. Commun. 2022, 13, 3409. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- Nicin, L.; Schroeter, S.M.; Glaser, S.F.; Schulze-Brüning, R.; Pham, M.-D.; Hille, S.S.; Yekelchyk, M.; Kattih, B.; Abplanalp, W.T.; Tombor, L.; et al. A human cell atlas of the pressure-induced hypertrophic heart. Nat. Cardiovasc. Res. 2022, 1, 174–185. [Google Scholar] [CrossRef]
- Kuppe, C.; Flores, R.O.R.; Li, Z.; Hayat, S.; Levinson, R.T.; Liao, X.; Hannani, M.T.; Tanevski, J.; Wünnemann, F.; Nagai, J.S.; et al. Spatial multi-omic map of human myocardial infarction. Nature 2022, 608, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H934–H949. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Umemura, M.; Narikawa, M.; Hikichi, M.; Osaw, K.; Fujita, T.; Yokoyama, U.; Ishigami, T.; Tamura, K.; Ishikawa, Y. Reactive fibrosis precedes doxorubicin-induced heart failure through sterile inflammation. ESC Heart Fail. 2020, 7, 588–603. [Google Scholar] [CrossRef]
- Rood, J.E.; Maartens, A.; Hupalowska, A.; Teichmann, S.A.; Regev, A. Impact of the Human Cell Atlas on medicine. Nat. Med. 2022, 28, 2486–2496. [Google Scholar] [CrossRef]
- Marmisolle, I.; Martínez, J.; Liu, J.; Mastrogiovanni, M.; Fergusson, M.M.; Rovira, I.I.; Castro, L.; Trostchansky, A.; Moreno, M.; Cao, L.; et al. Reciprocal regulation of acetyl-CoA carboxylase 1 and senescence in human fibroblasts involves oxidant mediated p38 MAPK activation. Arch. Biochem. Biophys. 2017, 613, 12–22. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; Karabekmez, M.E.; Eraslan, S.; Kırdar, B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci. Rep. 2018, 8, 13672. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telesca, M.; Donniacuo, M.; Bellocchio, G.; Riemma, M.A.; Mele, E.; Dell’Aversana, C.; Sgueglia, G.; Cianflone, E.; Cappetta, D.; Torella, D.; et al. Initial Phase of Anthracycline Cardiotoxicity Involves Cardiac Fibroblasts Activation and Metabolic Switch. Cancers 2024, 16, 53. https://doi.org/10.3390/cancers16010053
Telesca M, Donniacuo M, Bellocchio G, Riemma MA, Mele E, Dell’Aversana C, Sgueglia G, Cianflone E, Cappetta D, Torella D, et al. Initial Phase of Anthracycline Cardiotoxicity Involves Cardiac Fibroblasts Activation and Metabolic Switch. Cancers. 2024; 16(1):53. https://doi.org/10.3390/cancers16010053
Chicago/Turabian StyleTelesca, Marialucia, Maria Donniacuo, Gabriella Bellocchio, Maria Antonietta Riemma, Elena Mele, Carmela Dell’Aversana, Giulia Sgueglia, Eleonora Cianflone, Donato Cappetta, Daniele Torella, and et al. 2024. "Initial Phase of Anthracycline Cardiotoxicity Involves Cardiac Fibroblasts Activation and Metabolic Switch" Cancers 16, no. 1: 53. https://doi.org/10.3390/cancers16010053
APA StyleTelesca, M., Donniacuo, M., Bellocchio, G., Riemma, M. A., Mele, E., Dell’Aversana, C., Sgueglia, G., Cianflone, E., Cappetta, D., Torella, D., Altucci, L., Castaldo, G., Rossi, F., Berrino, L., Urbanek, K., & De Angelis, A. (2024). Initial Phase of Anthracycline Cardiotoxicity Involves Cardiac Fibroblasts Activation and Metabolic Switch. Cancers, 16(1), 53. https://doi.org/10.3390/cancers16010053