
Cell-Free DNA Genomic Profiling and Its Clinical Implementation in Advanced Prostate Cancer

 , , ,
, , ,  , , , , , and
, , , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Liquid Biopsy Preparation
2.3. cfDNA Extraction
2.4. DNA Extraction from Tissue Biopsies
2.5. DNA Quantification
2.6. Custom-Designed AmpliSeq HD cfDNA Panel
2.7. Spike-in Validation of Custom-Designed AmpliSeq HD Targeted cfDNA PCa Panel
2.8. Library Preparation and Sequencing
2.9. NGS Data Analysis
2.10. Copy Number Analysis by Targeted cfDNA Panel
2.11. Statistical Analysis
3. Results
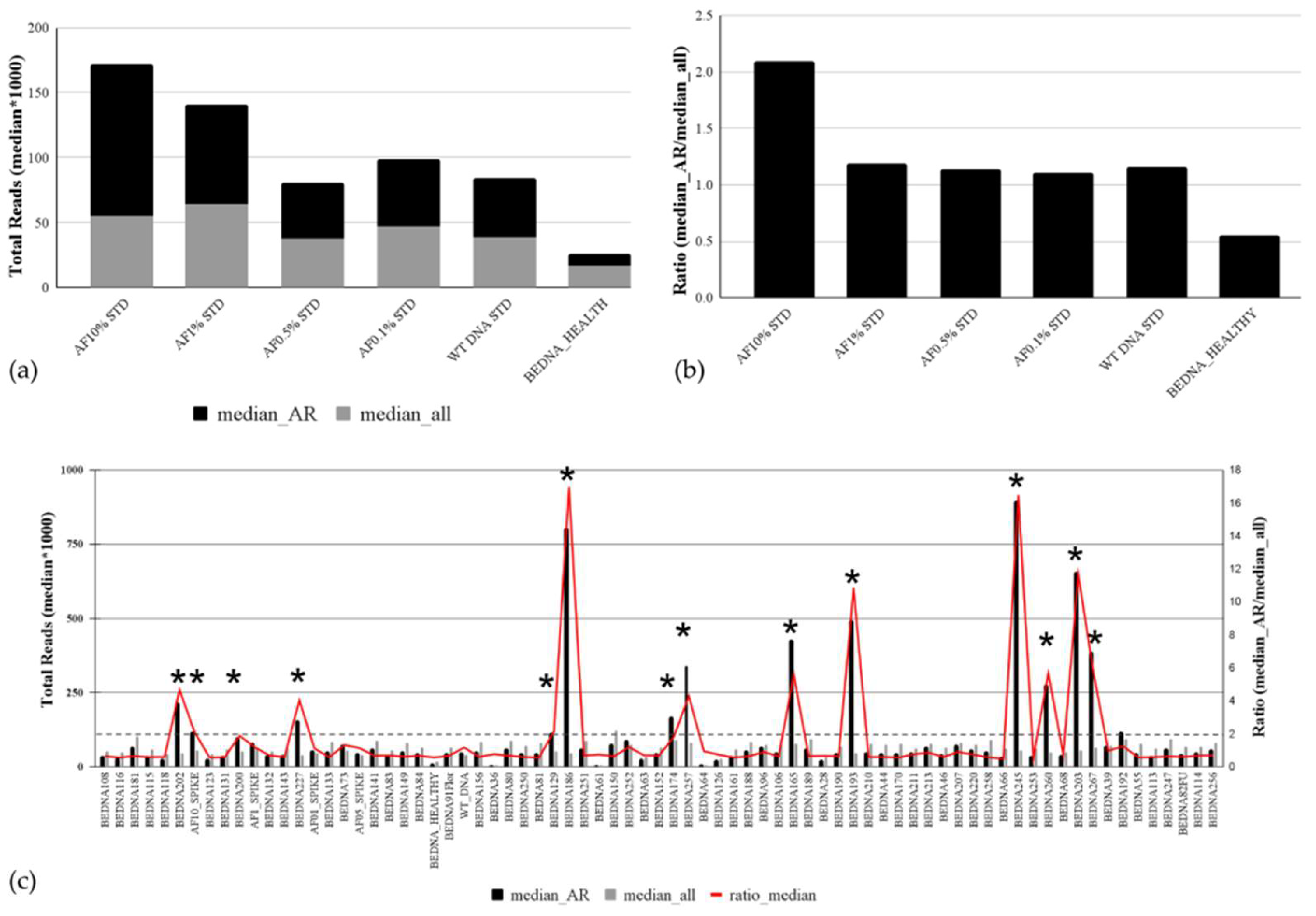
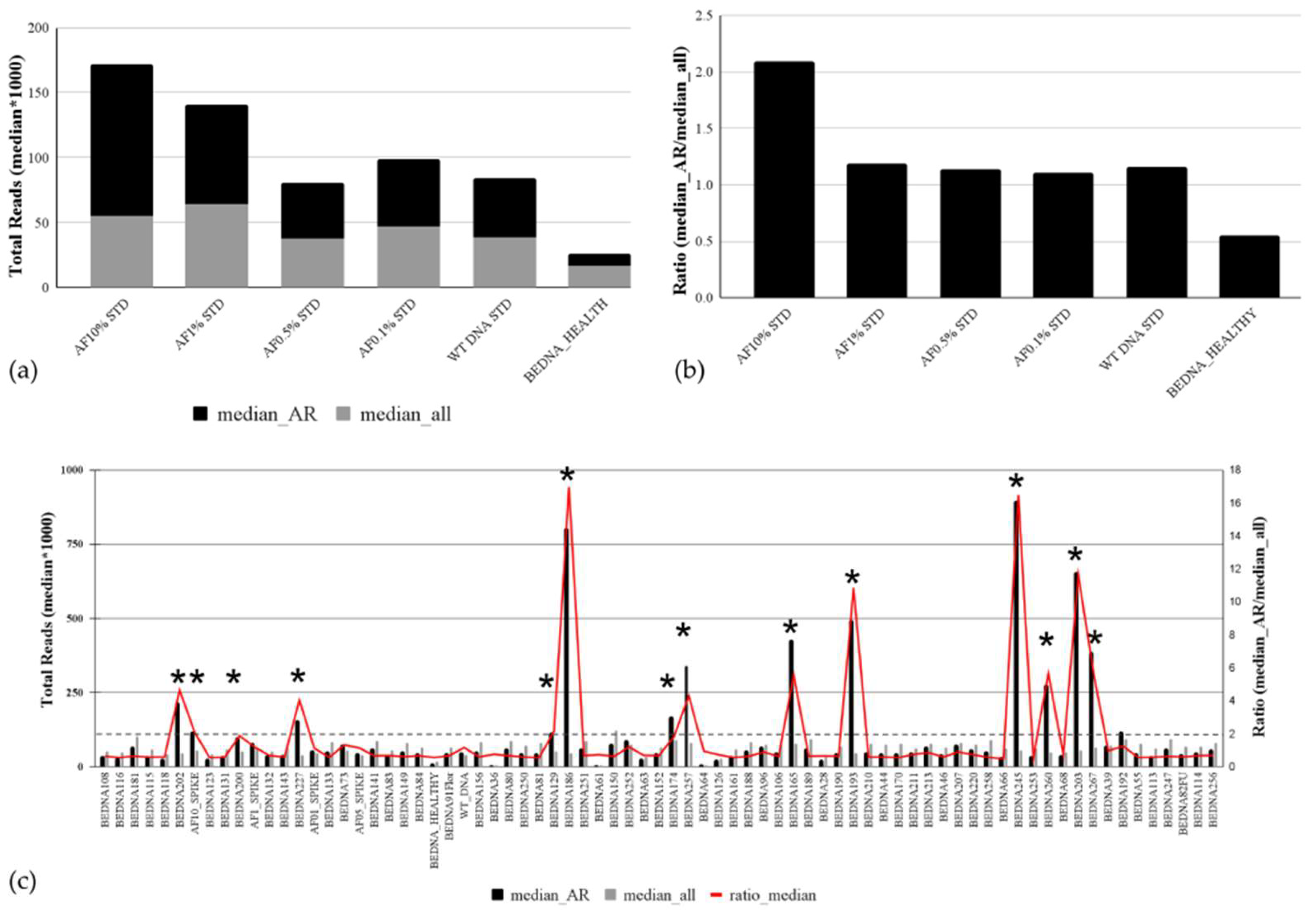
3.1. Sensitivity of the Custom-Designed Targeted cfDNA PCa NGS Panel Based on AmpliSeq HD Technology
3.2. Patient Cohort
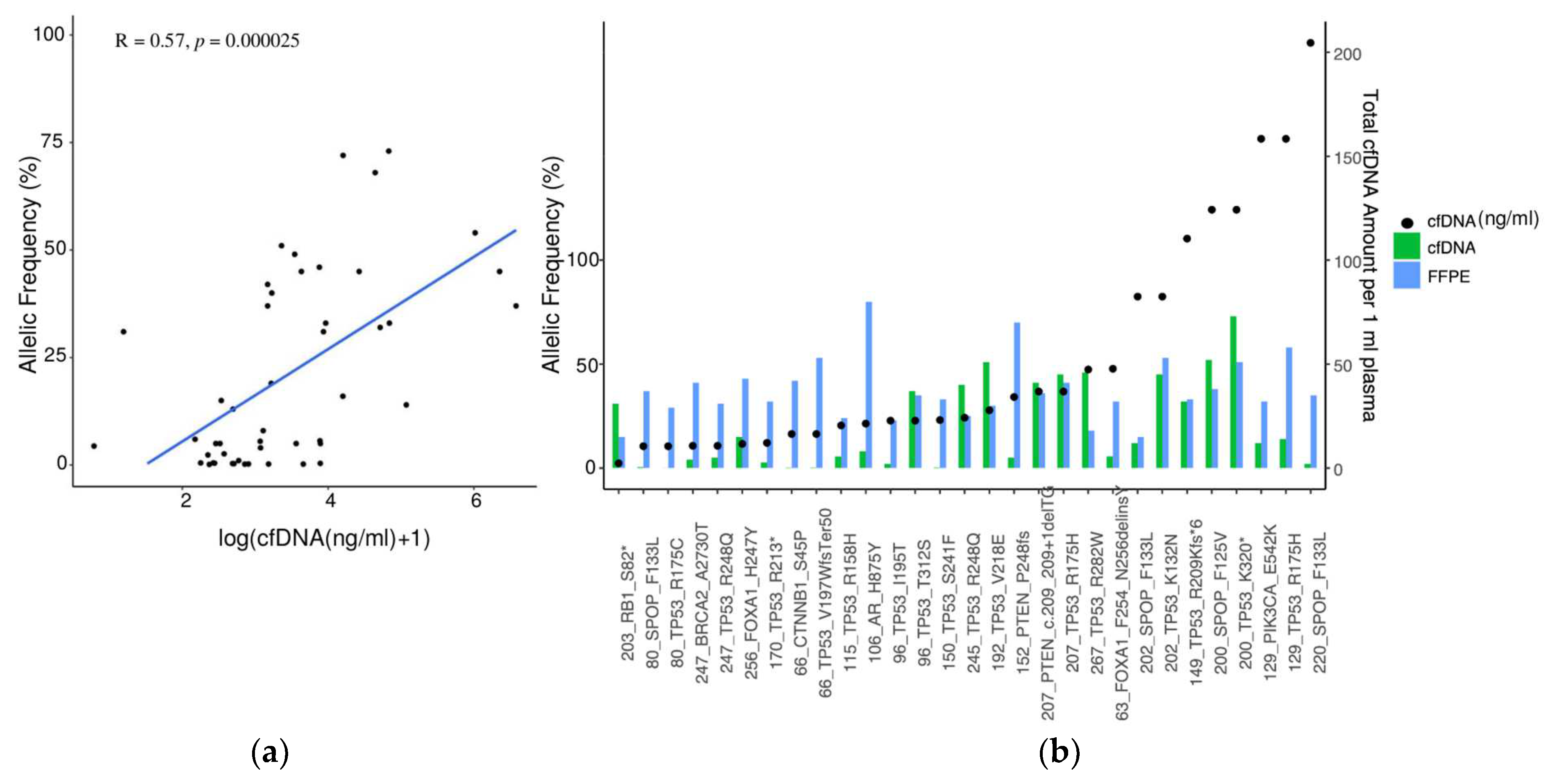
3.3. Detection of Mutations in Tumor Biopsy and Matched cfDNA
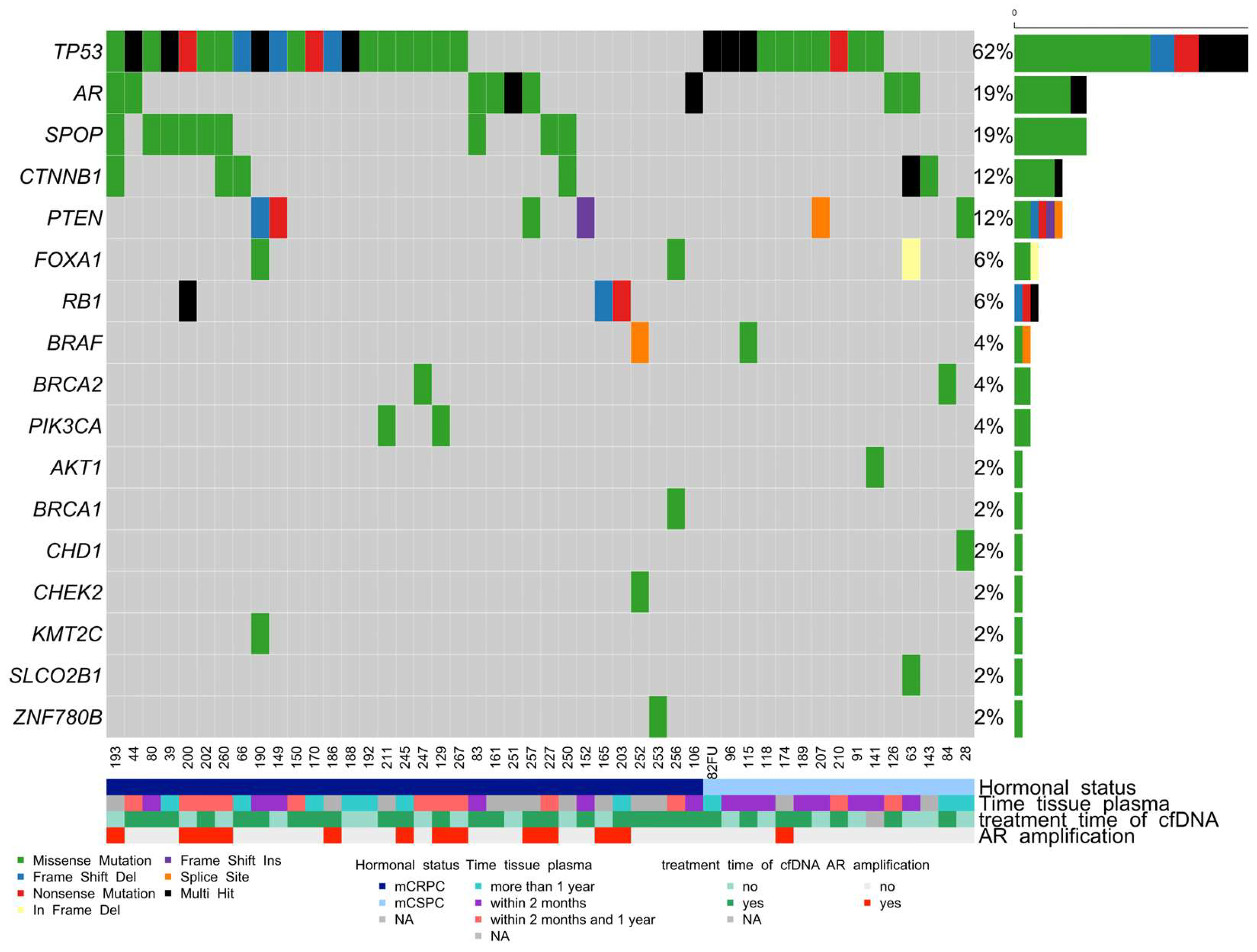
3.4. Somatic Mutation Landscape in cfDNA in Advanced mCRPC and mCSPC PCa Patients
3.5. Analytical Validation of the Copy Number Variation Detection
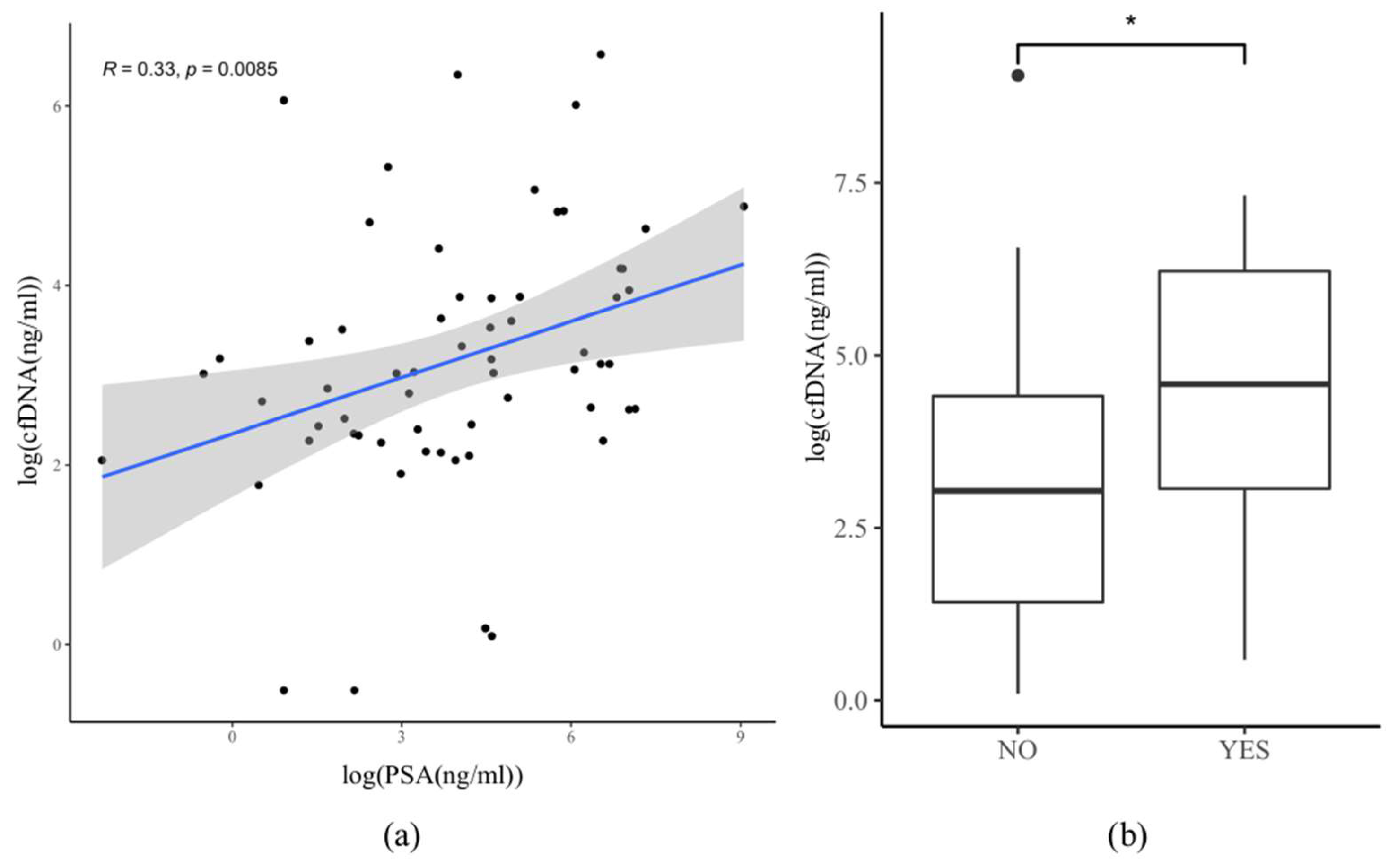
3.6. Correlation between PSA and cfDNA Amount in Plasma
3.7. Correlation between cfDNA Amount, the Onset of Therapy, and Hormonal Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primer 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Egan, J.; Marhamati, S.; Galfano, A.; Kowalczyk, K.J. Retzius-Sparing Robot-Assisted Robotic Prostatectomy: Past, Present, and Future. Urol. Clin. N. Am. 2021, 48, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Omlin, A.; Spahn, M.; Beyer, J.; Eberli, D.; Gillessen, S.; Jochum, W.; Kueng, M.; Nitzsche, E.; Rentsch, C.A.; Roggero, E.; et al. Treatment of Advanced Prostate Carcinoma—An Interdisciplinary Recommendation. Praxis 2018, 107, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Block, N.L.; Rick, F.G. Discovery of LHRH and Development of LHRH Analogs for Prostate Cancer Treatment. Prostate 2017, 77, 1036–1054. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, E.N.; Zhang, T.; Armstrong, A.J. Metastatic Hormone-Sensitive Prostate Cancer: A Review of the Current Treatment Landscape. Cancer J. Sudbury Mass. 2020, 26, 64–75. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Spears, M.R.; Clarke, N.W.; Dearnaley, D.P.; De Bono, J.S.; Gale, J.; Hetherington, J.; Hoskin, P.J.; Jones, R.J.; Laing, R.; et al. Survival with Newly Diagnosed Metastatic Prostate Cancer in the “Docetaxel Era”: Data from 917 Patients in the Control Arm of the STAMPEDE Trial (MRC PR08, CRUK/06/019). Eur. Urol. 2015, 67, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Tatkovic, A.; McBean, R.; Wong, D. Lu177-PSMA Therapy for Men with Advanced Prostate Cancer: 18 Months Survival Analysis in a Single Australian Tertiary Institution. J. Med. Imaging Radiat. Oncol. 2021, 65, 740–747. [Google Scholar] [CrossRef]
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The Genomic Landscape of Metastatic Castration-Resistant Prostate Cancers Reveals Multiple Distinct Genotypes with Potential Clinical Impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J. Natl. Cancer Inst. 2017, 109, djx118. [Google Scholar] [CrossRef]
- Kwan, E.M.; Wyatt, A.W. Androgen Receptor Genomic Alterations and Treatment Resistance in Metastatic Prostate Cancer. Prostate 2022, 82, S25–S36. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Crocetto, F.; Russo, G.; Di Zazzo, E.; Pisapia, P.; Mirto, B.F.; Palmieri, A.; Pepe, F.; Bellevicine, C.; Russo, A.; La Civita, E.; et al. Liquid Biopsy in Prostate Cancer Management—Current Challenges and Future Perspectives. Cancers 2022, 14, 3272. [Google Scholar] [CrossRef] [PubMed]
- Sutera, P.; Van Der Eecken, K.; Kishan, A.U.; Hamid, A.; Grist, E.; Attard, G.; Lotan, T.; Mendes, A.A.; Paller, C.J.; Carducci, M.A.; et al. Definitions of Disease Burden across the Spectrum of Metastatic Castration-Sensitive Prostate Cancer: Comparison by Disease Outcomes and Genomics. Prostate Cancer Prostatic Dis. 2022, 25, 713–719. [Google Scholar] [CrossRef]
- Swami, U.; Graf, R.P.; Nussenzveig, R.H.; Fisher, V.; Tukachinsky, H.; Schrock, A.B.; Li, G.; Ross, J.S.; Sayegh, N.; Tripathi, N.; et al. SPOP Mutations as a Predictive Biomarker for Androgen Receptor Axis-Targeted Therapy in De Novo Metastatic Castration-Sensitive Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 4917–4925. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The Long Tail of Oncogenic Drivers in Prostate Cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Herberts, C.; Murtha, A.J.; Fu, S.; Wang, G.; Schönlau, E.; Xue, H.; Lin, D.; Gleave, A.; Yip, S.; Angeles, A.; et al. Activating AKT1 and PIK3CA Mutations in Metastatic Castration-Resistant Prostate Cancer. Eur. Urol. 2020, 78, 834–844. [Google Scholar] [CrossRef]
- Nguyen, B.; Mota, J.M.; Nandakumar, S.; Stopsack, K.H.; Weg, E.; Rathkopf, D.; Morris, M.J.; Scher, H.I.; Kantoff, P.W.; Gopalan, A.; et al. Pan-Cancer Analysis of CDK12 Alterations Identifies a Subset of Prostate Cancers with Distinct Genomic and Clinical Characteristics. Eur. Urol. 2020, 78, 671–679. [Google Scholar] [CrossRef]
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-Resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 2019, 76, 452–458. [Google Scholar] [CrossRef]
- Salami, S.S.; Hovelson, D.H.; Kaplan, J.B.; Mathieu, R.; Udager, A.M.; Curci, N.E.; Lee, M.; Plouffe, K.R.; De La Vega, L.L.; Susani, M.; et al. Transcriptomic Heterogeneity in Multifocal Prostate Cancer. JCI Insight 2018, 3, e123468. [Google Scholar] [CrossRef] [PubMed]
- Garofoli, A.; Paradiso, V.; Montazeri, H.; Jermann, P.M.; Roma, G.; Tornillo, L.; Terracciano, L.M.; Piscuoglio, S.; Ng, C.K.Y. PipeIT: A Singularity Container for Molecular Diagnostic Somatic Variant Calling on the Ion Torrent Next-Generation Sequencing Platform. J. Mol. Diagn. JMD 2019, 21, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.J.; Chen, Y.-H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.-N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Gulati, R.; Beightol, M.; Konnick, E.Q.; Cheng, H.H.; Klemfuss, N.; De Sarkar, N.; Yu, E.Y.; Montgomery, R.B.; Nelson, P.S.; et al. Clinical Determinants for Successful Circulating Tumor DNA Analysis in Prostate Cancer. Prostate 2019, 79, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.G.; Malapelle, U.; André, F.; Paz-Ares, L.; Schuler, M.; Thomas, D.M.; Vainer, G.; Yoshino, T.; Rolfo, C. Practical Considerations for the Use of Circulating Tumor DNA in the Treatment of Patients with Cancer: A Narrative Review. JAMA Oncol. 2022, 8, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.S.; Wyatt, A.W. Building Confidence in Circulating Tumour DNA Assays for Metastatic Castration-Resistant Prostate Cancer. Nat. Rev. Urol. 2021, 18, 255–256. [Google Scholar] [CrossRef]
- De Laere, B.; Crippa, A.; Discacciati, A.; Larsson, B.; Oldenburg, J.; Mortezavi, A.; Ost, P.; Eklund, M.; Lindberg, J.; Grönberg, H.; et al. Clinical Trial Protocol for ProBio: An Outcome-Adaptive and Randomised Multiarm Biomarker-Driven Study in Patients with Metastatic Prostate Cancer. Eur. Urol. Focus 2022, 8, 1617–1621. [Google Scholar] [CrossRef]
- Herberts, C.; Annala, M.; Sipola, J.; Ng, S.W.S.; Chen, X.E.; Nurminen, A.; Korhonen, O.V.; Munzur, A.D.; Beja, K.; Schönlau, E.; et al. Deep Whole-Genome ctDNA Chronology of Treatment-Resistant Prostate Cancer. Nature 2022, 608, 199–208. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef]
- Deveson, I.W.; Gong, B.; Lai, K.; LoCoco, J.S.; Richmond, T.A.; Schageman, J.; Zhang, Z.; Novoradovskaya, N.; Willey, J.C.; Jones, W.; et al. Evaluating the Analytical Validity of Circulating Tumor DNA Sequencing Assays for Precision Oncology. Nat. Biotechnol. 2021, 39, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Seyedolmohadessin, S.M.; Akbari, M.T.; Nourmohammadi, Z.; Basiri, A.; Pourmand, G. Assessing the Diagnostic Value of Plasma-Free DNA in Prostate Cancer Screening. Iran. Biomed. J. 2018, 22, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Madison, R.W.; Chung, J.H.; Gjoerup, O.V.; Severson, E.A.; Dennis, L.; Fendler, B.J.; Morley, S.; Zhong, L.; Graf, R.P.; et al. Genomic Analysis of Circulating Tumor DNA in 3334 Patients with Advanced Prostate Cancer Identifies Targetable BRCA Alterations and AR Resistance Mechanisms. Clin. Cancer Res. 2021, 27, 3094–3105. [Google Scholar] [CrossRef] [PubMed]
- Tolmeijer, S.H.; Boerrigter, E.; Sumiyoshi, T.; Kwan, E.M.; Ng, S.; Annala, M.; Donnellan, G.; Herberts, C.; Benoist, G.E.; Hamberg, P.; et al. Early On-Treatment Changes in Circulating Tumor DNA Fraction and Response to Enzalutamide or Abiraterone in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023, 29, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-Free DNA Profiling of Metastatic Prostate Cancer Reveals Microsatellite Instability, Structural Rearrangements and Clonal Hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Vandekerkhove, G.; Struss, W.J.; Annala, M.; Kallio, H.M.L.; Khalaf, D.; Warner, E.W.; Herberts, C.; Ritch, E.; Beja, K.; Loktionova, Y.; et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur. Urol. 2019, 75, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Pastor, B.; André, T.; Henriques, J.; Trouilloud, I.; Tournigand, C.; Jary, M.; Mazard, T.; Louvet, C.; Azan, S.; Bauer, A.; et al. Monitoring Levels of Circulating Cell-free DNA in Patients with Metastatic Colorectal Cancer as a Potential Biomarker of Responses to Regorafenib Treatment. Mol. Oncol. 2021, 15, 2401–2411. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Tierno, M.; Fisher, V.; Tukachinsky, H.; Alexander, S.; Hamdani, O.; Hiemenz, M.C.; Huang, R.S.P.; Oxnard, G.R.; Graf, R.P. Clinical and Pathological Features Associated with Circulating Tumor DNA Content in Real-world Patients with Metastatic Prostate Cancer. Prostate 2022, 82, 867–875. [Google Scholar] [CrossRef]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-Institutional Prospective Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Cotter, K.; Rubin, M.A. The Evolving Landscape of Prostate Cancer Somatic Mutations. Prostate 2022, 82, S13–S24. [Google Scholar] [CrossRef]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.-K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, T.; Stürm, S.; Rey, S.; Zlobec, I.; Gsponer, J.R.; Rentsch, C.A.; Terracciano, L.M.; Bachmann, A.; Bubendorf, L.; Ruiz, C. Estrogen Receptor β Expression and Androgen Receptor Phosphorylation Correlate with a Poor Clinical Outcome in Hormone-Naive Prostate Cancer and Are Elevated in Castration-Resistant Disease. Endocr. Relat. Cancer 2013, 20, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Casadei, C.; Scarpi, E.; Brighi, N.; Schepisi, G.; Lolli, C.; Gurioli, G.; Toma, I.; Poti, G.; Farolfi, A.; et al. Baseline Plasma Tumor DNA (ctDNA) Correlates with PSA Kinetics in Metastatic Castration-Resistant Prostate Cancer (mCRPC) Treated with Abiraterone or Enzalutamide. Cancers 2022, 14, 2219. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; van den Broek, D.; Denis, M.G.; Hofman, P.; Hubank, M.; Mouliere, F.; Paz-Ares, L.; Schuuring, E.; Sültmann, H.; Vainer, G.; et al. Recommendations for a Practical Implementation of Circulating Tumor DNA Mutation Testing in Metastatic Non-Small-Cell Lung Cancer. ESMO Open 2022, 7, 100399. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | High Volume | Low Volume | |
|---|---|---|---|
| Number 1 | 68 | 43 | 24 |
| Age mean value (range) | 77 (56–92) | 75 (56–92) | 78 (58–92) |
| Gleason score (%) 2 | 65 (100) | 42 | 23 |
| ≤7 (%) | 14 (19) | 9 | 5 |
| 8 (%) | 11 (16) | 8 | 3 |
| ≥9 (%) | 40 (59) | 24 | 15 |
| PSA at the time of cfDNA (ng/mL) sampling: 3 | 62 | 38 | 22 |
| (median; 1st–3rd Quartile) | (57; 9.9–410) | (58; 14–335) | (48; 4–382) |
| Disease status at the time of cfDNA 4 | 68 | ||
| mCSPC (%) | 28 (41) | 14 | 14 |
| mCRPC (%) | 40 (59) | 29 | 10 |
| Therapy at the time of cfDNA sampling (%) 5 | 45 (66) | 28 | 16 |
| Matched Samples FFPE—Liquid Biopsy | FFPE Tumor Biopsy No Mutation Detected | Concordant Cases FFPE—cfDNA | Discordant Cases FFPE—cfDNA |
| mCRPC | 5 | 17 | 0 |
| mCSPC | 9 | 4 | 7 |
| Total | 14 | 21 | 7 |
| ClinicalParameter | Total Samples (n) | ctDNA Positive (n) | ctDNA Positive(%) | Average AF (%) | Fisher’s Exact Test p Value | |
|---|---|---|---|---|---|---|
| Treatment | YES | 43 | 29 | 67 | 12 | 0.14 |
| NO | 22 | 19 | 86 | 17 | ||
| Hormonal Status | mCRPC | 40 | 33 | 83 | 17 | 0.08 |
| mCSPC | 25 | 15 | 60 | 10 | ||
| Tumor Volume | High | 42 | 33 | 79 | 19 | 0.24 |
| Low | 22 | 14 | 64 | 8 | ||
| PSA | Normal (<4 ng/mL) | 8 | 3 | 38 | 14 | 0.02 |
| High | 50 | 40 | 80 | 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bratic Hench, I.; Roma, L.; Conticelli, F.; Bubendorf, L.; Calgua, B.; Le Magnen, C.; Piscuoglio, S.; Rubin, M.A.; Chirindel, A.; Nicolas, G.P.; et al. Cell-Free DNA Genomic Profiling and Its Clinical Implementation in Advanced Prostate Cancer. Cancers 2024, 16, 45. https://doi.org/10.3390/cancers16010045
Bratic Hench I, Roma L, Conticelli F, Bubendorf L, Calgua B, Le Magnen C, Piscuoglio S, Rubin MA, Chirindel A, Nicolas GP, et al. Cell-Free DNA Genomic Profiling and Its Clinical Implementation in Advanced Prostate Cancer. Cancers. 2024; 16(1):45. https://doi.org/10.3390/cancers16010045
Chicago/Turabian StyleBratic Hench, Ivana, Luca Roma, Floriana Conticelli, Lenard Bubendorf, Byron Calgua, Clémentine Le Magnen, Salvatore Piscuoglio, Mark A. Rubin, Alin Chirindel, Guillaume P. Nicolas, and et al. 2024. "Cell-Free DNA Genomic Profiling and Its Clinical Implementation in Advanced Prostate Cancer" Cancers 16, no. 1: 45. https://doi.org/10.3390/cancers16010045
APA StyleBratic Hench, I., Roma, L., Conticelli, F., Bubendorf, L., Calgua, B., Le Magnen, C., Piscuoglio, S., Rubin, M. A., Chirindel, A., Nicolas, G. P., Vlajnic, T., Zellweger, T., Templeton, A. J., Stenner, F., Ruiz, C., Rentsch, C., & Bubendorf, L. (2024). Cell-Free DNA Genomic Profiling and Its Clinical Implementation in Advanced Prostate Cancer. Cancers, 16(1), 45. https://doi.org/10.3390/cancers16010045