

Current Landscape of Immune Checkpoint Inhibitors for Metastatic Urothelial Carcinoma: Is There a Role for Additional T-Cell Blockade? †

Abstract
:Simple Summary
Abstract
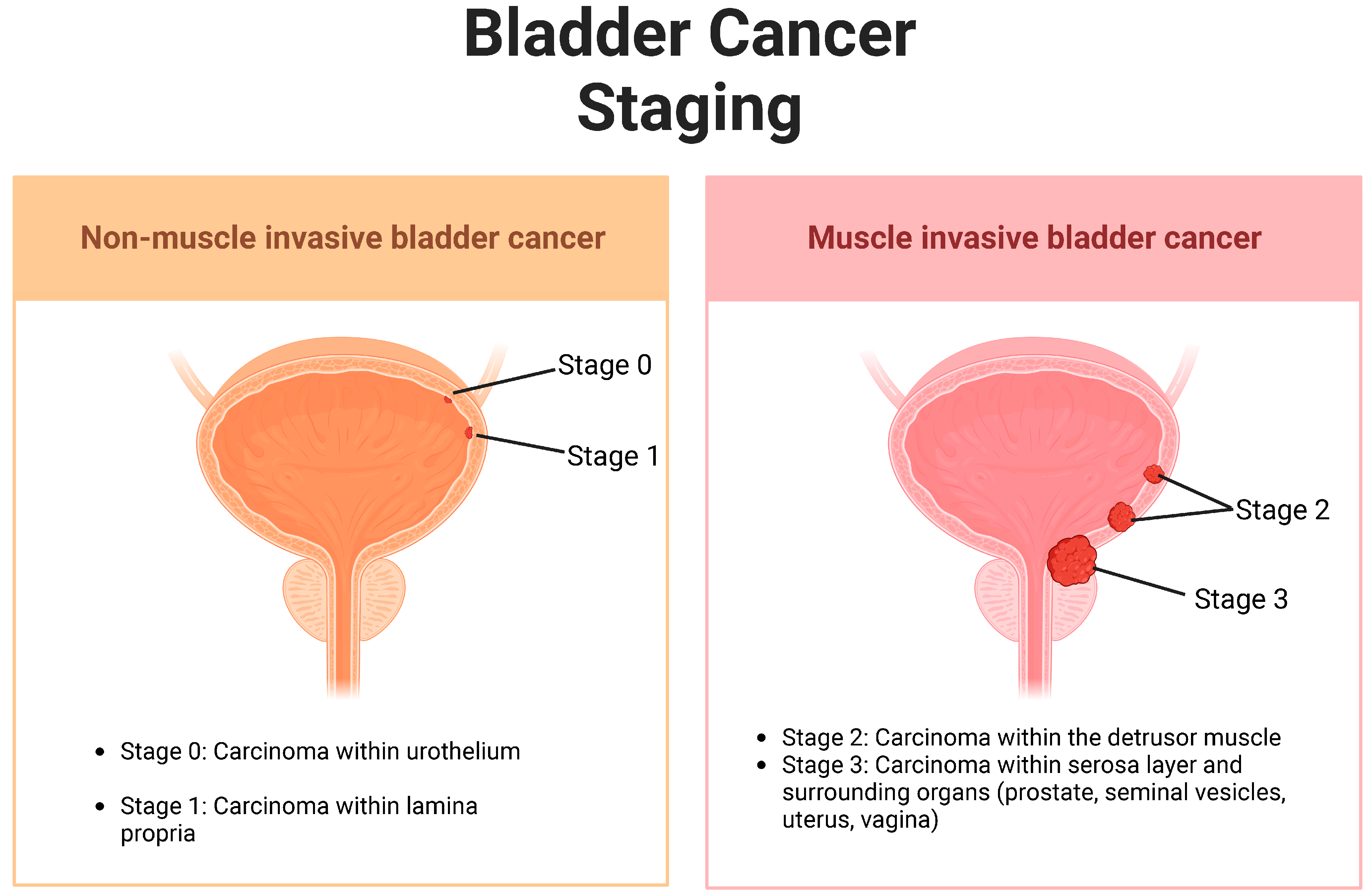
1. Introduction: Urothelial Carcinoma
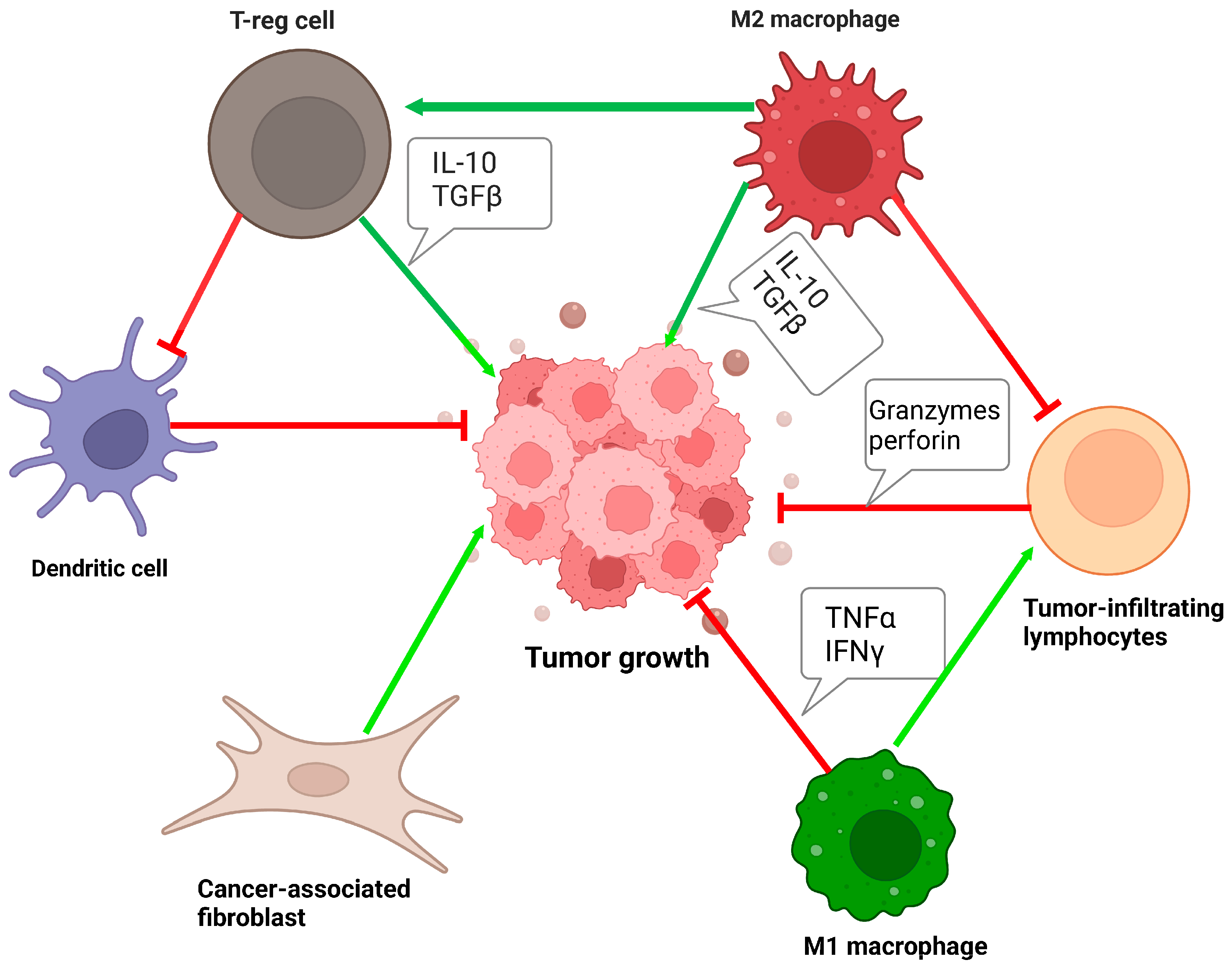
1.1. Tumor Microenvironment in Urothelial Carcinoma
1.2. Tumor Mutational Burden in Bladder Cancer
1.3. Tumor-Infiltrating Immune Cells in Bladder Cancer
2. Immune Checkpoints in Urothelial Carcinoma: Mechanism of Action
2.1. Program Death 1 (PD-1)
2.2. Programmed Death-Ligand 1 (PD-L1)
2.3. Cytotoxic T-Lymphocyte-Associated Protein-4 (CTLA-4)
3. FDA-Approved Immune Checkpoint Inhibitors in Urothelial Carcinoma
3.1. Pembrolizumab
3.2. Nivolumab
3.3. Avelumab
4. ICI Combination Therapies
4.1. Approved Combination Therapies in Other Cancers
4.2. Clinical Studies in UC
4.3. Future Directions and Controversies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jubber, I.; Ong, S.; Bukavina, L.; Black, P.C.; Comperat, E.; Kamat, A.M.; Kiemeney, L.; Lawrentschuk, N.; Lerner, S.P.; Meeks, J.J.; et al. Epidemiology of Bladder Cancer in 2023: A Systematic Review of Risk Factors. Eur. Urol. 2023, 84, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.; Afferi, L.; Moschini, M.; Mostafid, H.; Porten, S.; Psutka, S.P.; Gupta, S.; Smith, A.B.; Williams, S.B.; Lotan, Y. Epidemiology, Screening, and Prevention of Bladder Cancer. Eur. Urol. Oncol. 2022, 5, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Rani, B.; Ignatz-Hoover, J.J.; Rana, P.S.; Driscoll, J.J. Current and Emerging Strategies to Treat Urothelial Carcinoma. Cancers 2023, 15, 4886. [Google Scholar] [CrossRef] [PubMed]
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non–T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol. Res. 2016, 4, 563–568. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Bladder Cancer Stages—NCI; National Cancer Institute: Bethesda, MA, USA, 2023. [Google Scholar]
- Rhea, L.P.; Aragon-Ching, J.B. Advances and Controversies With Checkpoint Inhibitors in Bladder Cancer. Clin. Med. Insights Oncol. 2021, 15, 117955492110449. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.-X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef] [PubMed]
- Rouanne, M.; Roumiguié, M.; Houédé, N.; Masson-Lecomte, A.; Colin, P.; Pignot, G.; Larré, S.; Xylinas, E.; Rouprêt, M.; Neuzillet, Y. Development of immunotherapy in bladder cancer: Present and future on targeting PD(L)1 and CTLA-4 pathways. World J. Urol. 2018, 36, 1727–1740. [Google Scholar] [CrossRef]
- Meng, J.; Lu, X.; Zhou, Y.; Zhang, M.; Ge, Q.; Zhou, J.; Hao, Z.; Gao, S.; Yan, F.; Liang, C. Tumor immune microenvironment-based classifications of bladder cancer for enhancing the response rate of immunotherapy. Mol. Ther. Oncolytics 2021, 20, 410–421. [Google Scholar] [CrossRef]
- Tran, L.; Xiao, J.-F.; Agarwal, N.; Duex, J.E.; Theodorescu, D. Advances in bladder cancer biology and therapy. Nat. Rev. Cancer 2021, 21, 104–121. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Segersten, U.; Leiss, L.W.; Malmström, P.-U.; Hatina, J.; Östman, A.; Strell, C. Fibroblasts in urothelial bladder cancer define stroma phenotypes that are associated with clinical outcome. Sci. Rep. 2020, 10, 281. [Google Scholar] [CrossRef]
- Calvete, J.; Larrinaga, G.; Errarte, P.; Martín, A.M.; Dotor, A.; Esquinas, C.; Nunes-Xavier, C.E.; Pulido, R.; López, J.I.; Angulo, J.C. The coexpression of fibroblast activation protein (FAP) and basal-type markers (CK 5/6 and CD44) predicts prognosis in high-grade invasive urothelial carcinoma of the bladder. Hum. Pathol. 2019, 91, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERβ/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Goutas, D.; Palamaris, K.; Stofas, A.; Politakis, N.; Despotidi, A.; Giannopoulou, I.; Goutas, N.; Vlachodimitropoulos, D.; Kavantzas, N.; Lazaris, A.C.; et al. Immunohistochemical Study of Bladder Cancer Molecular Subtypes and Their Association with PD-L1 Expression. Cancers 2022, 15, 188. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e525. [Google Scholar] [CrossRef]
- Leão, R.; Lee, D.; Figueiredo, A.; Hermanns, T.; Wild, P.; Komosa, M.; Lau, I.; Mistry, M.; Nunes, N.M.; Price, A.J.; et al. Combined genetic and epigenetic alterations of the TERT promoter affect clinical and biological behavior of bladder cancer. Int. J. Cancer 2019, 144, 1676–1684. [Google Scholar] [CrossRef]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef]
- Deng, B.; Park, J.-H.; Ren, L.; Yew, P.Y.; Kiyotani, K.; Antic, T.; O’Connor, K.; O’Donnell, P.H.; Nakamura, Y. CD8 lymphocytes in tumors and nonsynonymous mutational load correlate with prognosis of bladder cancer patients treated with immune checkpoint inhibitors. Cancer Rep. 2018, 1, e1002. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.C.; Mason, A.S.; Slip, R.G.; Eriksson, P.; Sjödahl, G.; Trejdosiewicz, L.K.; Southgate, J. The Urothelial Transcriptomic Response to Interferon Gamma: Implications for Bladder Cancer Prognosis and Immunotherapy. Cancers 2022, 14, 5295. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Bolzenius, J.K.; Eteleeb, A.M.; Su, X.; Maher, C.A.; Sehn, J.K.; Arora, V.K. CD4+ T cells induce rejection of urothelial tumors after immune checkpoint blockade. JCI Insight 2018, 3, e121062. [Google Scholar] [CrossRef] [PubMed]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma. Cancers 2020, 12, 3401. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef]
- Mancini, M.; Righetto, M.; Noessner, E. Checkpoint Inhibition in Bladder Cancer: Clinical Expectations, Current Evidence, and Proposal of Future Strategies Based on a Tumor-Specific Immunobiological Approach. Cancers 2021, 13, 6016. [Google Scholar] [CrossRef]
- Wong, R.M.; Cameron, R.B. Immune Checkpoint Blockade and Adaptive Immune Resistance in Cancer; InTech: London, UK, 2017. [Google Scholar]
- Zhang, W.; Shi, L.; Zhao, Z.; Du, P.; Ye, X.; Li, D.; Cai, Z.; Han, J.; Cai, J. Disruption of CTLA-4 expression on peripheral blood CD8 + T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother. Pharmacol. 2019, 83, 911–920. [Google Scholar] [CrossRef]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.-J.; Liu, Z.; Cheng, Q. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Jang, B.-C.; Lee, S.-W.; Yang, Y.-I.; Suh, S.-I.; Park, Y.-M.; Oh, S.; Shin, J.-G.; Yao, S.; Chen, L.; et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006, 580, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, Q.; Xie, K.; Cheng, L.; Peng, S.; Xie, R.; Liu, L.; Zhang, Y.; Dong, W.; Han, J.; et al. Targeting WD repeat domain 5 enhances chemosensitivity and inhibits proliferation and programmed death-ligand 1 expression in bladder cancer. J. Exp. Clin. Cancer Res. 2021, 40, 203. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Tao, H.; Si, X.; Chen, Q. The Histone H3 Lysine 4 Presenter WDR5 as an Oncogenic Protein and Novel Epigenetic Target in Cancer. Front. Oncol. 2018, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Hunter, E.; Salter, M.; Powell, R.; Dring, A.; Naithani, T.; Chatziioannou, M.E.; Gebregzabhar, A.; Issa, M.; Green, J.; Ng, S.; et al. Development and Validation of Blood-Based Predictive Biomarkers for Response to PD-1/PD-L1 Checkpoint Inhibitors: Evidence of a Universal Systemic Core of 3D Immunogenetic Profiling across Multiple Oncological Indications. Cancers 2023, 15, 2696. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Crist, M.; Iyer, G.; Hsu, M.; Huang, W.C.; Balar, A.V. Pembrolizumab in the treatment of locally advanced or metastatic urothelial carcinoma: Clinical trial evidence and experience. Ther. Adv. Urol. 2019, 11, 175628721983928. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cimadamore, A.; Blanca, A.; Massari, F.; Vau, N.; Scarpelli, M.; Cheng, L.; Montironi, R. Immune Checkpoint Inhibitors for the Treatment of Bladder Cancer. Cancers 2021, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; De Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; De Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Bajorin, D.F.; Witjes, J.A.; Gschwend, J.E.; Schenker, M.; Valderrama, B.P.; Tomita, Y.; Bamias, A.; Lebret, T.; Shariat, S.F.; Park, S.H.; et al. Adjuvant Nivolumab versus Placebo in Muscle-Invasive Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 2102–2114. [Google Scholar] [CrossRef]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Lebbé, C.; Meyer, N.; Mortier, L.; Marquez-Rodas, I.; Robert, C.; Rutkowski, P.; Menzies, A.M.; Eigentler, T.; Ascierto, P.A.; Smylie, M.; et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J. Clin. Oncol. 2019, 37, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Van Der Heijden, M.S.; Castellano, D.; Galsky, M.D.; Loriot, Y.; Petrylak, D.P.; Ogawa, O.; Park, S.H.; Lee, J.-L.; De Giorgi, U.; et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1574–1588. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siefker-Radtke, A.; De Braud, F.; Basso, U.; Calvo, E.; Bono, P.; Morse, M.A.; Ascierto, P.A.; Lopez-Martin, J.; Brossart, P.; et al. Nivolumab Alone and With Ipilimumab in Previously Treated Metastatic Urothelial Carcinoma: CheckMate 032 Nivolumab 1 mg/kg Plus Ipilimumab 3 mg/kg Expansion Cohort Results. J. Clin. Oncol. 2019, 37, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3′s Enigmatic Mechanism of Action. Front. Immunol. 2020, 11, 615317. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) Modulates the Ability of CD4 T-cells to Be Suppressed In Vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef]
- Zeng, H.; Zhou, Q.; Wang, Z.; Zhang, H.; Liu, Z.; Huang, Q.; Wang, J.; Chang, Y.; Bai, Q.; Xia, Y.; et al. Stromal LAG-3+ cells infiltration defines poor prognosis subtype muscle-invasive bladder cancer with immunoevasive contexture. J. Immunother. Cancer 2020, 8, e000651. [Google Scholar] [CrossRef]
- Vanguri, R.S.; Smithy, J.W.; Li, Y.; Zhuang, M.; Maher, C.A.; Aleynick, N.; Peng, X.; Al-Ahmadie, H.; Funt, S.A.; Rosenberg, J.E.; et al. Integration of peripheral blood- and tissue-based biomarkers of response to immune checkpoint blockade in urothelial carcinoma. J. Pathol. 2023, 261, 349–360. [Google Scholar] [CrossRef]
- Attalla, K.; Farkas, A.M.; Anastos, H.; Audenet, F.; Galsky, M.D.; Bhardwaj, N.; Sfakianos, J.P. TIM-3 and TIGIT are possible immune checkpoint targets in patients with bladder cancer. Urol. Oncol. 2022, 40, 403–406. [Google Scholar] [CrossRef]
- Galsky, M.D.; Balar, A.V.; Black, P.C.; Campbell, M.T.; Dykstra, G.S.; Grivas, P.; Gupta, S.; Hoimes, C.J.; Lopez, L.P.; Meeks, J.J.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immunotherapy for the treatment of urothelial cancer. J. Immunother. Cancer 2021, 9, e002552. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Sun, J.; Wang, L.; Li, Z.; Wang, L.; Li, Z. Prognostic and Clinicopathological Significance of PD-L1 in Patients with Bladder Cancer: A Meta-Analysis. Front. Pharmacol. 2019, 10, 962. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Cai, X.; Xie, Z.; Zhou, L.; Cheng, B.; Zhong, R.; Xiong, S.; Li, J.; Chen, Z.; et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. eClinicalMedicine 2021, 41, 101134. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Rosenberg, J.E. The biology and rationale of targeting nectin-4 in urothelial carcinoma. Nat. Rev. Urol. 2021, 18, 93–103. [Google Scholar] [CrossRef]
- Powles, T.B.; Perez Valderrama, B.; Gupta, S.; Bedke, J.; Kikuchi, E.; Hoffman-Censits, J.; Iyer, G.; Vulsteke, C.; Park, S.H.; Shin, S.J.; et al. LBA6 EV-302/KEYNOTE-A39: Open-label, randomized phase III study of enfortumab vedotin in combination with pembrolizumab (EV+P) vs chemotherapy (Chemo) in previously untreated locally advanced metastatic urothelial carcinoma (la/mUC). Ann. Oncol. 2023, 34, S1340. [Google Scholar] [CrossRef]
- Zengin, Z.B.; Chehrazi-Raffle, A.; Salgia, N.J.; Muddasani, R.; Ali, S.; Meza, L.; Pal, S.K. Targeted therapies: Expanding the role of FGFR3 inhibition in urothelial carcinoma. Urol. Oncol. 2022, 40, 25–36. [Google Scholar] [CrossRef]
- Loriot, Y.; Matsubara, N.; Park, S.H.; Huddart, R.A.; Burgess, E.F.; Houede, N.; Banek, S.; Guadalupi, V.; Ku, J.H.; Valderrama, B.P.; et al. Erdafitinib or Chemotherapy in Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2023, 389, 1961–1971. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Treatment | Name and Year | Phase | Line of Therapy | N | Findings |
|---|---|---|---|---|---|
| Pembrolizumab vs. chemotherapy | Keynote-045, 2017 | III | Second | 542 | OS: 10.3 vs. 7.4 months (all patients) OS: 8 vs. 5.2 months (for PD-L1 ≥ 10%) |
| Nivolumab post platinum-based chemotherapy | CheckMate 275, 2017 | II | Second | 270 | ORR: 19.6% (52/265) |
| Avelumab maintenance vs. best supportive care | Javelin Bladder 100, 2020 | III | First | 700 | OS: 21.4 vs. 14.3 months |
| Cancer Type | Treatment | Phase | Findings | Adverse Effects |
|---|---|---|---|---|
| NSLC—stage IV/recurrent | Nivolumab (360 mg IV) + ipilimumab (1 mg/kg) + chemotherapy vs. chemotherapy alone | III | OS: 14.1 vs. 10.7 months | Grade 3–4: 47% vs. 38% |
| RCC—advanced stage | Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) vs. sunitinib | III | ORR: 42% vs. 27% | Grade 3–4: 46% vs. 63% |
| Melanoma—stages III and IV | Nivolumab (1 mg/kg) + ipilimumab (3 mg/kg) vs. nivolumab alone (3 mg/kg) vs. ipilimumab alone (3 mg/kg) | III | PFS: 11.5 vs. 6.9 vs. 2.9 months | Grade 3–4: 59% vs. 22% vs. 28% |
| Melanoma—advanced stage | Nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) vs. nivolumab (1 mg/kg) + ipilimumab (3 mg/kg) | IIIb/IV | ORR: 50.6% vs. 48%PFS: 9.9 months vs. 8.9 months | Grade 3–5: 34% vs. 48% |
| Urothelial carcinoma—metastatic | Nivolumab (3 mg/kg) vs. nivolumab (3 mg/kg) + ipilimumab (1 mg/kg) vs. nivolumab (1 mg/kg) + ipilimumab (3 mg/kg) | I/II | ORR: 25.6% vs. 26.9% vs. 38% | Grade 3–4: 26.9% vs. 30.8% vs. 39.1% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogbuji, V.; Paster, I.C.; Recio-Boiles, A.; Carew, J.S.; Nawrocki, S.T.; Chipollini, J. Current Landscape of Immune Checkpoint Inhibitors for Metastatic Urothelial Carcinoma: Is There a Role for Additional T-Cell Blockade? Cancers 2024, 16, 131. https://doi.org/10.3390/cancers16010131
Ogbuji V, Paster IC, Recio-Boiles A, Carew JS, Nawrocki ST, Chipollini J. Current Landscape of Immune Checkpoint Inhibitors for Metastatic Urothelial Carcinoma: Is There a Role for Additional T-Cell Blockade? Cancers. 2024; 16(1):131. https://doi.org/10.3390/cancers16010131
Chicago/Turabian StyleOgbuji, Vanessa, Irasema C. Paster, Alejandro Recio-Boiles, Jennifer S. Carew, Steffan T. Nawrocki, and Juan Chipollini. 2024. "Current Landscape of Immune Checkpoint Inhibitors for Metastatic Urothelial Carcinoma: Is There a Role for Additional T-Cell Blockade?" Cancers 16, no. 1: 131. https://doi.org/10.3390/cancers16010131
APA StyleOgbuji, V., Paster, I. C., Recio-Boiles, A., Carew, J. S., Nawrocki, S. T., & Chipollini, J. (2024). Current Landscape of Immune Checkpoint Inhibitors for Metastatic Urothelial Carcinoma: Is There a Role for Additional T-Cell Blockade? Cancers, 16(1), 131. https://doi.org/10.3390/cancers16010131