
Potential Immunotherapy Targets for Liver-Directed Therapies, and the Current Scope of Immunotherapeutics for Liver-Related Malignancies
 , , , , ,
, , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Liver Tolerogenic Environment
1.2. Hepatic Immune Dysfunction in the Setting of Underlying Fibrosis
1.3. Innate Immune System Dysfunction
1.4. Adaptive Immune System Dysfunction
1.5. Effector Cell Dysfunction
1.6. Liver Tumor Immunobiology: Primary and Metastatic
2. Systemic Therapies for Liver Primary and Metastatic Cancers
2.1. Primary Liver Cancer: HCC
2.2. Primary Liver Cancer: Cholangiocarcinoma
2.3. Colorectal Liver Metastasis
3. Targeted Immunotherapy
3.1. Targeted Immunotherapy versus Non-Specific Systemic Therapy
3.2. Immune Checkpoint Inhibitor Monotherapy
3.3. CTLA-4 Pathway
3.4. PD-1/PD-L1 Pathway
3.5. T-Cell Immunoglobulin and Mucin-Domain Containing Protein-3
3.6. Lymphocyte Activation Gene-3
3.7. Co-Stimulatory Molecules
4. Epigenetics and Targets
4.1. DNA Methylation
4.2. Noncoding RNAs
4.3. Chromatin Modifiers
4.4. Histone Deacetylation
5. Combination Immune Checkpoint Inhibitors and Targeted Therapies
5.1. Angiogenesis and Immunosuppression
5.2. Combination ICIs + ICIs
5.3. Combination ICIs + Chemotherapy
5.4. Combination ICIs + Epigenetic Treatments
6. The Role of Interventional Radiology in Targeted Immunotherapy
6.1. Ablation Plus ICIs
6.2. TACE Plus ICIs
6.3. TARE Pus ICIs
7. Looking Ahead
7.1. Cabozantinib
7.2. Capmatinib
7.3. Decitabine
7.4. Oncolytic Viruses
7.5. Mesenchymal Stem Cells
7.6. Interferon Alpha
7.7. Looking Forward as an Interventional Radiologist
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Trends in Liver Cancer Mortality Among Adults Aged 25 and Over in the United States, 2000–2016. NCHS Data Brief 2018, 314, 1–8. [Google Scholar]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Keenan, B.P.; Fong, L.; Kelley, R.K. Immunotherapy in hepatocellular carcinoma: The complex interface between inflammation, fibrosis, and the immune response. J. Immunother. Cancer 2019, 7, 267. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef]
- Wilkinson, A.L.; Qurashi, M.; Shetty, S. The Role of Sinusoidal Endothelial Cells in the Axis of Inflammation and Cancer Within the Liver. Front. Physiol. 2020, 11, 990. [Google Scholar] [CrossRef]
- Bamboat, Z.M.; Stableford, J.A.; Plitas, G.; Burt, B.M.; Nguyen, H.M.; Welles, A.P.; Gonen, M.; Young, J.W.; DeMatteo, R.P. Human Liver Dendritic Cells Promote T Cell Hyporesponsiveness. J. Immunol. 2009, 182, 1901–1911. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Kumar, S.; Wang, J.; Thomson, A.W.; Gandhi, C.R. Hepatic stellate cells increase the immunosuppressive function of natural Foxp3+ regulatory T cells via IDO-induced AhR activation. J. Leukoc. Biol. 2017, 101, 429–438. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Tanwar, S.; Rhodes, F.; Srivastava, A.; Trembling, P.M.; Rosenberg, W. Inflammation and fibrosis in chronic liver diseases including non-alcoholic fatty liver disease and hepatitis C. World J. Gastroenterol. 2020, 26, 109–133. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ge, W.; Zhou, J.; Gao, B.; Qian, X.; Wang, W. The Role of Tumor Associated Macrophages in Hepatocellular Carcinoma. J. Cancer 2021, 12, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; He, K.; Pan, Y.; Wang, H.; Luo, Y.; Xia, Q. The role of tumor-associated macrophages in primary hepatocellular carcinoma and its related targeting therapy. Int. J. Med. Sci. 2021, 18, 2109–2116. [Google Scholar] [CrossRef]
- Lu, L.-C.; Chang, C.-J.; Hsu, C.-H. Targeting myeloid-derived suppressor cells in the treatment of hepatocellular carcinoma: Current state and future perspectives. J. Hepatocell. Carcinoma 2019, 6, 71–84. [Google Scholar] [CrossRef]
- Jayant, K.; Habib, N.; Huang, K.W.; Warwick, J.; Arasaradnam, R. Recent Advances: The Imbalance of Immune Cells and Cytokines in the Pathogenesis of Hepatocellular Carcinoma. Diagnostics 2020, 10, 338. [Google Scholar] [CrossRef]
- Lurje, I.; Hammerich, L.; Tacke, F. Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer. Int. J. Mol. Sci. 2020, 21, 7378. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Fujiwara, K.; Higashi, T.; Nouso, K.; Nakatsukasa, H.; Kobayashi, Y.; Uemura, M.; Nakamura, S.-I.; Sato, S.; Hanafusa, T.; Yumoto, Y.; et al. Decreased expression of B7 costimulatory molecules and major histocompatibility complex class-I in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2004, 19, 1121–1127. [Google Scholar] [CrossRef]
- Tatsumi, T.; Takehara, T.; Katayama, K.; Mochizuki, K.; Yamamoto, M.; Kanto, T.; Sasaki, Y.; Kasahara, A.; Hayashi, N. Expression of costimulatory molecules B7-1 (CD80) and B7-2 (CD86) on human hepatocellular carcinoma. Hepatology 1997, 25, 1108–1114. [Google Scholar] [CrossRef]
- Saviano, A.; Roehlen, N.; Virzì, A.; Suarez, A.A.R.; Hoshida, Y.; Lupberger, J.; Baumert, T.F. Stromal and Immune Drivers of Hepatocarcinogenesis. In Hepatocellular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Humana Press: Cham, Switzerland, 2019; pp. 317–331. [Google Scholar]
- Cariani, E.; Missale, G. Immune landscape of hepatocellular carcinoma microenvironment: Implications for prognosis and therapeutic applications. Liver Int. 2019, 39, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Lin, J.; Long, J.; Yang, X.; Yang, X.; Lu, X.; Sang, X.; Zhao, H. T lymphocytes in hepatocellular carcinoma immune microenvironment: Insights into human immunology and immunotherapy. Am. J. Cancer Res. 2020, 10, 4585–4606. [Google Scholar] [PubMed]
- Mantovani, S.; Oliviero, B.; Varchetta, S.; Mele, D.; Mondelli, M.U. Natural Killer Cell Responses in Hepatocellular Carcinoma: Implications for Novel Immunotherapeutic Approaches. Cancers 2020, 12, 926. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Pedrosa, L.; Esposito, F.; Thomson, T.M.; Maurel, J. The Tumor Microenvironment in Colorectal Cancer Therapy. Cancers 2019, 11, 1172. [Google Scholar] [CrossRef]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: Challenges and opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef]
- Wang, J.; Ilyas, S. Targeting the tumor microenvironment in cholangiocarcinoma: Implications for therapy. Expert Opin. Investig. Drugs 2021, 30, 429–438. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2006, 10, 25–34. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Y.; Liu, S. The new insight of treatment in Cholangiocarcinoma. J. Cancer 2022, 13, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Weigt, J.; Malfertheiner, P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 395–397. [Google Scholar] [CrossRef]
- Morizane, C.; Okusaka, T.; Mizusawa, J.; Katayama, H.; Ueno, M.; Ikeda, M.; Ozaka, M.; Okano, N.; Sugimori, K.; Fukutomi, A.; et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: The FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann. Oncol. 2019, 30, 1950–1958. [Google Scholar] [CrossRef]
- Ioka, T.; Kanai, M.; Kobayashi, S.; Sakai, D.; Eguchi, H.; Baba, H.; Seo, S.; Taketomi, A.; Takayama, T.; Yamaue, H.; et al. Randomized phase III study of gemcitabine, cisplatin plus S-1 versus gemcitabine, cisplatin for advanced biliary tract cancer (KHBO1401- MITSUBA). J. Hepatobiliary Pancreat. Sci. 2023, 30, 102–110. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Valle, J.W.; Van Cutsem, E.; Rimassa, L.; Furuse, J.; Ioka, T.; Melisi, D.; Macarulla, T.; Bridgewater, J.; Wasan, H.; et al. FIGHT-302: First-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Future Oncol. 2020, 16, 2385–2399. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Frega, G.; Ricci, A.D.; Palloni, A.; Abbati, F.; De Lorenzo, S.; Deserti, M.; Tavolari, S.; Brandi, G. Anti-EGFR Monoclonal Antibodies in Advanced Biliary Tract Cancer: A Systematic Review and Meta-analysis. Vivo 2020, 34, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Nordlinger, B.; Sorbye, H.; Glimelius, B.; Poston, G.J.; Schlag, P.M.; Rougier, P.; Bechstein, W.O.; Primrose, J.N.; Walpole, E.T.; Finch-Jones, M.; et al. Perioperative FOLFOX4 chemotherapy and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC 40983): Long-term results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2013, 14, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Gruenberger, B.; Scheithauer, W.; Punzengruber, R.; Zielinski, C.; Tamandl, D.; Gruenberger, T. Importance of response to neoadjuvant chemotherapy in potentially curable colorectal cancer liver metastases. BMC Cancer 2008, 8, 120. [Google Scholar] [CrossRef]
- Khoo, E.; O’Neill, S.; Brown, E.; Wigmore, S.J.; Harrison, E.M. Systematic review of systemic adjuvant, neoadjuvant and perioperative chemotherapy for resectable colorectal-liver metastases. HPB 2016, 18, 485–493. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Lin, X.; Wang, F.-H.; Goodman, K.; Cai, P.-Q.; Kong, L.-H.; Fang, Y.-J.; Gao, Y.-H.; Lin, J.-Z.; Wan, D.-S.; et al. Short term results of neoadjuvant chemoradiotherapy with fluoropyrimidine alone or in combination with oxaliplatin in locally advanced rectal cancer: A meta analysis. Eur. J. Cancer 2013, 49, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Saltz, L.B.; Cox, J.V.; Blanke, C.; Rosen, L.S.; Fehrenbacher, L.; Moore, M.J.; Maroun, J.A.; Ackland, S.P.; Locker, P.K.; Pirotta, N.; et al. Irinotecan plus Fluorouracil and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef]
- Falcone, A.; Ricci, S.; Brunetti, I.; Pfanner, E.; Allegrini, G.; Barbara, C.; Crinò, L.; Benedetti, G.; Evangelista, W.; Fanchini, L.; et al. Phase III Trial of Infusional Fluorouracil, Leucovorin, Oxaliplatin, and Irinotecan (FOLFOXIRI) Compared With Infusional Fluorouracil, Leucovorin, and Irinotecan (FOLFIRI) As First-Line Treatment for Metastatic Colorectal Cancer: The Gruppo Oncologico Nord Ovest. J. Clin. Oncol. 2007, 25, 1670–1676. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, Leucovorin, and Oxaliplatin With and Without Cetuximab in the First-Line Treatment of Metastatic Colorectal Cancer. J. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Saltz, L.B.; Clarke, S.; Diaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.-S.; Rivera, F.; et al. Bevacizumab in Combination With Oxaliplatin-Based Chemotherapy As First-Line Therapy in Metastatic Colorectal Cancer: A Randomized Phase III Study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef]
- Al Bandar, M.H.; Kim, N.K. Current status and future perspectives on treatment of liver metastasis in colorectal cancer (Review). Oncol. Rep. 2017, 37, 2553–2564. [Google Scholar] [CrossRef] [PubMed]
- Knuth, A.; Wölfel, T.; Klehmann, E.; Boon, T.; Büschenfelde, K.H.M.Z. Cytolytic T-cell clones against an autologous human melanoma: Specificity study and definition of three antigens by immunoselection. Proc. Natl. Acad. Sci. USA 1989, 86, 2804–2808. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Nowak, A.K. Immunological checkpoint inhibitors enter adolescence. Lancet Oncol. 2013, 14, 1035–1037. [Google Scholar] [CrossRef]
- Walker, L.S.K.; Sansom, D. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kroemer, G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology 2012, 1, 1223–1225. [Google Scholar] [CrossRef]
- Munir, S.; Andersen, G.H.; Svane, I.M.; Andersen, M.H. The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4+ T cells. Oncoimmunology 2013, 2, e23991. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.M.; Buqué, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.-P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef]
- Lacy, M.Q.; Gertz, M.A.; Dispenzieri, A.; Hayman, S.R.; Geyer, S.; Kabat, B.; Zeldenrust, S.R.; Kumar, S.; Greipp, P.R.; Fonseca, R.; et al. Long-term Results of Response to Therapy, Time to Progression, and Survival With Lenalidomide Plus Dexamethasone in Newly Diagnosed Myeloma. Mayo Clin. Proc. 2007, 82, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A. Prognostic factors in multiple myeloma. Stem Cells 1995, 13, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Meserve, J.; Facciorusso, A.; Holmer, A.K.; Annese, V.; Sandborn, W.J.; Singh, S. Systematic review with meta-analysis: Safety and tolerability of immune checkpoint inhibitors in patients with pre-existing inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2020, 53, 374–382. [Google Scholar]
- Amiot, A.; Laharie, D.; Malamut, G.; Serrero, M.; Poullenot, F.; Peyrin-Biroulet, L.; Bourreille, A.; Vuitton, L.; Bouguen, G.; Abitbol, V.; et al. Management of immune checkpoint inhibitor in patients with cancer and pre-existing inflammatory bowel disease: Recommendations from the GETAID. Dig. Liver Dis. 2022, 54, 1162–1167. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Magistrelli, G.; Jeannin, P.; Herbault, N.; De Coignac, A.B.; Gauchat, J.F.; Bonnefoy, J.Y.; Delneste, Y. A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur. J. Immunol. 1999, 29, 3596–3602. [Google Scholar] [CrossRef]
- Lee, K.-M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular Basis of T Cell Inactivation by CTLA-4. Science 1998, 282, 2263–2266. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- O’Neill, R.E.; Cao, X. Chapter Three—Co-stimulatory and co-inhibitory pathways in cancer immunotherapy. In Advances in Cancer Research; Wang, X.-Y., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 145–194. [Google Scholar]
- Fritz, J.M.; Lenardo, M.J. Development of immune checkpoint therapy for cancer. J. Exp. Med. 2019, 216, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, D.W.-M.; Lim, H.Y.; Yau, T.; et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Guo, X.J.; Lu, J.-C.; Zeng, H.-Y.; Zhou, R.; Sun, Q.-M.; Yang, G.-H.; Pei, Y.-Z.; Meng, X.-L.; Shen, Y.-H.; Zhang, P.-F.; et al. CTLA-4 Synergizes With PD1/PD-L1 in the Inhibitory Tumor Microenvironment of Intrahepatic Cholangiocarcinoma. Front. Immunol. 2021, 12, 705378. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann. N. Y. Acad. Sci. 2011, 1217, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune Dilated Cardiomyopathy in PD-1 Receptor-Deficient Mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Harkus, U.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Immune checkpoint inhibitors in HCC: Cellular, molecular and systemic data. Semin. Cancer Biol. 2022, 86, 799–815. [Google Scholar] [CrossRef]
- Sukowati, C.H.C.; El-Khobar, K.E.; Tiribelli, C. Immunotherapy against programmed death-1/programmed death ligand 1 in hepatocellular carcinoma: Importance of molecular variations, cellular heterogeneity, and cancer stem cells. World J. Stem Cells 2021, 13, 795–824. [Google Scholar] [CrossRef]
- Pedoeem, A.; Azoulay-Alfaguter, I.; Strazza, M.; Silverman, G.J.; Mor, A. Programmed death-1 pathway in cancer and autoimmunity. Clin. Immunol. 2014, 153, 145–152. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef] [PubMed]
- Ohigashi, Y.; Sho, M.; Yamada, Y.; Tsurui, Y.; Hamada, K.; Ikeda, N.; Mizuno, T.; Yoriki, R.; Kashizuka, H.; Yane, K.; et al. Clinical Significance of Programmed Death-1 Ligand-1 and Programmed Death-1 Ligand-2 Expression in Human Esophageal Cancer. Clin. Cancer Res. 2005, 11, 2947–2953. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.-Z.; Liu, Z.-W.; Zhang, J.-Y.; Yang, Y.-P.; Tien, P.; Wang, F.-S. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.I.; Jeong, D.; Ji, S.; Ahn, T.S.; Bae, S.H.; Chin, S.; Chung, J.C.; Kim, H.C.; Lee, M.S.; Baek, M.-J. Overexpression of PD-L1 and PD-L2 Is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. Cancer Res. Treat. 2017, 49, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.-S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship With clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xue, J.; Hu, J.; Yang, S.-L.; Chen, G.G.; Lai, P.B.; Yu, C.; Zeng, C.; Fang, X.; Pan, X.; et al. Positive Expression of Programmed Death Ligand 1 in Peritumoral Liver Tissue is Associated with Poor Survival after Curative Resection of Hepatocellular Carcinoma. Transl. Oncol. 2017, 10, 511–517. [Google Scholar] [CrossRef]
- Semaan, A.; Dietrich, D.; Bergheim, D.; Dietrich, J.; Kalff, J.C.; Branchi, V.; Matthaei, H.; Kristiansen, G.; Fischer, H.-P.; Goltz, D. CXCL12 expression and PD-L1 expression serve as prognostic biomarkers in HCC and are induced by hypoxia. Virchows Arch. 2017, 470, 185–196. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Segal, N.H.; Jaeger, D.; Lee, K.-H.; Marshall, J.; Antonia, S.J.; Butler, M.; Sanborn, R.E.; Nemunaitis, J.J.; Carlson, C.A.; et al. Safety and clinical activity of durvalumab monotherapy in patients with hepatocellular carcinoma (HCC). J. Clin. Oncol. 2017, 35, 4071. [Google Scholar] [CrossRef]
- Shi, Y.; Su, H.; Song, Y.; Jiang, W.; Sun, X.; Qian, W.; Zhang, W.; Gao, Y.; Jin, Z.; Zhou, J.; et al. Circulating tumor DNA predicts response in Chinese patients with relapsed or refractory classical hodgkin lymphoma treated with sintilimab. Ebiomedicine 2020, 54, 102731. [Google Scholar] [CrossRef]
- Wang, J.; Fei, K.; Jing, H.; Wu, Z.; Wu, W.; Zhou, S.; Ni, H.; Chen, B.; Xiong, Y.; Liu, Y.; et al. Durable blockade of PD-1 signaling links preclinical efficacy of sintilimab to its clinical benefit. mAbs 2019, 11, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-M.; Jia, R.; Wang, Y.; Liu, R.; Zhao, C.; Zhou, H.; Xu, L.; Kong, X. A first-in-human phase 1a trial of sintilimab (IBI308), a monoclonal antibody targeting programmed death-1 (PD-1), in Chinese patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, e15125. [Google Scholar] [CrossRef]
- Tao, R.; Fan, L.; Song, Y.; Hu, Y.; Zhang, W.; Wang, Y.; Xu, L.; Zhou, H.; Li, J. Sintilimab for relapsed/refractory (r/r) extranodal NK/T-cell lymphoma (ENKTL): A multicenter, single-arm, phase 2 trial (ORIENT-4). J. Clin. Oncol. 2019, 37, 7504. [Google Scholar] [CrossRef]
- Zheng, R.; Chen, X.; Wang, C.; Qin, P.; Tan, H.; Luo, X. Triplet Therapy with PD-1 Blockade, Histone Deacetylase Inhibitor, and DNA Methyltransferase Inhibitor Achieves Radiological Response in Refractory Double-Expressor Diffuse Large B-cell Lymphoma with 17p Deletion. Case Rep. Hematol. 2020, 2020, 8879448. [Google Scholar] [CrossRef]
- Gao, S.; Li, N.; Gao, S.; Xue, Q.; Ying, J.; Wang, S.; Tao, X.; Zhao, J.; Mao, Y.; Wang, B.; et al. Neoadjuvant PD-1 inhibitor (Sintilimab) in NSCLC. J. Thorac. Oncol. 2020, 15, 816–826. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Chaft, J.E.; William, W.N., Jr.; Rusch, V.; Pisters, K.M.W.; Kalhor, N.; Pataer, A.; Travis, W.D.; Swisher, S.G.; Kris, M.G.; et al. Pathological response after neoadjuvant chemotherapy in resectable non-small-cell lung cancers: Proposal for the use of major pathological response as a surrogate endpoint. Lancet Oncol. 2014, 15, e42–e50. [Google Scholar] [CrossRef]
- Jiang, H.; Zheng, Y.; Qian, J.; Mao, C.; Xu, X.; Li, N.; Xiao, C.; Wang, H.; Teng, L.; Zhou, H.; et al. Safety and efficacy of sintilimab combined with oxaliplatin/capecitabine as first-line treatment in patients with locally advanced or metastatic gastric/gastroesophageal junction adenocarcinoma in a phase Ib clinical trial. BMC Cancer 2020, 20, 760. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y.; Fan, Q.; Shu, Y.; Wu, Z.; Cui, T.; Gu, K.; Tao, M.; Wang, X.; Cui, C.; et al. Sintilimab in patients with advanced esophageal squamous cell carcinoma refractory to previous chemotherapy: A randomized, open-label phase II trial (ORIENT-2). J. Clin. Oncol. 2020, 38, 4511. [Google Scholar] [CrossRef]
- Duan, X.; Zhang, H.; Zhou, L.; Jiang, B.; Mao, X. Complete response to the combination of sintilimab and IBI305 for a patient with HBV-associated hepatocellular carcinoma with multiple lung metastasis. Dig. Liver Dis. 2020, 52, 794–796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, L.; Liu, J.; Lin, M. A metastatic intrahepatic cholangiocarcinoma treated with programmed cell death 1 inhibitor: A case report and literature review. Immunotherapy 2020, 12, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, C.; Cao, L.; Li, S. Treatment of advanced intrahepatic cholangiocarcinoma with sintilimab combined with tegafur-gimeracil-oteracil potassium capsules (S-1): A case report. Ann. Palliat. Med. 2020, 9, 497–503. [Google Scholar] [CrossRef]
- Grenga, I.; Donahue, R.N.; Lepone, L.M.; Richards, J.; Schlom, J. A fully human IgG1 anti-PD-L1 MAb in an in vitro assay enhances antigen-specific T-cell responses. Clin. Transl. Immunol. 2016, 5, e83. [Google Scholar] [CrossRef]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-Dependent Cellular Cytotoxicity Activity of a Novel Anti–PD-L1 Antibody Avelumab (MSB0010718C) on Human Tumor CellsAn Anti–PD-L1 mAb That Mediates ADCC of Human Tumor Cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef]
- Heery, C.R.; O’Sullivan-Coyne, G.; Madan, R.A.; Cordes, L.; Rajan, A.; Rauckhorst, M.; Lamping, E.; Oyelakin, I.; Marté, J.L.; Lepone, L.M.; et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): A phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 2017, 18, 587–598. [Google Scholar] [CrossRef]
- Gulley, J.L.; Rajan, A.; Spigel, D.R.; Iannotti, N.; Chandler, J.; Wong, D.J.L.; Leach, J.; Edenfield, W.J.; Wang, D.; Grote, H.J.; et al. Avelumab for patients with previously treated metastatic or recurrent non-small-cell lung cancer (JAVELIN Solid Tumor): Dose-expansion cohort of a multicentre, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 599–610. [Google Scholar] [CrossRef]
- Rajan, A.; Gulley, J.L.; Spigel, D.R.; Iannotti, N.; Chandler, J.C.; Wong, D.J.L.; Leach, J.W.; Edenfield, W.J.; Wang, D.; Redfern, C.H.; et al. Avelumab (anti–PD-L1) in patients with platinum-treated advanced NSCLC: 2.5-year follow-up from the JAVELIN Solid Tumor trial. J. Clin. Oncol. 2018, 36, 9090. [Google Scholar] [CrossRef]
- Shaw, A.T.; Lee, S.-H.; Ramalingam, S.S.; Bauer, T.M.; Boyer, M.J.; Costa, E.C.; Felip, E.; Han, J.-Y.; Hida, T.; Hughes, B.G.M.; et al. Avelumab (anti–PD-L1) in combination with crizotinib or lorlatinib in patients with previously treated advanced NSCLC: Phase 1b results from JAVELIN Lung 101. J. Clin. Oncol. 2018, 36, 9008. [Google Scholar] [CrossRef]
- Keilholz, U.; Mehnert, J.M.; Bauer, S.; Bourgeois, H.P.; Patel, M.R.; Gravenor, D.; Nemunaitis, J.J.; Taylor, M.H.; Wyrwicz, L.; Lee, K.-W.; et al. Avelumab in patients with previously treated metastatic melanoma: Phase 1b results from the JAVELIN Solid Tumor trial. J. Clin. Oncol. 2018, 36, 191. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Larkin, J.; Oya, M.; Thistlethwaite, F.; Martignoni, M.; Nathan, P.; Powles, T.; McDermott, D.; Robbins, P.B.; Chism, D.D.; et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): An open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol. 2018, 19, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Arkenau, H.-T.; Wyrwicz, L.; Oh, D.-Y.; Lee, K.-W.; Infante, J.R.; Lee, S.S.; Lee, J.; Keilholz, U.; Mita, A.C.; et al. Avelumab (MSB0010718C; anti-PD-L1) in patients with advanced gastric or gastroesophageal junction cancer from JAVELIN solid tumor phase Ib trial: Analysis of safety and clinical activity. J. Clin. Oncol. 2016, 34, 4009. [Google Scholar] [CrossRef]
- Chung, H.C.; Arkenau, H.-T.; Lee, J.; Rha, S.Y.; Oh, D.-Y.; Wyrwicz, L.; Kang, Y.-K.; Lee, K.-W.; Bauer, T.M.; Lee, S.S.; et al. Abstract CT111: Avelumab (anti-PD-L1) as first-line maintenance (1L mn) or second-line (2L) therapy in patients with advanced gastric or gastroesophageal junction cancer (GC/GEJC): Updated phase Ib results from the JAVELIN Solid Tumor trial. Cancer Res. 2018, 78, CT111. [Google Scholar] [CrossRef]
- Disis, M.L.; Patel, M.R.; Pant, S.; Hamilton, E.P.; Lockhart, A.C.; Kelly, K.; Beck, J.T.; Gordon, M.S.; Weiss, G.J.; Taylor, M.H.; et al. Avelumab (MSB0010718C; anti-PD-L1) in patients with recurrent/refractory ovarian cancer from the JAVELIN Solid Tumor phase Ib trial: Safety and clinical activity. J. Clin. Oncol. 2016, 34, 5533. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Fujiwara, K.; Dychter, S.S.; Devgan, G.; Monk, B.J. Avelumab (anti-PD-L1) in platinum-resistant/refractory ovarian cancer: JAVELIN Ovarian 200 Phase III study design. Futur. Oncol. 2018, 14, 2103–2113. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef]
- Mayer, E.; Wander, S.; Regan, M.; DeMichele, A.; Forero, A.; Rimawi, M.; Ma, C.; Cristofanilli, M.; Anders, C.; Bartlett, C.H.; et al. Abstract OT3-05-11: Palbociclib after CDK inhibitor and endocrine therapy (PACE): A randomized phase II study of fulvestrant versus palbociclib plus fulvestrant, with and without avelumab, for CDK inhibitor pre-treated HR+/HER2- metastatic breast cancer. Cancer Res. 2018, 78, OT3-05. [Google Scholar] [CrossRef]
- Ben Salama, L.; Duerinck, J.; Du Four, S.; Awada, G.; Fischbuch, L.; De Cremer, J.; Rogiers, A.; Neyns, B. Safety of axitinib plus avelumab in patients with recurrent glioblastoma. J. Clin. Oncol. 2018, 36, e14082. [Google Scholar] [CrossRef]
- Hassan, R.; Thomas, A.; Nemunaitis, J.J.; Patel, M.R.; Bennouna, J.; Chen, F.; Delord, J.-P.; Dowlati, A.; Taylor, M.H.; Powderly, J.D.; et al. Avelumab in patients with previously treated mesothelioma: Updated phase 1b results from the JAVELIN Solid Tumor trial. J. Clin. Oncol. 2018, 36, 166. [Google Scholar] [CrossRef]
- Elbers, J.B.W.; Zuur, C.L.; Tesselaar, M.E.; Lange, C.; Al-Mamgani, A.; De Boer, J.P. Radiotherapy with concurrent Avelumab and Cetuximab as primary treatment in patients with locally advanced squamous cell carcinoma of the head and neck: A phase-IB feasibility trial in patients unfit for cisplatin (NCT02938273). J. Clin. Oncol. 2018, 36, e18019. [Google Scholar] [CrossRef]
- Tao, Y.; Auperin, A.; Sun, X.S.; Sire, C.; Martin, L.; Bera, G.; Coutte, A.; Miroir, J.; Lafond, C.; Colin-Batailhou, N.; et al. Avelumab-cetuximab-radiotherapy (RT) versus standards of care (SoC) in locally advanced squamous cell carcinoma of the head and neck (SCCHN): Safety phase of the randomized trial GORTEC 2017-01 (REACH). J. Clin. Oncol. 2018, 36, 6076. [Google Scholar] [CrossRef]
- Stein, A.; Binder, M.; Al-Batran, S.-E.; Hinke, A.; Waberer, L.; Goekkurt, E.; Meyer, T.; Statovci, D.; Depenbusch, R.; Riera-Knorrenschild, J.; et al. Avelumab and cetuximab in combination with FOLFOX in patients with previously untreated metastatic colorectal cancer (MCRC): Results of the safety run-in phase of the phase II AVETUX trial (AIO-KRK-0216). J. Clin. Oncol. 2018, 36, 3561. [Google Scholar] [CrossRef]
- Lee, D.-W.; Cho, E.J.; Lee, J.-H.; Yu, S.J.; Kim, Y.J.; Yoon, J.-H.; Kim, T.-Y.; Han, S.-W.; Oh, D.-Y.; Im, S.-A.; et al. Phase II Study of Avelumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib. Clin. Cancer Res. 2021, 27, 713–718. [Google Scholar] [CrossRef]
- Español-Rego, M.; Fernández-Martos, C.; Elez, E.; Foguet, C.; Pedrosa, L.; Rodríguez, N.; Ruiz-Casado, A.; Pineda, E.; Cid, J.; Cabezón, R.; et al. A Phase I-II multicenter trial with Avelumab plus autologous dendritic cell vaccine in pre-treated mismatch repair-proficient (MSS) metastatic colorectal cancer patients; GEMCAD 1602 study. Cancer Immunol. Immunother. 2022, 72, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Motomura, K.; Wada, Y.; Inaba, Y.; Sakamoto, Y.; Kurosaki, M.; Umeyama, Y.; Kamei, Y.; Yoshimitsu, J.; Fujii, Y.; et al. Avelumab in Combination with Axitinib as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma: Results from the Phase 1b VEGF Liver 100 Trial. Liver Cancer 2021, 10, 249–259. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Hastings, W.D.; Anderson, D.E.; Kassam, N.; Koguchi, K.; Greenfield, E.A.; Kent, S.C.; Zheng, X.X.; Strom, T.B.; Hafler, D.A.; Kuchroo, V.K. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur. J. Immunol. 2009, 39, 2492–2501. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and Is Associated with Lung Cancer Progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.-J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Lian, J.; Yang, H.; Li, F.; Zhao, S.; Qi, Y.; Zhang, Y.; Huang, L. TNF-α-induced Tim-3 expression marks the dysfunction of infiltrating natural killer cells in human esophageal cancer. J. Transl. Med. 2019, 17, 165. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Li, Z.; Li, N.; Li, F.; Zhou, Z.; Sang, J.; Chen, Y.; Han, Q.; Lv, Y.; Liu, Z. Immune checkpoint proteins PD-1 and TIM-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with HBV-related hepatocellular carcinoma. Medicine 2016, 95, e5749. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Liu, J.-F.; Wu, L.; Yang, L.-L.; Deng, W.-W.; Mao, L.; Wu, H.; Zhang, W.-F.; Sun, Z.-J. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J. Exp. Clin. Cancer Res. 2018, 37, 44. [Google Scholar] [CrossRef]
- Zhang, H.; Song, Y.; Yang, H.; Liu, Z.; Gao, L.; Liang, X.; Ma, C. Tumor cell-intrinsic Tim-3 promotes liver cancer via NF-κB/IL-6/STAT3 axis. Oncogene 2018, 37, 2456–2468. [Google Scholar] [CrossRef]
- Moghaddam, Y.; Andalib, A.; Mohammad-Ganji, M.; Homayouni, V.; Sharifi, M.; Ganjalikhani-Hakemi, M. Evaluation of the effect of TIM-3 suppression by miR-498 and its effect on apoptosis and proliferation rate of HL-60 cell line. Pathol. Res. Pract. 2018, 214, 1482–1488. [Google Scholar] [CrossRef]
- Fooladinezhad, H.; Khanahmad, H.; Ganjalikhani-Hakemi, M.; Doosti, A. Negative regulation of TIM-3 expression in AML cell line (HL-60) using miR-330-5p. Br. J. Biomed. Sci. 2016, 73, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Ganji, M. Silencing of TIM-3 Expression by miR-326 Affects Apoptosis and Proliferation of Human HL-60 Leukemia Cell Line. Int. J. Hematol. Oncol. 2018, 28, 112–122. [Google Scholar]
- Homayouni, V.; Ganjalikhani-Hakemi, M.; Rezaei, A.; Khanahmad, H.; Behdani, M.; Lomedasht, F.K. Preparation and characterization of a novel nanobody against T-cell immunoglobulin and mucin-3 (TIM-3). Iran. J. Basic. Med. Sci. 2016, 19, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Ruan, Z. Tim-3 and Tim-4 as the potential targets for antitumor therapy. Hum. Vaccines Immunother. 2015, 11, 2458–2462. [Google Scholar] [CrossRef]
- Sakuishi, K.; Jayaraman, P.; Behar, S.M.; Anderson, A.C.; Kuchroo, V.K. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011, 32, 345–349. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef]
- Silva, I.G.; Gibbs, B.F.; Bardelli, M.; Varani, L.; Sumbayev, V.V. Differential expression and biochemical activity of the immune receptor Tim-3 in healthy and malignant human myeloid cells. Oncotarget 2015, 6, 33823–33833. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, L.-F.; Liang, X.-H.; Ma, C.-H. Role of Tim-3 in hepatitis B virus infection: An overview. World J. Gastroenterol. 2016, 22, 2294–2303. [Google Scholar] [CrossRef]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Sharpe, A.H. Mechanisms of costimulation. Immunol. Rev. 2009, 229, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Ganjalikhani Hakemi, M.; Jafarinia, M.; Azizi, M.; Rezaeepoor, M.; Isayev, O.; Bazhin, A.V. The Role of TIM-3 in Hepatocellular Carcinoma: A Promising Target for Immunotherapy? Front. Oncol. 2020, 10, 601661. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef]
- Workman, C.J.; Wang, Y.; El Kasmi, K.C.; Pardoll, D.M.; Murray, P.J.; Drake, C.G.; Vignali, D.A.A. LAG-3 Regulates Plasmacytoid Dendritic Cell Homeostasis. J. Immunol. 2009, 182, 1885–1891. [Google Scholar] [CrossRef]
- Huard, B.; Tournier, M.; Hercend, T.; Triebel, F.; Faure, F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur. J. Immunol. 1994, 24, 3216–3221. [Google Scholar] [CrossRef]
- Workman, C.J.; Vignali, D.A.A. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol. 2003, 33, 970–979. [Google Scholar] [CrossRef]
- Workman, C.J.; Cauley, L.S.; Kim, I.-J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A.A. Lymphocyte Activation Gene-3 (CD223) Regulates the Size of the Expanding T Cell Population Following Antigen Activation In Vivo. J. Immunol. 2004, 172, 5450–5455. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Okazaki, I.-M.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Xu, L.; Wu, H.; Liao, H.; Luo, L.; Liao, M.; Gong, J.; Deng, Y.; Yuan, K.; Wu, H.; et al. OX40 expression in hepatocellular carcinoma is associated with a distinct immune microenvironment, specific mutation signature, and poor prognosis. Oncoimmunology 2018, 7, e1404214. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-J.; Zhang, Y.; Jin, G.-X.; Yao, L.; Wu, D.-Q. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8+ T cell in HCC patients. Immunol. Lett. 2013, 150, 116–122. [Google Scholar] [CrossRef]
- Legat, A.; Hajjami, H.M.-E.; Baumgaertner, P.; Cagnon, L.; Maillard, S.A.; Geldhof, C.; Iancu, E.M.; Lebon, L.; Guillaume, P.; Dojcinovic, D.; et al. Vaccination with LAG-3Ig (IMP321) and Peptides Induces Specific CD4 and CD8 T-Cell Responses in Metastatic Melanoma Patients—Report of a Phase I/IIa Clinical Trial. Clin. Cancer Res. 2016, 22, 1330–1340. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Ward-Kavanagh, L.K.; Lin, W.W.; Sedy, J.R.; Ware, C.F. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef]
- Bour-Jordan, H.; Esensten, J.H.; Martinez-Llordella, M.; Penaranda, C.; Stumpf, M.; Bluestone, J.A. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol. Rev. 2011, 241, 180–205. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Wu, L.; Garza, K.M.; La Rose, J.; Khoo, W.; Odermatt, B.; Mak, T.W.; Ohashi, P.S.; Rottapel, R. A point mutation in CD28 distinguishes proliferative signals from survival signals. Nat. Immunol. 2001, 2, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Hünig, T. The storm has cleared: Lessons from the CD28 superagonist TGN1412 trial. Nat. Rev. Immunol. 2012, 12, 317–318. [Google Scholar] [CrossRef]
- Tabares, P.; Berr, S.; Römer, P.S.; Chuvpilo, S.; Matskevich, A.A.; Tyrsin, D.; Fedotov, Y.; Einsele, H.; Tony, H.-P.; Hünig, T. Human regulatory T cells are selectively activated by low-dose application of the CD28 superagonist TGN1412/TAB08. Eur. J. Immunol. 2014, 44, 1225–1236. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of Co-stimulation in Cancer Immunotherapy Directed Against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef] [PubMed]
- de Jong, R.; Loenen, W.A.; Brouwer, M.; van Emmerik, L.; de Vries, E.F.; Borst, J.; van Lier, R.A. Regulation of expression of CD27, a T cell-specific member of a novel family of membrane receptors. J. Immunol. 1991, 146, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Nolte, M.A.; van Olffen, R.W.; van Gisbergen, K.P.J.M.; van Lier, R.A.W. Timing and tuning of CD27-CD70 interactions: The impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol. Rev. 2009, 229, 216–231. [Google Scholar] [CrossRef]
- Borst, J.; Hendriks, J.; Xiao, Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 2005, 17, 275–281. [Google Scholar] [CrossRef]
- Takeda, K.; Oshima, H.; Hayakawa, Y.; Akiba, H.; Atsuta, M.; Kobata, T.; Kobayashi, K.; Ito, M.; Yagita, H.; Okumura, K. CD27-Mediated Activation of Murine NK Cells. J. Immunol. 2000, 164, 1741–1745. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Tesselaar, K.; Van Oers, M.H.J.; van Lier, R.A.W. Control of lymphocyte function through CD27–CD70 interactions. Semin. Immunol. 1998, 10, 491–499. [Google Scholar] [CrossRef]
- Hendriks, J.; Xiao, Y.; Borst, J. CD27 Promotes Survival of Activated T Cells and Complements CD28 in Generation and Establishment of the Effector T Cell Pool. J. Exp. Med. 2003, 198, 1369–1380. [Google Scholar] [CrossRef]
- van Oosterwijk, M.F.; Juwana, H.; Arens, R.; Tesselaar, K.; van Oers, M.H.J.; Eldering, E.; van Lier, R.A.W. CD27-CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int. Immunol. 2007, 19, 713–718. [Google Scholar] [CrossRef]
- Dolfi, D.V.; Boesteanu, A.C.; Petrovas, C.; Xia, D.; Butz, E.A.; Katsikis, P.D. Late Signals from CD27 Prevent Fas-Dependent Apoptosis of Primary CD8+ T Cells. J. Immunol. 2008, 180, 2912–2921. [Google Scholar] [CrossRef]
- French, R.R.; Taraban, V.Y.; Crowther, G.R.; Rowley, T.F.; Gray, J.; Johnson, P.W.; Tutt, A.L.; Al-Shamkhani, A.; Glennie, M.J. Eradication of lymphoma by CD8 T cells following anti-CD40 monoclonal antibody therapy is critically dependent on CD27 costimulation. Blood 2007, 109, 4810–4815. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Flinn, I.; Taylor, M.H.; Sikic, B.I.; Brody, J.; Nemunaitis, J.; Feldman, A.; Hawthorne, T.R.; Rawls, T.; Keler, T.; et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, for hematologic malignancies. Blood Adv. 2020, 4, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Tesselaar, K.; Gravestein, L.A.; van Schijndel, G.M.; Borst, J.; van Lier, R.A. Characterization of murine CD70, the ligand of the TNF receptor family member CD27. J. Immunol. 1997, 159, 4959–4965. [Google Scholar] [CrossRef]
- Grewal, I.S. CD70 as a therapeutic target in human malignancies. Expert Opin. Ther. Targets 2008, 12, 341–351. [Google Scholar] [CrossRef]
- Silence, K.; Dreier, T.; Moshir, M.; Ulrichts, P.; Gabriels, S.M.E.; Saunders, M.; Wajant, H.; Brouckaert, P.; Huyghe, L.; Van Hauwermeiren, T.; et al. ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. mAbs 2014, 6, 523–532. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Drillenburg, P.; Drijver, B.F.A.D.; Van Schijndel, G.; Pals, S.T.; Van Lier, R.A.W.; Van Oers, M.H.J. Aberrant expression and reverse signalling of CD70 on malignant B cells. Br. J. Haematol. 1999, 106, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Aftimos, P.; Rolfo, C.; Rottey, S.; Offner, F.; Bron, D.; Maerevoet, M.; Soria, J.-C.; Moshir, M.; Dreier, T.; Van Rompaey, L.; et al. Phase I Dose-Escalation Study of the Anti-CD70 Antibody ARGX-110 in Advanced Malignancies. Clin. Cancer Res. 2017, 23, 6411–6420. [Google Scholar] [CrossRef]
- Matter, M.; Odermatt, B.; Yagita, H.; Nuoffer, J.-M.; Ochsenbein, A.F. Elimination of chronic viral infection by blocking CD27 signaling. J. Exp. Med. 2006, 203, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Rasheed, A.U.; Iyer, S.S.; Yagita, H.; Blazar, B.R.; Ahmed, R. Opposing Effects of CD70 Costimulation during Acute and Chronic Lymphocytic Choriomeningitis Virus Infection of Mice. J. Virol. 2011, 85, 6168–6174. [Google Scholar] [CrossRef] [PubMed]
- Claus, C.; Riether, C.; Schürch, C.; Matter, M.S.; Hilmenyuk, T.; Ochsenbein, A.F. CD27 Signaling Increases the Frequency of Regulatory T Cells and Promotes Tumor Growth. Cancer Res. 2012, 72, 3664–3676. [Google Scholar] [CrossRef] [PubMed]
- van Kooten, C.; Banchereau, J. Functions of CD40 on B cells, dendritic cells and other cells. Curr. Opin. Immunol. 1997, 9, 330–337. [Google Scholar] [CrossRef]
- Bishop, G.A.; Moore, C.R.; Xie, P.; Stunz, L.L.; Kraus, Z.J. TRAF Proteins in CD40 Signaling. Adv. Exp. Med. Biol. 2007, 597, 131–151. [Google Scholar] [CrossRef]
- Quezada, S.A.; Jarvinen, L.Z.; Lind, E.F.; Noelle, R.J. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 2004, 22, 307–328. [Google Scholar] [CrossRef]
- Frleta, D.; Lin, J.T.; Quezada, S.A.; Wade, T.K.; Barth, R.J.; Noelle, R.J.; Wade, W.F. Distinctive maturation of in vitro versus in vivo anti-CD40 mAb-matured dendritic cells in mice. J. Immunother. 2003, 26, 72–84. [Google Scholar] [CrossRef]
- Akiyama, T.; Shimo, Y.; Yanai, H.; Qin, J.; Ohshima, D.; Maruyama, Y.; Asaumi, Y.; Kitazawa, J.; Takayanagi, H.; Penninger, J.; et al. The Tumor Necrosis Factor Family Receptors RANK and CD40 Cooperatively Establish the Thymic Medullary Microenvironment and Self-Tolerance. Immunity 2008, 29, 423–437. [Google Scholar] [CrossRef]
- Peters, A.L.; Stunz, L.L.; Bishop, G.A. CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol. 2009, 21, 293–300. [Google Scholar] [CrossRef]
- Baxendale, A.J.; Dawson, C.W.; Stewart, S.E.; Mudaliar, V.; Reynolds, G.; Gordon, J.; Murray, P.G.; Young, L.S.; Eliopoulos, A.G. Constitutive activation of the CD40 pathway promotes cell transformation and neoplastic growth. Oncogene 2005, 24, 7913–7923. [Google Scholar] [CrossRef]
- Choi, M.S.; Boise, L.H.; Gottschalk, A.R.; Quintans, J.; Thompson, C.B.; Klaus, G.G. The role of bcl-XL in CD40-mediated rescue from anti-mu-induced apoptosis in WEHI-231 B lymphoma cells. Eur. J. Immunol. 1995, 25, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.W.; Papayoti, M.H.; Netto, G.; Armstrong, D.T.; Ordonez, G.; Lawson, J.M.; Stone, M.J. Growth-inhibitory effects of CD40 ligand (CD154) and its endogenous expression in human breast cancer. Clin. Cancer Res. 2001, 7, 691–703. [Google Scholar]
- Song, M.-A.; Tiirikainen, M.; Kwee, S.; Okimoto, G.; Yu, H.; Wong, L.L. Elucidating the Landscape of Aberrant DNA Methylation in Hepatocellular Carcinoma. PLoS ONE 2013, 8, e55761. [Google Scholar] [CrossRef] [PubMed]
- Hama, N.; Totoki, Y.; Miura, F.; Tatsuno, K.; Saito-Adachi, M.; Nakamura, H.; Arai, Y.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Epigenetic landscape influences the liver cancer genome architecture. Nat. Commun. 2018, 9, 1643. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Lu, X.; Wang, X.; Liu, Y.; Guo, B.; Zhang, Y.; Zhang, W.; Nie, J.; Feng, K.; Chen, M.; et al. Low-Dose Decitabine-Based Chemoimmunotherapy for Patients with Refractory Advanced Solid Tumors: A Phase I/II Report. J. Immunol. Res. 2014, 2014, 371087. [Google Scholar] [CrossRef]
- Mei, Q.; Chen, M.; Lu, X.; Li, X.; Duan, F.; Wang, M.; Luo, G.; Han, W. An open-label, single-arm, phase I/II study of lower-dose decitabine based therapy in patients with advanced hepatocellular carcinoma. Oncotarget 2015, 6, 16698–16711. [Google Scholar] [CrossRef]
- El-Khoueiry, A.; Mulcahy, M.F.; Bekaii-Saab, T.; Kim, R.; Denlinger, C.; Goel, R.; Gupta, S.; Jueliger, S.; Oganesian, A.; Keer, H.; et al. Abstract 2947: Pharmacodynamic (PD) and pharmacokinetic (PK) results of the second-generation hypomethylating agent, SGI-110, in patients with hepatocellular carcinoma (HCC) after progression on sorafenib. Cancer Res 2015, 75, 2947. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics of hepatocellular carcinoma. Clin. Transl. Med. 2019, 8, 13. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe–Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef]
- Hämmerle, M.; Gutschner, T.; Uckelmann, H.; Ozgur, S.; Fiskin, E.; Gross, M.; Skawran, B.; Geffers, R.; Longerich, T.; Breuhahn, K.; et al. Posttranscriptional destabilization of the liver-specific long noncoding RNA HULC by the IGF2 mRNA-binding protein 1 (IGF2BP1). Hepatology 2013, 58, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-P.; Xu, H.-X.; Yu, Y.; He, J.-D.; Wang, Z.; Xu, Y.-J.; Wang, C.-Y.; Zhang, H.-M.; Zhang, R.-X.; Zhang, J.-J.; et al. LncRNA HULC enhances epithelial-mesenchymal transition to promote tumorigenesis and metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1 signaling pathway. Oncotarget 2016, 7, 42431–42446. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xiao, Z.; Liu, F.; Cui, M.; Li, W.; Yang, Z.; Li, J.; Ye, L.; Zhang, X. Long non-coding RNA HULC promotes tumor angiogenesis in liver cancer by up-regulating sphingosine kinase 1 (SPHK1). Oncotarget 2016, 7, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large Intervening Non-Coding RNA HOTAIR is Associated with Hepatocellular Carcinoma Progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, L.; Wu, L.-M.; Lai, M.-C.; Xie, H.-Y.; Zhang, F.; Zheng, S.-S. Overexpression of Long Non-coding RNA HOTAIR Predicts Tumor Recurrence in Hepatocellular Carcinoma Patients Following Liver Transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Y.; Liu, W.; Zhang, W.; Xu, J. EZH2-mediated loss of miR-622 determines CXCR4 activation in hepatocellular carcinoma. Nat. Commun. 2015, 6, 8494. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.-C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 32, 574–589.e6. [Google Scholar] [CrossRef]
- Oba, A.; Shimada, S.; Akiyama, Y.; Nishikawaji, T.; Mogushi, K.; Ito, H.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; et al. ARID2 modulates DNA damage response in human hepatocellular carcinoma cells. J. Hepatol. 2017, 66, 942–951. [Google Scholar] [CrossRef]
- Wu, L.-M.; Yang, Z.; Zhou, L.; Zhang, F.; Xie, H.-Y.; Feng, X.-W.; Wu, J.; Zheng, S.-S. Identification of Histone Deacetylase 3 as a Biomarker for Tumor Recurrence Following Liver Transplantation in HBV-Associated Hepatocellular Carcinoma. PLoS ONE 2010, 5, e14460. [Google Scholar] [CrossRef]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, srep43864. [Google Scholar] [CrossRef]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.F.; Koh, J.; Rha, S.Y.; et al. Epigenetic Therapy Using Belinostat for Patients With Unresectable Hepatocellular Carcinoma: A Multicenter Phase I/II Study With Biomarker and Pharmacokinetic Analysis of Tumors From Patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Wörns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.-G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a Multitargeted Inhibitor of Histone Deacetylase, Epidermal Growth Factor Receptor, and Human Epidermal Growth Factor Receptor 2, Exerts Potent Anticancer Activity. Cancer Res 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Brunetti, O.; Gnoni, A.; Licchetta, A.; Delcuratolo, S.; Memeo, R.; Solimando, A.G.; Argentiero, A. Emerging Role of Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Medicina 2019, 55, 698. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, H.; Yuan, X.; Fan, X.; Zhang, C. Advances in Immune Checkpoint Inhibitors for Advanced Hepatocellular Carcinoma. Front. Immunol. 2022, 13, 896752. [Google Scholar] [CrossRef]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef]
- Manzoni, M.; Rovati, B.; Ronzoni, M.; Loupakis, F.; Mariucci, S.; Ricci, V.; Gattoni, E.; Salvatore, L.; Tinelli, C.; Villa, E.; et al. Immunological Effects of Bevacizumab-Based Treatment in Metastatic Colorectal Cancer. Oncology 2010, 79, 187–196. [Google Scholar] [CrossRef]
- Martino, E.; Misso, G.; Pastina, P.; Costantini, S.; Vanni, F.; Gandolfo, C.; Botta, C.; Capone, F.; Lombardi, A.; Pirtoli, L.; et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov. 2016, 2, 16025. [Google Scholar] [CrossRef]
- Siegel, A.B.; Cohen, E.I.; Ocean, A.; Lehrer, D.; Goldenberg, A.; Knox, J.J.; Chen, H.; Clark-Garvey, S.; Weinberg, A.; Mandeli, J.; et al. Phase II Trial Evaluating the Clinical and Biologic Effects of Bevacizumab in Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2008, 26, 2992–2998. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.-H.; He, A.R.; Ryoo, B.-Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Huo, H.; Dai, S.; Li, S. Efficacy and safety of immune checkpoint inhibitors-combined antiangiogenic drugs in the treatment of hepatocellular carcinoma: A systematic review and meta analysis. Front. Oncol. 2022, 12, 964779. [Google Scholar] [CrossRef]
- Kudo, M. Durvalumab Plus Tremelimumab: A Novel Combination Immunotherapy for Unresectable Hepatocellular Carcinoma. Liver Cancer 2022, 11, 87–93. [Google Scholar] [CrossRef]
- Li, H.; He, Q.; Zhou, G.-M.; Wang, W.-J.; Shi, P.-P.; Wang, Z.-H. Potential biomarkers for the prognosis and treatment of HCC immunotherapy. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 2027–2046. [Google Scholar]
- Oh, D.-Y.; Lee, K.-H.; Lee, D.-W.; Yoon, J.; Kim, T.-Y.; Bang, J.-H.; Nam, A.-R.; Oh, K.-S.; Kim, J.-M.; Lee, Y.; et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: An open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 2022, 7, 522–532. [Google Scholar] [CrossRef]
- Zheng, J.; Shao, M.; Yang, W.; Ren, J.; Chen, X.; Yang, H. Benefits of combination therapy with immune checkpoint inhibitors and predictive role of tumour mutation burden in hepatocellular carcinoma: A systematic review and meta-analysis. Int. Immunopharmacol. 2022, 112, 109244. [Google Scholar] [CrossRef]
- Zhang, B.; Tao, B.; Li, Y.; Yi, C.; Lin, Z.; Ma, Y.; Han, J.; Shao, W.; Chen, Z.; Lin, J.; et al. Dual immune checkpoint inhibitors or combined with anti-VEGF agents in advanced, unresectable hepatocellular carcinoma. Eur. J. Intern. Med. 2022, 111, 37–46. [Google Scholar] [CrossRef]
- Tao, S.; Liang, S.; Zeng, T.; Yin, D. Epigenetic modification-related mechanisms of hepatocellular carcinoma resistance to immune checkpoint inhibition. Front. Immunol. 2022, 13, 1043667. [Google Scholar] [CrossRef]
- Cossío, F.P.; Esteller, M.; Berdasco, M. Towards a more precise therapy in cancer: Exploring epigenetic complexity. Curr. Opin. Chem. Biol. 2020, 57, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.H.; Wang, L.; Goldberg, M.S. Improving cancer immunotherapy with DNA methyltransferase inhibitors. Cancer Immunol. Immunother. 2016, 65, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Denman, C.J.; Cobanoglu, Z.S.; Kiany, S.; Lau, C.C.; Gottschalk, S.M.; Hughes, D.P.M.; Kleinerman, E.S.; Lee, D. The Narrow-Spectrum HDAC Inhibitor Entinostat Enhances NKG2D Expression Without NK Cell Toxicity, Leading to Enhanced Recognition of Cancer Cells. Pharm. Res. 2015, 32, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Costantini, B.; Kordasti, S.Y.; Kulasekararaj, A.G.; Jiang, J.; Seidl, T.; Abellan, P.P.; Mohamedali, A.; Thomas, N.S.B.; Farzaneh, F.; Mufti, G.J. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica 2013, 98, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, C.; Jiang, H.; Zhang, Y.; Lin, W.; Mo, J.; Jin, C. Combination of Ablation and Immunotherapy for Hepatocellular Carcinoma: Where We Are and Where to Go. Front. Immunol. 2021, 12, 792781. [Google Scholar] [CrossRef] [PubMed]
- Bo, X.-W.; Sun, L.-P.; Yu, S.-Y.; Xu, H.-X. Thermal ablation and immunotherapy for hepatocellular carcinoma: Recent advances and future directions. World J. Gastrointest. Oncol. 2021, 13, 1397–1411. [Google Scholar] [CrossRef]
- Singh, P.; Toom, S.; Avula, A.; Kumar, V.; Rahma, O.E. The Immune Modulation Effect of Locoregional Therapies and Its Potential Synergy with Immunotherapy in Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2020, 7, 11–17. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Ueda, T.; Arihara, F.; Kagaya, T.; Yamashita, T.; Fushimi, K.; Kaneko, S. Enhancement of tumor-associated antigen-specific T cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology 2013, 57, 1448–1457. [Google Scholar] [CrossRef]
- Huang, S.; Li, T.; Chen, Y.; Liu, J.; Wang, Y.; Yang, C.; Wang, C.; Ju, S.; Bai, Y.; Yao, W.; et al. Microwave ablation combined with anti-PD-1 therapy enhances systemic antitumor immunity in a multitumor murine model of Hepa1-6. Int. J. Hyperth. 2022, 39, 278–286. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, X.; Cai, H.; Zhuang, X. Effects of microwave ablation on T-cell subsets and cytokines of patients with hepatocellular carcinoma. Minim. Invasive Ther. Allied Technol. 2017, 26, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.W.; Zhang, J.; Liang, P.; Yu, X.L.; Su, L.; Yu, D.J.; Ji, X.L.; Yu, G. Sequential pathological and immunologic analysis of percutaneous microwave coagulation therapy of hepatocellular carcinoma. Int. J. Hyperth. 2003, 19, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Dumolard, L.; Ghelfi, J.; Roth, G.; Decaens, T.; Jilkova, Z.M. Percutaneous Ablation-Induced Immunomodulation in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 4398. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.C.; van Hillegersberg, R.; Schoots, I.G.; Levi, M.; Beek, J.F.; Crezee, J.; van Gulik, T.M. Cryoablation induces greater inflammatory and coagulative responses than radiofrequency ablation or laser induced thermotherapy in a rat liver model. Surgery 2010, 147, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, F.; Zhou, L.; Zhang, M.-N.; Chen, Y.; Chang, X.-J.; Lu, Y.-Y.; Bai, W.-L.; Qu, J.-H.; Wang, C.-P.; et al. Upregulation of circulating PD-L1/PD-1 is associated with poor post-cryoablation prognosis in patients with HBV-related hepatocellular carcinoma. PLoS ONE 2011, 6, e23621. [Google Scholar] [CrossRef]
- Kohles, N.; Nagel, D.; Jüngst, D.; Stieber, P.; Holdenrieder, S. Predictive value of immunogenic cell death biomarkers HMGB1, sRAGE, and DNase in liver cancer patients receiving transarterial chemoembolization therapy. Tumor Biol. 2012, 33, 2401–2409. [Google Scholar] [CrossRef]
- Liao, J.; Xiao, J.; Zhou, Y.; Liu, Z.; Wang, C. Effect of transcatheter arterial chemoembolization on cellular immune function and regulatory T cells in patients with hepatocellular carcinoma. Mol. Med. Rep. 2015, 12, 6065–6071. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, B.; Huang, Z.-L.; Shi, M.; Yu, X.-J.; Zheng, L.; Li, S.; Li, L. Increased Circulating Th17 Cells after Transarterial Chemoembolization Correlate with Improved Survival in Stage III Hepatocellular Carcinoma: A Prospective Study. PLoS ONE 2013, 8, e60444. [Google Scholar] [CrossRef]
- Kim, M.J.; Jang, J.W.; Oh, B.S.; Kwon, J.H.; Chung, K.W.; Jung, H.S.; Jekarl, D.W.; Lee, S. Change in inflammatory cytokine profiles after transarterial chemotherapy in patients with hepatocellular carcinoma. Cytokine 2013, 64, 516–522. [Google Scholar] [CrossRef]
- Rivoltini, L.; Bhoori, S.; Camisaschi, C.; Bergamaschi, L.; Lalli, L.; Frati, P.; Citterio, D.; Castelli, C.; Mazzaferro, V. Y90-radioembolisation in hepatocellular carcinoma induces immune responses calling for early treatment with multiple checkpoint blockers. Gut 2023, 72, 406–407. [Google Scholar] [CrossRef]
- Chew, V.; Lee, Y.H.; Pan, L.; Nasir, N.J.M.; Lim, C.J.; Chua, C.; Lai, L.; Hazirah, S.N.; Lim, T.K.H.; Goh, B.K.P.; et al. Immune activation underlies a sustained clinical response to Yttrium-90 radioembolisation in hepatocellular carcinoma. Gut 2019, 68, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.H.; Malagari, K.; Kulik, L.M. Role of locoregional therapies in the wake of systemic therapy. J. Hepatol. 2020, 72, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Leppelmann, K.S.; Mooradian, M.J.; Ganguli, S.; Uppot, R.N.; Yamada, K.; Irani, Z.; Wehrenberg-Klee, E.P.; Zubiri, L.; Reynolds, K.L.; Arellano, R.S.; et al. Thermal Ablation, Embolization, and Selective Internal Radiation Therapy Combined with Checkpoint Inhibitor Cancer Immunotherapy: Safety Analysis. J. Vasc. Interv. Radiol. 2021, 32, 187–195. [Google Scholar] [CrossRef]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.-Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.-H.; Tak, W.Y.; et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015, 16, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A.; El Aziz, M.A.A.; Cincione, I.; Cea, U.V.; Germini, A.; Granieri, S.; Cotsoglou, C.; Sacco, R. Angiotensin Receptor 1 Blockers Prolong Time to Recurrence after Radiofrequency Ablation in Hepatocellular Carcinoma patients: A Retrospective Study. Biomedicines 2020, 8, 399. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A.; Muscatiello, N.; Di Leo, A.; Barone, M. Combination Therapy With Sorafenib and Radiofrequency Ablation for Hepatocellular Carcinoma: A Glimmer of Light after the Storm Trial? Am. J. Gastroenterol. 2015, 110, 770–771. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, G.; Chen, S.; Bi, H.; Xia, F.; Feng, K.; Ma, K.; Ni, B. Combination therapy with PD-1 blockade and radiofrequency ablation for recurrent hepatocellular carcinoma: A propensity score matching analysis. Int. J. Hyperth. 2021, 38, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Liang, P.; Dong, B.; Yu, X.; Han, Z.; Xu, Y. Phase Ⅰ clinical study of combination therapy with microwave ablation and cellular immunotherapy in hepatocellular carcinoma. Cancer Biol. Ther. 2011, 11, 450–456. [Google Scholar] [CrossRef]
- Lyu, N.; Kong, Y.; Li, X.; Mu, L.; Deng, H.; Chen, H.; He, M.; Lai, J.; Li, J.; Tang, H.; et al. Ablation Reboots the Response in Advanced Hepatocellular Carcinoma With Stable or Atypical Response During PD-1 Therapy: A Proof-of-Concept Study. Front. Oncol. 2020, 10, 580241. [Google Scholar] [CrossRef]
- Li, X.; Xu, J.; Gu, X.; Chen, L.; Wu, Q.; Li, H.; Bai, H.; Yang, J.; Qian, J. Case Report: Antiangiogenic Therapy Plus Immune Checkpoint Inhibitors Combined With Intratumoral Cryoablation for Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 740790. [Google Scholar] [CrossRef]
- Niu, L.-Z.; Li, J.-L.; Zeng, J.-Y.; Mu, F.; Liao, M.-T.; Yao, F.; Li, L.; Liu, C.-Y.; Chen, J.-B.; Zuo, J.-S.; et al. Combination treatment with comprehensive cryoablation and immunotherapy in metastatic hepatocellular cancer. World J. Gastroenterol. 2013, 19, 3473–3480. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.; Zhou, C.; Yang, C.; Wang, C.; Liu, J.; Wang, Y.; Huang, S.; Li, T.; Chen, Y.; Bai, Y.; et al. Apatinib Plus Camrelizumab With/Without Chemoembolization for Hepatocellular Carcinoma: A Real-World Experience of a Single Center. Front. Oncol. 2021, 11, 835889. [Google Scholar] [CrossRef] [PubMed]
- Agdashian, D.; ElGindi, M.; Xie, C.; Sandhu, M.; Pratt, D.; Kleiner, D.E.; Figg, W.D.; Rytlewski, J.A.; Sanders, C.; Yusko, E.C.; et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. 2019, 68, 599–608. [Google Scholar] [CrossRef]
- Marinelli, B.; Cedillo, M.; Pasik, S.D.; Charles, D.; Murthy, S.; Patel, R.S.; Fischman, A.; Ranade, M.; Bishay, V.; Nowakowski, S.; et al. Safety and Efficacy of Locoregional Treatment during Immunotherapy with Nivolumab for Hepatocellular Carcinoma: A Retrospective Study of 41 Interventions in 29 Patients. J. Vasc. Interv. Radiol. 2020, 31, 1729–1738.e1. [Google Scholar] [CrossRef]
- Liou, H.; Mody, K.; Boyle, A.W.; Keaveny, A.P.; Croome, K.P.; Burns, J.M.; Harnois, D.M.; Patel, T.C.; Toskich, B. Neoadjuvant Radiation Lobectomy and Immunotherapy for Angioinvasive HCC Resulting in Complete Pathologic Response. Hepatology 2021, 74, 525–527. [Google Scholar] [CrossRef]
- Kudo, M.; Matilla, A.; Santoro, A.; Melero, I.; Gracián, A.C.; Acosta-Rivera, M.; Choo, S.-P.; El-Khoueiry, A.B.; Kuromatsu, R.; El-Rayes, B.; et al. CheckMate 040 cohort 5: A phase I/II study of nivolumab in patients with advanced hepatocellular carcinoma and Child-Pugh B cirrhosis. J. Hepatol. 2021, 75, 600–609. [Google Scholar] [CrossRef]
- de la Torre-Aláez, M.; Matilla, A.; Varela, M.; Iñarrairaegui, M.; Reig, M.; Lledó, J.L.; Arenas, J.I.; Lorente, S.; Testillano, M.; Márquez, L.; et al. Nivolumab after selective internal radiation therapy for the treatment of hepatocellular carcinoma: A phase 2, single-arm study. J. Immunother. Cancer 2022, 10, e005457. [Google Scholar] [CrossRef]
- Tai, D.; Loke, K.; Gogna, A.; Kaya, N.A.; Tan, S.H.; Hennedige, T.; Ng, D.; Irani, F.; Lee, J.; Lim, J.Q.; et al. Radioembolisation with Y90-resin microspheres followed by nivolumab for advanced hepatocellular carcinoma (CA 209-678): A single arm, single centre, phase 2 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 1025–1035. [Google Scholar] [CrossRef]
- Cochin, V.; Gross-Goupil, M.; Ravaud, A.; Godbert, Y.; Le Moulec, S. Cabozantinib: Mechanism of action, efficacy and indications. Bull. Cancer 2017, 104, 393–401. [Google Scholar] [CrossRef]
- Liao, W.; Huang, J.; Hutton, D.; Zhu, G.; Wu, Q.; Wen, F.; Bai, L.; Li, Q. Cost-effectiveness analysis of cabozantinib as second-line therapy in advanced hepatocellular carcinoma. Liver Int. 2019, 39, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chan, S.L.; Sukeepaisarnjaroen, W.; Han, G.; Choo, S.P.; Sriuranpong, V.; Pan, H.; Yau, T.; Guo, Y.; Chen, M.; et al. A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919889001. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.; Su, W.; Schuler, M.; Nam, D.; Lim, W.T.; Bauer, T.M.; Azaro, A.; Poon, R.T.P.; Hong, D.; Lin, C.; et al. Phase 1 study of capmatinib in MET-positive solid tumor patients: Dose escalation and expansion of selected cohorts. Cancer Sci. 2020, 111, 536–547. [Google Scholar] [CrossRef]
- Luo, Y.; Lin, C.; Ren, W.; Ju, F.; Xu, Z.; Liu, H.; Yu, Z.; Chen, J.; Zhang, J.; Liu, P.; et al. Intravenous Injections of a Rationally Selected Oncolytic Herpes Virus as a Potent Virotherapy for Hepatocellular Carcinoma. Mol. Ther. Oncolytics 2019, 5, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Hummel, J.L.; Safroneeva, E.; Mossman, K.L. The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Mol. Ther. 2005, 12, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.-H.; Yun, X.-J.; Xue, Y.; Zhou, T.; Sun, X.; Gao, Y.-J. A novel oncolytic adenovirus inhibits hepatocellular carcinoma growth. J. Zhejiang Univ. Sci. B 2019, 20, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Song, B.; Du, X.; Wei, Z.; Huo, T. Effect of bone-marrow-derived mesenchymal stem cells on high-potential hepatocellular carcinoma in mouse models: An intervention study. Eur. J. Med. Res. 2013, 18, 34. [Google Scholar] [CrossRef]
- Qiao, L.; Xu, Z.; Zhao, T.; Zhao, Z.; Shi, M.; Zhao, R.C.; Ye, L.; Zhang, X. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008, 18, 500–507. [Google Scholar] [CrossRef]
- Mohamed, Y.; Basyony, M.A.; El-Desouki, N.I.; Abdo, W.S.; El-Magd, M.A. The potential therapeutic effect for melatonin and mesenchymal stem cells on hepatocellular carcinoma. Biomed. Pharmacother. 2019, 9, 24. [Google Scholar] [CrossRef]
- Wu, L.; Tang, Q.; Yin, X.; Yan, D.; Tang, M.; Xin, J.; Pan, Q.; Ma, C.; Yan, S. The Therapeutic Potential of Adipose Tissue-Derived Mesenchymal Stem Cells to Enhance Radiotherapy Effects on Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2019, 7, 267. [Google Scholar] [CrossRef]
- Sun, H.-C.; Tang, Z.-Y.; Wang, L.; Qin, L.-X.; Ma, Z.-C.; Ye, Q.-H.; Zhang, B.-H.; Qian, Y.-B.; Wu, Z.-Q.; Fan, J.; et al. Postoperative interferon α treatment postponed recurrence and improved overall survival in patients after curative resection of HBV-related hepatocellular carcinoma: A randomized clinical trial. J. Cancer Res. Clin. Oncol. 2006, 132, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, T.-Q.; Zhang, T.; Wu, Q.; Kong, D.-L.; Li, Q.; Sun, H.-C. Adjuvant interferon for early or late recurrence of hepatocellular carcinoma and mortality from hepatocellular carcinoma following curative treatment: A meta-analysis with comparison of different types of hepatitis. Mol. Clin. Oncol. 2014, 2, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, V.; Romito, R.; Schiavo, M.; Mariani, L.; Camerini, T.; Bhoori, S.; Capussotti, L.; Calise, F.; Pellicci, R.; Belli, G.; et al. Prevention of hepatocellular carcinoma recurrence with alpha-interferon after liver resection in HCV cirrhosis. Hepatology 2006, 44, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Chen, X.-P.; Zhu, P.; Ding, L.; Guan, J.; Shi, Z.-L. Inhibitory effect of interferon-α-2b on expression of cyclooxygenase-2 and vascular endothelial growth factor in human hepatocellular carcinoma inoculated in nude mice. World J. Gastroenterol. 2008, 14, 6802–6807. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Shi, J.; Budhu, A.; Yu, Z.; Forgues, M.; Roessler, S.; Ambs, S.; Chen, Y.; Meltzer, P.S.; Croce, C.M.; et al. MicroRNA Expression, Survival, and Response to Interferon in Liver Cancer. N. Engl. J. Med. 2009, 361, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Golfieri, R.; Giampalma, E.; Renzulli, M.; Cioni, R.; Bargellini, I.; Bartolozzi, C.; Breatta, A.D.; Gandini, G.; Nani, R.; on behalf of the Precision Italia Study Group; et al. Randomised controlled trial of doxorubicin-eluting beads vs conventional chemoembolisation for hepatocellular carcinoma. Br. J. Cancer 2014, 111, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]



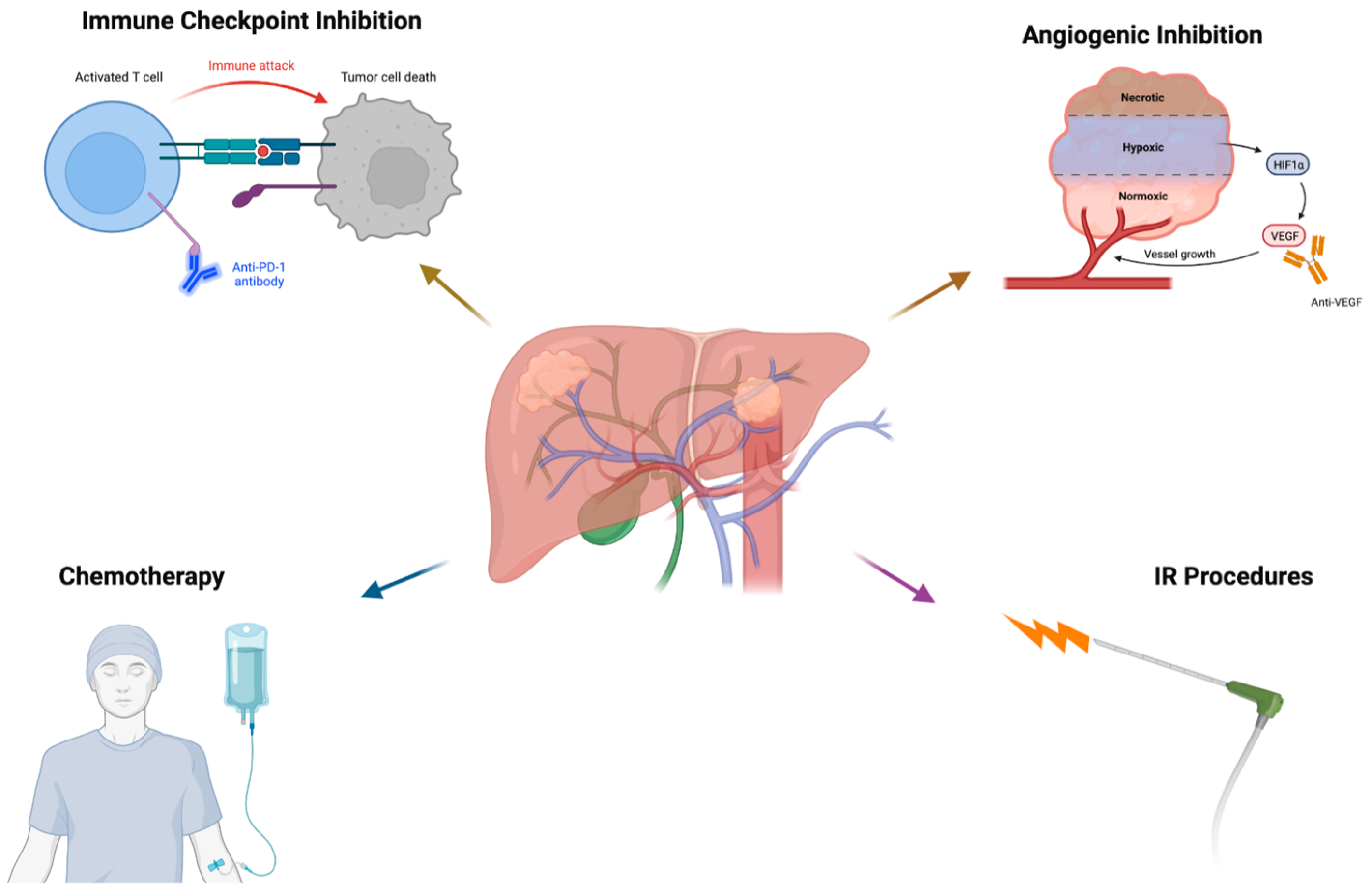{kind=link}
{kind=link}
| Drug | Targets | First/ Second Line | OS (Months) | PFS (Months) | n | Year | Phase III Trial |
|---|---|---|---|---|---|---|---|
| Sorafenib | VEGFRs 1-3, PDGFR-β, Raf-1, B-Raf | First | 10.7 vs. 4.9 (placebo) 6.5 vs. 4.2 (placebo) | 5.5 vs. 2.8 2.8 vs. 1.4 | 602 226 | 2008 2009 | SHARP ORIENTAL |
| Lenvatinib | VEGFRs 1–3, FGFRs 1–4, PDGFR-α, RET, KIT | First | 13.6 vs. 12.3 (sorafenib) | 7.4 vs. 3.7 | 954 | 2018 | REFLECT |
| Atezolizumab and Bevacizumab | PD-L1 and VEGF | First | 19.2 vs. 13.4 (sorafenib) | 6.8 vs. 4.3 | 501 | 2020 | IMBrave 150 |
| Tremelimumab and Durvalumab | CTLA-4 and PD-L1 | First | 16.4 vs. 13.8 (sorafenib) | 3.8 vs. 4.1 | 782 | 2022 | HIMALAYA |
| Regorafenib | VEGFR, FGFR, PDGFR, B-RAF, RET and KIT | Second | 10.6 months vs. 7.8 (placebo) | 3.1 vs. 1.5 | 573 | 2017 | RESOURCE |
| Pembrolizumab | PD-1 | Second | 13.9 vs. 10.6 (placebo) | 3.0 vs. 2.8 | 413 | 2018 | KEYNOTE 240 |
| Cabozantinib | VEGFR, AXL, c-MET, KIT, RET | Second | 10.2 vs. 8.0 (placebo) | 5.2 vs. 1.9 | 707 | 2019 | CELESTIAL |
| Ramucirumab | VEGFR 2 | Second | 8.5 vs. 7.3 (placebo) | 2.8 vs. 1.6 | 292 | 2019 | REACH-2 |
| Nivolumab and Ipilimumab | PD-1 and CTLA-4 | Second | Arm A: 22.8 Arm B: 12.5 Arm C: 12.7 | Arm A: 17.0 Arm B: 22.2 Arm C: 16.6 | 148 | 2020 | CHECK-MATE 040 |
| Study Name | Phase | Disease | Therapy | n | ORR (%) | OS | mPFS | Grade ≥ 3 AEs (%) | Trial Number |
|---|---|---|---|---|---|---|---|---|---|
| IMMUTACE | II | intermediate-stage HCC | TACE + nivolumab | 49 | 71.4 | NA | 7.2 | 34.7 | NCT03572582 |
| PETAL | Ib | intermediate-stage HCC | TACE + pembrolizumab | 14 | NA | NA | 10.8 | 21.0 | NCT03397654 |
| NA | I | BCLC Stage B HCC + Child Pugh A cirrhosis | DEB-TACE + nivolumab | 9 | NA | 1-year: 71% | NA | 66.7 | NCT03143270 |
| NA | NA | Intermediate-advanced HCC | TACE + camrelizumab | 101 | 57.7 | 1-year: 61.9 | 9.7 | 40.6 | ChiCTR1900026163 |
| START-FIT | II | Locally advanced HCC | TACE + SBRT + Avelumab | 33 | 62.5 | Median: 30.3 | 30.3 | 30.3 | NCT03817736 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charles, J.; Vrionis, A.; Mansur, A.; Mathias, T.; Shaikh, J.; Ciner, A.; Jiang, Y.; Nezami, N. Potential Immunotherapy Targets for Liver-Directed Therapies, and the Current Scope of Immunotherapeutics for Liver-Related Malignancies. Cancers 2023, 15, 2624. https://doi.org/10.3390/cancers15092624
Charles J, Vrionis A, Mansur A, Mathias T, Shaikh J, Ciner A, Jiang Y, Nezami N. Potential Immunotherapy Targets for Liver-Directed Therapies, and the Current Scope of Immunotherapeutics for Liver-Related Malignancies. Cancers. 2023; 15(9):2624. https://doi.org/10.3390/cancers15092624
Chicago/Turabian StyleCharles, Jonathan, Andrea Vrionis, Arian Mansur, Trevor Mathias, Jamil Shaikh, Aaron Ciner, Yixing Jiang, and Nariman Nezami. 2023. "Potential Immunotherapy Targets for Liver-Directed Therapies, and the Current Scope of Immunotherapeutics for Liver-Related Malignancies" Cancers 15, no. 9: 2624. https://doi.org/10.3390/cancers15092624
APA StyleCharles, J., Vrionis, A., Mansur, A., Mathias, T., Shaikh, J., Ciner, A., Jiang, Y., & Nezami, N. (2023). Potential Immunotherapy Targets for Liver-Directed Therapies, and the Current Scope of Immunotherapeutics for Liver-Related Malignancies. Cancers, 15(9), 2624. https://doi.org/10.3390/cancers15092624