
Neuroendocrine Tumors: Genomics and Molecular Biomarkers with a Focus on Metastatic Disease

Simple Summary
Abstract
1. Introduction
2. Genomics
2.1. Inherited Disorders
2.1.1. MEN Type 1
2.1.2. MEN Type 2
2.2. Sporadic Mutations
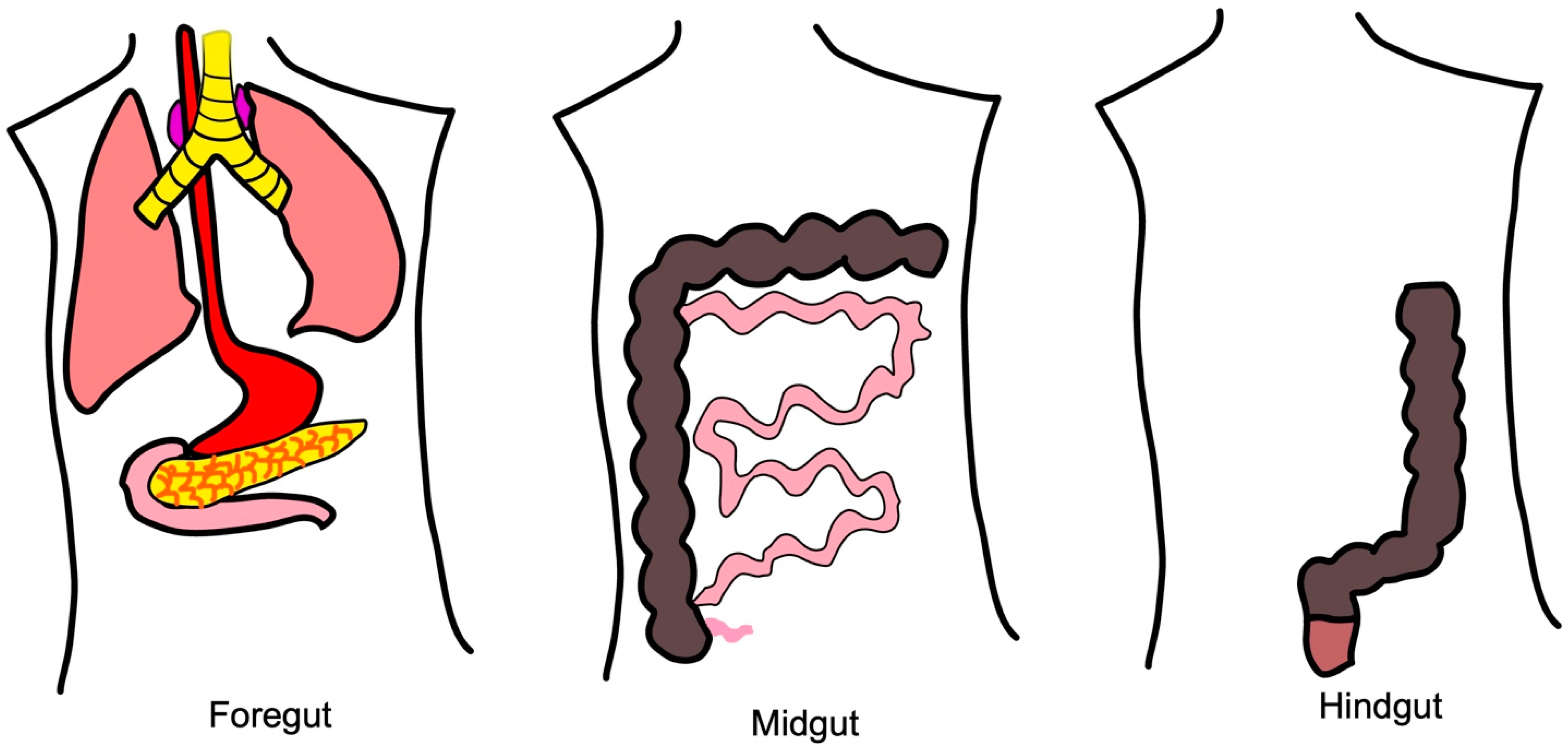
2.2.1. Molecular Pathways of Foregut NETs
2.2.2. Molecular Pathways of Midgut NETs
2.2.3. Molecular Pathways of Hindgut NETs
3. Diagnostic Biomarkers
3.1. Chromogranin A
3.2. Synaptophysin
3.3. 5-HIAA
3.4. NETest
3.5. Somatostatin Receptor Imaging
4. Prognostic Biomarkers and Genes
4.1. Ki-67 Proliferation Index
4.2. Chromogranin A
4.3. 5-HIAA
4.4. DAXX or ATRX Mutations
5. Biomarkers Associated with Treatment Response
5.1. Somatostatin Synthetic Analogs
5.2. Somatostatin Receptor Imaging
5.3. Cytotoxic Chemotherapy
5.3.1. mTOR Inhibitors
5.3.2. Alkylating Agents
5.3.3. Platinum Chemotherapy
5.4. Interventional Radiology
5.4.1. Ablation Therapy
5.4.2. Embolization Therapies
5.5. Surgical Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Man, D.; Wu, J.; Shen, Z.; Zhu, X. Prognosis of patients with neuroendocrine tumor: A SEER database analysis. Cancer Manag. Res. 2018, 10, 5629–5638. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Sandler, M. The classification of carcinoid tum ours. Lancet 1963, 1, 238–239. [Google Scholar] [CrossRef] [PubMed]
- Kloppel, G.; Perren, A.; Heitz, P.U. The gastroenteropancreatic neuroendocrine cell system and its tumors: The WHO classification. Ann. N. Y. Acad. Sci. USA 2004, 1014, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef]
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Pavel, M.; Baudin, E.; Couvelard, A.; Krenning, E.; Oberg, K.; Steinmuller, T.; Anlauf, M.; Wiedenmann, B.; Salazar, R.; Barcelona Consensus Conference Participants. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012, 95, 157–176. [Google Scholar] [CrossRef]
- Veenendaal, L.M.; Borel Rinkes, I.H.; Lips, C.J.; van Hillegersberg, R. Liver metastases of neuroendocrine tumours; early reduction of tumour load to improve life expectancy. World J. Surg. Oncol. 2006, 4, 35. [Google Scholar] [CrossRef]
- Oates, J.A. The carcinoid syndrome. N. Engl. J. Med. 1986, 315, 702–704. [Google Scholar] [CrossRef]
- Cloyd, J.M.; Ejaz, A.; Konda, B.; Makary, M.S.; Pawlik, T.M. Neuroendocrine liver metastases: A contemporary review of treatment strategies. Hepatobiliary Surg. Nutr. 2020, 9, 440–451. [Google Scholar] [CrossRef]
- Leotlela, P.D.; Jauch, A.; Holtgreve-Grez, H.; Thakker, R.V. Genetics of neuroendocrine and carcinoid tumours. Endocr. Relat. Cancer 2003, 10, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Gut, P.; Komarowska, H.; Czarnywojtek, A.; Waligorska-Stachura, J.; Baczyk, M.; Ziemnicka, K.; Fischbach, J.; Wrotkowska, E.; Ruchala, M. Familial syndromes associated with neuroendocrine tumours. Contemp. Oncol. 2015, 19, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Gortz, B.; Roth, J.; Krahenmann, A.; de Krijger, R.R.; Muletta-Feurer, S.; Rutimann, K.; Saremaslani, P.; Speel, E.J.; Heitz, P.U.; Komminoth, P. Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am. J. Pathol. 1999, 154, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.; Skogseid, B.; Oberg, K.; Nakamura, Y.; Nordenskjold, M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988, 332, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M.; Kwok, J.B.; Healey, C.S.; Elsdon, M.J.; Eng, C.; Gardner, E.; Love, D.R.; Mole, S.E.; Moore, J.K.; Papi, L.; et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363, 458–460. [Google Scholar] [CrossRef]
- van Treijen, M.J.C.; de Vries, L.H.; Hertog, D.; Vriens, M.R.; Verrijn Stuart, A.A.; van Nesselrooij, B.P.M.; Valk, G.D. Multiple Endocrine Neoplasia Type 2. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Schuffenecker, I.; Ginet, N.; Goldgar, D.; Eng, C.; Chambe, B.; Boneu, A.; Houdent, C.; Pallo, D.; Schlumberger, M.; Thivolet, C.; et al. Prevalence and parental origin of de novo RET mutations in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma. Le Groupe d’Etude des Tumeurs a Calcitonine. Am. J. Hum. Genet. 1997, 60, 233–237. [Google Scholar]
- Jakobovitz, O.; Nass, D.; DeMarco, L.; Barbosa, A.J.; Simoni, F.B.; Rechavi, G.; Friedman, E. Carcinoid tumors frequently display genetic abnormalities involving chromosome 11. J. Clin. Endocrinol. Metab. 1996, 81, 3164–3167. [Google Scholar]
- D’Adda, T.; Pizzi, S.; Azzoni, C.; Bottarelli, L.; Crafa, P.; Pasquali, C.; Davoli, C.; Corleto, V.D.; Delle Fave, G.; Bordi, C. Different patterns of 11q allelic losses in digestive endocrine tumors. Hum. Pathol. 2002, 33, 322–329. [Google Scholar] [CrossRef]
- Onuki, N.; Wistuba, I.I.; Travis, W.D.; Virmani, A.K.; Yashima, K.; Brambilla, E.; Hasleton, P.; Gazdar, A.F. Genetic changes in the spectrum of neuroendocrine lung tumors. Cancer 1999, 85, 600–607. [Google Scholar] [CrossRef]
- D’Adda, T.; Candidus, S.; Denk, H.; Bordi, C.; Hofler, H. Gastric neuroendocrine neoplasms: Tumour clonality and malignancy-associated large X-chromosomal deletions. J. Pathol. 1999, 189, 394–401. [Google Scholar] [CrossRef]
- Han, H.S.; Kim, H.S.; Woo, D.K.; Kim, W.H.; Kim, Y.I. Loss of heterozygosity in gastric neuroendocrine tumor. Anticancer Res. 2000, 20, 2849–2854. [Google Scholar] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Modica, I.; Klimstra, D.S.; Song, L.; Allen, P.J.; Brennan, M.F.; Levine, A.J.; Tang, L.H. Gene Amplifications in Well-Differentiated Pancreatic Neuroendocrine Tumors Inactivate the p53 Pathway. Genes Cancer 2010, 1, 360–368. [Google Scholar] [CrossRef]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; Demarzo, A.M.; et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef]
- Wang, G.G.; Yao, J.C.; Worah, S.; White, J.A.; Luna, R.; Wu, T.T.; Hamilton, S.R.; Rashid, A. Comparison of genetic alterations in neuroendocrine tumors: Frequent loss of chromosome 18 in ileal carcinoid tumors. Mod. Pathol. 2005, 18, 1079–1087. [Google Scholar] [CrossRef]
- Kytola, S.; Hoog, A.; Nord, B.; Cedermark, B.; Frisk, T.; Larsson, C.; Kjellman, M. Comparative genomic hybridization identifies loss of 18q22-qter as an early and specific event in tumorigenesis of midgut carcinoids. Am. J. Pathol. 2001, 158, 1803–1808. [Google Scholar] [CrossRef]
- Kulke, M.H.; Freed, E.; Chiang, D.Y.; Philips, J.; Zahrieh, D.; Glickman, J.N.; Shivdasani, R.A. High-resolution analysis of genetic alterations in small bowel carcinoid tumors reveals areas of recurrent amplification and loss. Genes Chromosomes Cancer 2008, 47, 591–603. [Google Scholar] [CrossRef]
- Kytola, S.; Nord, B.; Elder, E.E.; Carling, T.; Kjellman, M.; Cedermark, B.; Juhlin, C.; Hoog, A.; Isola, J.; Larsson, C. Alterations of the SDHD gene locus in midgut carcinoids, Merkel cell carcinomas, pheochromocytomas, and abdominal paragangliomas. Genes Chromosomes Cancer 2002, 34, 325–332. [Google Scholar] [CrossRef]
- Zhao, J.; de Krijger, R.R.; Meier, D.; Speel, E.J.; Saremaslani, P.; Muletta-Feurer, S.; Matter, C.; Roth, J.; Heitz, P.U.; Komminoth, P. Genomic alterations in well-differentiated gastrointestinal and bronchial neuroendocrine tumors (carcinoids): Marked differences indicating diversity in molecular pathogenesis. Am. J. Pathol. 2000, 157, 1431–1438. [Google Scholar] [CrossRef]
- Tonnies, H.; Toliat, M.R.; Ramel, C.; Pape, U.F.; Neitzel, H.; Berger, W.; Wiedenmann, B. Analysis of sporadic neuroendocrine tumours of the enteropancreatic system by comparative genomic hybridisation. Gut 2001, 48, 536–541. [Google Scholar] [CrossRef] [PubMed]
- van Riet, J.; van de Werken, H.J.G.; Cuppen, E.; Eskens, F.; Tesselaar, M.; van Veenendaal, L.M.; Klumpen, H.J.; Dercksen, M.W.; Valk, G.D.; Lolkema, M.P.; et al. The genomic landscape of 85 advanced neuroendocrine neoplasms reveals subtype-heterogeneity and potential therapeutic targets. Nat. Commun. 2021, 12, 4612. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tanaka, S.; Haruma, K.; Kitadai, Y.; Yoshihara, M.; Sumii, K.; Kajiyama, G.; Shimamoto, F. Growth characteristics of rectal carcinoid tumors. Oncology 2000, 59, 229–237. [Google Scholar] [CrossRef]
- Watanabe, H.; Yamazaki, Y.; Fujishima, F.; Izumi, K.; Imamura, M.; Hijioka, S.; Toriyama, K.; Yatabe, Y.; Kudo, A.; Motoi, F.; et al. O(6)-methylguanine DNA methyltransferase and glucose transporter 2 in foregut and hindgut gastrointestinal neuroendocrine neoplasms. BMC Cancer 2020, 20, 1195. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.; Oberg, K.; Stridsberg, M. Tumor markers in neuroendocrine tumors. Digestion 2000, 62 (Suppl. 1), 33–38. [Google Scholar] [CrossRef]
- Oberg, K.; Couvelard, A.; Delle Fave, G.; Gross, D.; Grossman, A.; Jensen, R.T.; Pape, U.F.; Perren, A.; Rindi, G.; Ruszniewski, P.; et al. ENETS Consensus Guidelines for Standard of Care in Neuroendocrine Tumours: Biochemical Markers. Neuroendocrinology 2017, 105, 201–211. [Google Scholar] [CrossRef]
- O’Connor, D.T.; Deftos, L.J. Secretion of chromogranin A by peptide-producing endocrine neoplasms. N. Engl. J. Med. 1986, 314, 1145–1151. [Google Scholar] [CrossRef]
- Nobels, F.R.; Kwekkeboom, D.J.; Coopmans, W.; Schoenmakers, C.H.; Lindemans, J.; De Herder, W.W.; Krenning, E.P.; Bouillon, R.; Lamberts, S.W. Chromogranin A as serum marker for neuroendocrine neoplasia: Comparison with neuron-specific enolase and the alpha-subunit of glycoprotein hormones. J. Clin. Endocrinol. Metab. 1997, 82, 2622–2628. [Google Scholar]
- Goebel, S.U.; Serrano, J.; Yu, F.; Gibril, F.; Venzon, D.J.; Jensen, R.T. Prospective study of the value of serum chromogranin A or serum gastrin levels in the assessment of the presence, extent, or growth of gastrinomas. Cancer 1999, 85, 1470–1483. [Google Scholar] [CrossRef]
- Modlin, I.M.; Gustafsson, B.I.; Moss, S.F.; Pavel, M.; Tsolakis, A.V.; Kidd, M. Chromogranin A--biological function and clinical utility in neuro endocrine tumor disease. Ann. Surg. Oncol. 2010, 17, 2427–2443. [Google Scholar] [CrossRef]
- Jensen, K.H.; Hilsted, L.; Jensen, C.; Mynster, T.; Rehfeld, J.F.; Knigge, U. Chromogranin A is a sensitive marker of progression or regression in ileo-cecal neuroendocrine tumors. Scand. J. Gastroenterol. 2013, 48, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M. Synaptophysin and neurofilament proteins as markers for neuroendocrine tumors. Arch. Pathol. Lab. Med. 1987, 111, 813–818. [Google Scholar] [PubMed]
- Wiedenmann, B.; Franke, W.W.; Kuhn, C.; Moll, R.; Gould, V.E. Synaptophysin: A marker protein for neuroendocrine cells and neoplasms. Proc. Natl. Acad. Sci. USA 1986, 83, 3500–3504. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Mete, O. Algorithmic approach to neuroendocrine tumors in targeted biopsies: Practical applications of immunohistochemical markers. Cancer Cytopathol. 2016, 124, 871–884. [Google Scholar] [CrossRef]
- Weissferdt, A.; Phan, A.; Suster, S.; Moran, C.A. Adrenocortical carcinoma: A comprehensive immunohistochemical study of 40 cases. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 24–30. [Google Scholar] [CrossRef]
- Notohara, K.; Hamazaki, S.; Tsukayama, C.; Nakamoto, S.; Kawabata, K.; Mizobuchi, K.; Sakamoto, K.; Okada, S. Solid-pseudopapillary tumor of the pancreas: Immunohistochemical localization of neuroendocrine markers and CD10. Am. J. Surg. Pathol. 2000, 24, 1361–1371. [Google Scholar] [CrossRef]
- Kriegsmann, K.; Zgorzelski, C.; Muley, T.; Christopoulos, P.; Thomas, M.; Winter, H.; Eichhorn, M.; Eichhorn, F.; von Winterfeld, M.; Herpel, E.; et al. Role of Synaptophysin, Chromogranin and CD56 in adenocarcinoma and squamous cell carcinoma of the lung lacking morphological features of neuroendocrine differentiation: A retrospective large-scale study on 1170 tissue samples. BMC Cancer 2021, 21, 486. [Google Scholar] [CrossRef]
- Lenchner, J.R.; Santos, C. Biochemistry, 5 Hydroxyindoleacetic Acid; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Oberg, K.; Modlin, I.M.; De Herder, W.; Pavel, M.; Klimstra, D.; Frilling, A.; Metz, D.C.; Heaney, A.; Kwekkeboom, D.; Strosberg, J.; et al. Consensus on biomarkers for neuroendocrine tumour disease. Lancet Oncol. 2015, 16, e435–e446. [Google Scholar] [CrossRef]
- Calanchini, M.; Tadman, M.; Krogh, J.; Fabbri, A.; Grossman, A.; Shine, B. Measurement of urinary 5-HIAA: Correlation between spot versus 24-h urine collection. Endocr. Connect. 2019, 8, 1082–1088. [Google Scholar] [CrossRef]
- Kema, I.P.; de Vries, E.G.; Muskiet, F.A. Clinical chemistry of serotonin and metabolites. J. Chromatogr. B Biomed. Sci. Appl. 2000, 747, 33–48. [Google Scholar] [CrossRef]
- Janson, E.T.; Holmberg, L.; Stridsberg, M.; Eriksson, B.; Theodorsson, E.; Wilander, E.; Oberg, K. Carcinoid tumors: Analysis of prognostic factors and survival in 301 patients from a referral center. Ann. Oncol. 1997, 8, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Kema, I.P.; Schellings, A.M.; Meiborg, G.; Hoppenbrouwers, C.J.; Muskiet, F.A. Influence of a serotonin- and dopamine-rich diet on platelet serotonin content and urinary excretion of biogenic amines and their metabolites. Clin. Chem. 1992, 38, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.M.; Lee, E.M. Serotonin content of foods: Effect on urinary excretion of 5-hydroxyindoleacetic acid. Am. J. Clin. Nutr. 1985, 42, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Modlin, I.M.; Kidd, M.; Malczewska, A.; Drozdov, I.; Bodei, L.; Matar, S.; Chung, K.M. The NETest: The Clinical Utility of Multigene Blood Analysis in the Diagnosis and Management of Neuroendocrine Tumors. Endocrinol. Metab. Clin. N. Am. 2018, 47, 485–504. [Google Scholar] [CrossRef]
- Drozdov, I.; Kidd, M.; Nadler, B.; Camp, R.L.; Mane, S.M.; Hauso, O.; Gustafsson, B.I.; Modlin, I.M. Predicting neuroendocrine tumor (carcinoid) neoplasia using gene expression profiling and supervised machine learning. Cancer 2009, 115, 1638–1650. [Google Scholar] [CrossRef]
- Malczewska, A.; Bodei, L.; Kidd, M.; Modlin, I.M. Blood mRNA Measurement (NETest) for Neuroendocrine Tumor Diagnosis of Image-Negative Liver Metastatic Disease. J. Clin. Endocrinol. Metab. 2019, 104, 867–872. [Google Scholar] [CrossRef]
- Modlin, I.M.; Kidd, M.; Falconi, M.; Filosso, P.L.; Frilling, A.; Malczewska, A.; Toumpanakis, C.; Valk, G.; Pacak, K.; Bodei, L.; et al. A multigenomic liquid biopsy biomarker for neuroendocrine tumor disease outperforms CgA and has surgical and clinical utility. Ann. Oncol. 2021, 32, 1425–1433. [Google Scholar] [CrossRef]
- Modlin, I.M.; Drozdov, I.; Kidd, M. The identification of gut neuroendocrine tumor disease by multiple synchronous transcript analysis in blood. PLoS ONE 2013, 8, e63364. [Google Scholar] [CrossRef]
- van Treijen, M.J.C.; van der Zee, D.; Heeres, B.C.; Staal, F.C.R.; Vriens, M.R.; Saveur, L.J.; Verbeek, W.H.M.; Korse, C.M.; Maas, M.; Valk, G.D.; et al. Blood Molecular Genomic Analysis Predicts the Disease Course of Gastroenteropancreatic Neuroendocrine Tumor Patients: A Validation Study of the Predictive Value of the NETest(R). Neuroendocrinology 2021, 111, 586–598. [Google Scholar] [CrossRef]
- Oberg, K.; Califano, A.; Strosberg, J.R.; Ma, S.; Pape, U.; Bodei, L.; Kaltsas, G.; Toumpanakis, C.; Goldenring, J.R.; Frilling, A.; et al. A meta-analysis of the accuracy of a neuroendocrine tumor mRNA genomic biomarker (NETest) in blood. Ann. Oncol. 2020, 31, 202–212. [Google Scholar] [CrossRef]
- Hofman, M.S.; Lau, W.F.; Hicks, R.J. Somatostatin receptor imaging with 68Ga DOTATATE PET/CT: Clinical utility, normal patterns, pearls, and pitfalls in interpretation. Radiographics 2015, 35, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Westlin, J.E.; Janson, E.T.; Arnberg, H.; Ahlstrom, H.; Oberg, K.; Nilsson, S. Somatostatin receptor scintigraphy of carcinoid tumours using the [111In-DTPA-D-Phe1]-octreotide. Acta Oncol. 1993, 32, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Smit Duijzentkunst, D.A.; Kwekkeboom, D.J.; Bodei, L. Somatostatin Receptor 2-Targeting Compounds. J. Nucl. Med. 2017, 58, 54S–60S. [Google Scholar] [CrossRef]
- Ambrosini, V.; Campana, D.; Bodei, L.; Nanni, C.; Castellucci, P.; Allegri, V.; Montini, G.C.; Tomassetti, P.; Paganelli, G.; Fanti, S. 68Ga-DOTANOC PET/CT clinical impact in patients with neuroendocrine tumors. J. Nucl. Med. 2010, 51, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M.; Decristoforo, C.; Kendler, D.; Dobrozemsky, G.; Heute, D.; Uprimny, C.; Kovacs, P.; Von Guggenberg, E.; Bale, R.; Virgolini, I.J. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: Comparison with somatostatin receptor scintigraphy and CT. J. Nucl. Med. 2007, 48, 508–518. [Google Scholar] [CrossRef]
- Kaemmerer, D.; Peter, L.; Lupp, A.; Schulz, S.; Sanger, J.; Prasad, V.; Kulkarni, H.; Haugvik, S.P.; Hommann, M.; Baum, R.P. Molecular imaging with (6)(8)Ga-SSTR PET/CT and correlation to immunohistochemistry of somatostatin receptors in neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1659–1668. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; Board, W.H.O.C.o.T.E. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef]
- Kulke, M.H.; Siu, L.L.; Tepper, J.E.; Fisher, G.; Jaffe, D.; Haller, D.G.; Ellis, L.M.; Benedetti, J.K.; Bergsland, E.K.; Hobday, T.J.; et al. Future directions in the treatment of neuroendocrine tumors: Consensus report of the National Cancer Institute Neuroendocrine Tumor clinical trials planning meeting. J. Clin. Oncol. 2011, 29, 934–943. [Google Scholar] [CrossRef]
- Hamilton, N.A.; Liu, T.C.; Cavatiao, A.; Mawad, K.; Chen, L.; Strasberg, S.S.; Linehan, D.C.; Cao, D.; Hawkins, W.G. Ki-67 predicts disease recurrence and poor prognosis in pancreatic neuroendocrine neoplasms. Surgery 2012, 152, 107–113. [Google Scholar] [CrossRef]
- Faggiano, A.; Carratu, A.C.; Guadagno, E.; Tafuto, S.; Tatangelo, F.; Riccardi, F.; Mocerino, C.; Palmieri, G.; Damiano, V.; Siciliano, R.; et al. Somatostatin analogues according to Ki67 index in neuroendocrine tumours: An observational retrospective-prospective analysis from real life. Oncotarget 2016, 7, 5538–5547. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Cwikla, J.B.; Phan, A.T.; Raderer, M.; Sedlackova, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Rose, S.; White, S.B.; El-Haddad, G.; Fidelman, N.; Yarmohammadi, H.; Hwang, W.; Sze, D.Y.; Kothary, N.; Stashek, K.; et al. Embolotherapy for Neuroendocrine Tumor Liver Metastases: Prognostic Factors for Hepatic Progression-Free Survival and Overall Survival. Cardiovasc. Intervent. Radiol. 2017, 40, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Singla, S.; LeVea, C.M.; Pokuri, V.K.; Attwood, K.M.; Wach, M.M.; Tomaszewski, G.M.; Kuvshinoff, B.; Iyer, R. Ki67 score as a potential predictor in the selection of liver-directed therapies for metastatic neuroendocrine tumors: A single institutional experience. J. Gastrointest. Oncol. 2016, 7, 441–448. [Google Scholar] [CrossRef]
- Tang, L.H.; Untch, B.R.; Reidy, D.L.; O’Reilly, E.; Dhall, D.; Jih, L.; Basturk, O.; Allen, P.J.; Klimstra, D.S. Well-Differentiated Neuroendocrine Tumors with a Morphologically Apparent High-Grade Component: A Pathway Distinct from Poorly Differentiated Neuroendocrine Carcinomas. Clin. Cancer Res. 2016, 22, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Wilke, A.; Rinke, A.; Mayer, C.; Kann, P.H.; Klose, K.J.; Scherag, A.; Hahmann, M.; Muller, H.H.; Barth, P. Plasma chromogranin A as marker for survival in patients with metastatic endocrine gastroenteropancreatic tumors. Clin. Gastroenterol. Hepatol. 2008, 6, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Kouvaraki, M.A.; Ajani, J.A.; Hoff, P.; Wolff, R.; Evans, D.B.; Lozano, R.; Yao, J.C. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J. Clin. Oncol. 2004, 22, 4762–4771. [Google Scholar] [CrossRef]
- Yao, J.C.; Lombard-Bohas, C.; Baudin, E.; Kvols, L.K.; Rougier, P.; Ruszniewski, P.; Hoosen, S.; St Peter, J.; Haas, T.; Lebwohl, D.; et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J. Clin. Oncol. 2010, 28, 69–76. [Google Scholar] [CrossRef]
- Hellman, P.; Ladjevardi, S.; Skogseid, B.; Akerstrom, G.; Elvin, A. Radiofrequency tissue ablation using cooled tip for liver metastases of endocrine tumors. World J. Surg. 2002, 26, 1052–1056. [Google Scholar] [CrossRef]
- Kolbeck, K.J.; Farsad, K. Catheter-based treatments for hepatic metastases from neuroendocrine tumors. AJR Am. J. Roentgenol. 2014, 203, 717–724. [Google Scholar] [CrossRef]
- Wedin, M.; Mehta, S.; Angeras-Kraftling, J.; Wallin, G.; Daskalakis, K. The Role of Serum 5-HIAA as a Predictor of Progression and an Alternative to 24-h Urine 5-HIAA in Well-Differentiated Neuroendocrine Neoplasms. Biology 2021, 10, 76. [Google Scholar] [CrossRef]
- Tirosh, A.; Nilubol, N.; Patel, D.; Kebebew, E. Prognostic Utility of 24-Hour Urinary 5-HIAA Doubling Time in Patients With Neuroendocrine Tumors. Endocr. Pract. 2018, 24, 710–717. [Google Scholar] [CrossRef]
- Martensson, H.; Nobin, A.; Bengmark, S.; Lunderquist, A.; Owman, T.; Sanden, G. Embolization of the liver in the management of metastatic carcinoid tumors. J. Surg. Oncol. 1984, 27, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Wangberg, B.; Westberg, G.; Tylen, U.; Tisell, L.; Jansson, S.; Nilsson, O.; Johansson, V.; Schersten, T.; Ahlman, H. Survival of patients with disseminated midgut carcinoid tumors after aggressive tumor reduction. World J. Surg. 1996, 20, 892–899; discussion 899. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, C.H.; Charnsangavej, C.; Ajani, J.; Samaan, N.A.; Richli, W.; Wallace, S. The carcinoid syndrome: Palliation by hepatic artery embolization. AJR Am. J. Roentgenol. 1986, 147, 149–154. [Google Scholar] [CrossRef]
- Hanssen, L.E.; Schrumpf, E.; Kolbenstvedt, A.N.; Tausjo, J.; Dolva, L.O. Treatment of malignant metastatic midgut carcinoid tumours with recombinant human alpha2b interferon with or without prior hepatic artery embolization. Scand. J. Gastroenterol. 1989, 24, 787–795. [Google Scholar] [CrossRef]
- Eriksson, J.; Stalberg, P.; Nilsson, A.; Krause, J.; Lundberg, C.; Skogseid, B.; Granberg, D.; Eriksson, B.; Akerstrom, G.; Hellman, P. Surgery and radiofrequency ablation for treatment of liver metastases from midgut and foregut carcinoids and endocrine pancreatic tumors. World J. Surg. 2008, 32, 930–938. [Google Scholar] [CrossRef]
- Pea, A.; Hruban, R.H.; Wood, L.D. Genetics of pancreatic neuroendocrine tumors: Implications for the clinic. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Marinoni, I.; Kurrer, A.S.; Vassella, E.; Dettmer, M.; Rudolph, T.; Banz, V.; Hunger, F.; Pasquinelli, S.; Speel, E.J.; Perren, A. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 2014, 146, 453–460.e455. [Google Scholar] [CrossRef]
- Cives, M.; Partelli, S.; Palmirotta, R.; Lovero, D.; Mandriani, B.; Quaresmini, D.; Pelle, E.; Andreasi, V.; Castelli, P.; Strosberg, J.; et al. DAXX mutations as potential genomic markers of malignant evolution in small nonfunctioning pancreatic neuroendocrine tumors. Sci. Rep. 2019, 9, 18614. [Google Scholar] [CrossRef]
- Ziv, E.; Rice, S.L.; Filtes, J.; Yarmohammadi, H.; Boas, F.E.; Erinjeri, J.P.; Petre, E.N.; Brody, L.A.; Brown, K.T.; Covey, A.M.; et al. DAXX Mutation Status of Embolization-Treated Neuroendocrine Tumors Predicts Shorter Time to Hepatic Progression. J. Vasc. Interv. Radiol. 2018, 29, 1519–1526. [Google Scholar] [CrossRef]
- Pavel, M. Translation of molecular pathways into clinical trials of neuroendocrine tumors. Neuroendocrinology 2013, 97, 99–112. [Google Scholar] [CrossRef]
- Oberg, K.; Lamberts, S.W. Somatostatin analogues in acromegaly and gastroenteropancreatic neuroendocrine tumours: Past, present and future. Endocr. Relat. Cancer 2016, 23, R551–R566. [Google Scholar] [CrossRef]
- Baldelli, R.; Barnabei, A.; Rizza, L.; Isidori, A.M.; Rota, F.; Di Giacinto, P.; Paoloni, A.; Torino, F.; Corsello, S.M.; Lenzi, A.; et al. Somatostatin analogs therapy in gastroenteropancreatic neuroendocrine tumors: Current aspects and new perspectives. Front. Endocrinol. 2014, 5, 7. [Google Scholar] [CrossRef]
- Patel, M.; Tena, I.; Jha, A.; Taieb, D.; Pacak, K. Somatostatin Receptors and Analogs in Pheochromocytoma and Paraganglioma: Old Players in a New Precision Medicine World. Front. Endocrinol. 2021, 12, 625312. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Muller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Blaker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef]
- Laskaratos, F.M.; Walker, M.; Naik, K.; Maragkoudakis, E.; Oikonomopoulos, N.; Grant, L.; Meyer, T.; Caplin, M.; Toumpanakis, C. Predictive factors of antiproliferative activity of octreotide LAR as first-line therapy for advanced neuroendocrine tumours. Br. J. Cancer 2016, 115, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Butturini, G.; Bettini, R.; Missiaglia, E.; Mantovani, W.; Dalai, I.; Capelli, P.; Ferdeghini, M.; Pederzoli, P.; Scarpa, A.; Falconi, M. Predictive factors of efficacy of the somatostatin analogue octreotide as first line therapy for advanced pancreatic endocrine carcinoma. Endocr. Relat. Cancer 2006, 13, 1213–1221. [Google Scholar] [CrossRef]
- Hope, T.A.; Bodei, L.; Chan, J.A.; El-Haddad, G.; Fidelman, N.; Kunz, P.L.; Mailman, J.; Menda, Y.; Metz, D.C.; Mittra, E.S.; et al. NANETS/SNMMI Consensus Statement on Patient Selection and Appropriate Use of (177)Lu-DOTATATE Peptide Receptor Radionuclide Therapy. J. Nucl. Med. 2020, 61, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kulkarni, H.R.; Singh, A.; Niepsch, K.; Muller, D.; Baum, R.P. Peptide Receptor Radionuclide Therapy in Grade 3 Neuroendocrine Neoplasms: Safety and Survival Analysis in 69 Patients. J. Nucl. Med. 2019, 60, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Stefanova, M.; Mavriopoulou, E.; Holland-Letz, T.; Dimitrakopoulou-Strauss, A.; Afshar-Oromieh, A.; Mier, W.; Haberkorn, U.; Giesel, F.L. SUV of [68Ga]DOTATOC-PET/CT Predicts Response Probability of PRRT in Neuroendocrine Tumors. Mol. Imaging Biol. 2015, 17, 313–318. [Google Scholar] [CrossRef]
- Baum, R.P.; Kulkarni, H.R.; Singh, A.; Kaemmerer, D.; Mueller, D.; Prasad, V.; Hommann, M.; Robiller, F.C.; Niepsch, K.; Franz, H.; et al. Results and adverse events of personalized peptide receptor radionuclide therapy with (90)Yttrium and (177)Lutetium in 1048 patients with neuroendocrine neoplasms. Oncotarget 2018, 9, 16932–16950. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q.; Singh, A.; Schuchardt, C.; Kulkarni, H.R.; Baum, R.P. Prognostic Value of (18)F-FDG PET/CT in a Large Cohort of Patients with Advanced Metastatic Neuroendocrine Neoplasms Treated with Peptide Receptor Radionuclide Therapy. J. Nucl. Med. 2020, 61, 1560–1569. [Google Scholar] [CrossRef]
- Katona, B.W.; Roccaro, G.A.; Soulen, M.C.; Yang, Y.X.; Bennett, B.J.; Riff, B.P.; Glynn, R.A.; Wild, D.; Nicolas, G.P.; Pryma, D.A.; et al. Efficacy of Peptide Receptor Radionuclide Therapy in a United States-Based Cohort of Metastatic Neuroendocrine Tumor Patients: Single-Institution Retrospective Analysis. Pancreas 2017, 46, 1121–1126. [Google Scholar] [CrossRef]
- Aalbersberg, E.A.; Huizing, D.M.V.; Walraven, I.; de Wit-van der Veen, B.J.; Kulkarni, H.R.; Singh, A.; Stokkel, M.P.M.; Baum, R.P. Parameters to Predict Progression-Free and Overall Survival After Peptide Receptor Radionuclide Therapy: A Multivariate Analysis in 782 Patients. J. Nucl. Med. 2019, 60, 1259–1265. [Google Scholar] [CrossRef]
- Ezziddin, S.; Attassi, M.; Yong-Hing, C.J.; Ahmadzadehfar, H.; Willinek, W.; Grunwald, F.; Guhlke, S.; Biersack, H.J.; Sabet, A. Predictors of long-term outcome in patients with well-differentiated gastroenteropancreatic neuroendocrine tumors after peptide receptor radionuclide therapy with 177Lu-octreotate. J. Nucl. Med. 2014, 55, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Kidd, M.; Modlin, I.M.; Severi, S.; Drozdov, I.; Nicolini, S.; Kwekkeboom, D.J.; Krenning, E.P.; Baum, R.P.; Paganelli, G. Measurement of circulating transcripts and gene cluster analysis predicts and defines therapeutic efficacy of peptide receptor radionuclide therapy (PRRT) in neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Liu, L.Z. Role of mTOR in anticancer drug resistance: Perspectives for improved drug treatment. Drug Resist Updat. 2008, 11, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Neychev, V.; Steinberg, S.M.; Cottle-Delisle, C.; Merkel, R.; Nilubol, N.; Yao, J.; Meltzer, P.; Pacak, K.; Marx, S.; Kebebew, E. Mutation-targeted therapy with sunitinib or everolimus in patients with advanced low-grade or intermediate-grade neuroendocrine tumours of the gastrointestinal tract and pancreas with or without cytoreductive surgery: Protocol for a phase II clinical trial. BMJ Open 2015, 5, e008248. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Goldner, W.S.; Halfdanarson, T.R.; Bergsland, E.; Berlin, J.D.; Halperin, D.; Chan, J.; Kulke, M.H.; Benson, A.B.; Blaszkowsky, L.S.; et al. NCCN Guidelines Insights: Neuroendocrine and Adrenal Tumors, Version 2.2018. J. Natl. Compr. Canc. Netw. 2018, 16, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Benslama, N.; Bollard, J.; Vercherat, C.; Massoma, P.; Roche, C.; Hervieu, V.; Peron, J.; Lombard-Bohas, C.; Scoazec, J.Y.; Walter, T. Prediction of response to everolimus in neuroendocrine tumors: Evaluation of clinical, biological and histological factors. Investig. New Drugs 2016, 34, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Pavel, M.; Phan, A.T.; Kulke, M.H.; Hoosen, S.; St Peter, J.; Cherfi, A.; Oberg, K.E. Chromogranin A and neuron-specific enolase as prognostic markers in patients with advanced pNET treated with everolimus. J. Clin. Endocrinol. Metab. 2011, 96, 3741–3749. [Google Scholar] [CrossRef] [PubMed]
- Gerson, S.L. Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. 2002, 20, 2388–2399. [Google Scholar] [CrossRef]
- Kulke, M.H.; Hornick, J.L.; Frauenhoffer, C.; Hooshmand, S.; Ryan, D.P.; Enzinger, P.C.; Meyerhardt, J.A.; Clark, J.W.; Stuart, K.; Fuchs, C.S.; et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin. Cancer Res. 2009, 15, 338–345. [Google Scholar] [CrossRef]
- Raj, N.; Klimstra, D.S.; Horvat, N.; Zhang, L.; Chou, J.F.; Capanu, M.; Basturk, O.; Do, R.K.G.; Allen, P.J.; Reidy-Lagunes, D. O6-Methylguanine DNA Methyltransferase Status Does Not Predict Response or Resistance to Alkylating Agents in Well-Differentiated Pancreatic Neuroendocrine Tumors. Pancreas 2017, 46, 758–763. [Google Scholar] [CrossRef]
- Girot, P.; Dumars, C.; Mosnier, J.F.; Muzellec, L.; Senellart, H.; Foubert, F.; Caroli-Bosc, F.X.; Cauchin, E.; Regenet, N.; Matysiak-Budnik, T.; et al. Short article: Evaluation of O6-methylguanine-DNA methyltransferase as a predicting factor of response to temozolomide-based chemotherapy in well-differentiated metastatic pancreatic neuroendocrine tumors. Eur. J. Gastroenterol. Hepatol. 2017, 29, 826–830. [Google Scholar] [CrossRef]
- Lemelin, A.; Barritault, M.; Hervieu, V.; Payen, L.; Peron, J.; Couvelard, A.; Cros, J.; Scoazec, J.Y.; Bin, S.; Villeneuve, L.; et al. O6-methylguanine-DNA methyltransferase (MGMT) status in neuroendocrine tumors: A randomized phase II study (MGMT-NET). Dig. Liver Dis. 2019, 51, 595–599. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Fine, R.L.; Choi, J.; Nasir, A.; Coppola, D.; Chen, D.T.; Helm, J.; Kvols, L. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer 2011, 117, 268–275. [Google Scholar] [CrossRef]
- Fine, R.L.; Gulati, A.P.; Krantz, B.A.; Moss, R.A.; Schreibman, S.; Tsushima, D.A.; Mowatt, K.B.; Dinnen, R.D.; Mao, Y.; Stevens, P.D.; et al. Capecitabine and temozolomide (CAPTEM) for metastatic, well-differentiated neuroendocrine cancers: The Pancreas Center at Columbia University experience. Cancer Chemother. Pharmacol. 2013, 71, 663–670. [Google Scholar] [CrossRef]
- Al-Toubah, T.; Pelle, E.; Valone, T.; Haider, M.; Strosberg, J.R. Efficacy and Toxicity Analysis of Capecitabine and Temozolomide in Neuroendocrine Neoplasms. J. Natl. Compr. Canc. Netw. 2021, 20, 29–36. [Google Scholar] [CrossRef]
- Das, S.; Al-Toubah, T.; Strosberg, J. Chemotherapy in Neuroendocrine Tumors. Cancers 2021, 13, 4872. [Google Scholar] [CrossRef] [PubMed]
- Soulen, M.C.; van Houten, D.; Teitelbaum, U.R.; Damjanov, N.; Cengel, K.A.; Metz, D.C. Safety and Feasibility of Integrating Yttrium-90 Radioembolization With Capecitabine-Temozolomide for Grade 2 Liver-Dominant Metastatic Neuroendocrine Tumors. Pancreas 2018, 47, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Raj, N.; Reidy-Lagunes, D. Exceptional Responses After Cessation of Therapy With Alkylating Agents for Pancreatic Neuroendocrine Tumors. Pancreas 2020, 49, e14–e16. [Google Scholar] [CrossRef] [PubMed]
- Girot, P.; Baudin, E.; Senellart, H.; Bouarioua, N.; Hentic, O.; Guimbaud, R.; Walter, T.; Ferru, A.; Roquin, G.; Cadiot, G.; et al. Oxaliplatin and 5-Fluorouracil in Advanced Well-Differentiated Digestive Neuroendocrine Tumors: A Multicenter National Retrospective Study from the French Group of Endocrine Tumors. Neuroendocrinology 2022, 112, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Oziel-Taieb, S.; Zemmour, C.; Raoul, J.L.; Mineur, L.; Poizat, F.; Charrier, N.; Piana, G.; Cavaglione, G.; Niccoli, P. Efficacy of FOLFOX Chemotherapy in Metastatic Enteropancreatic Neuroendocrine Tumors. Anticancer Res. 2021, 41, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.L.; Balise, R.R.; Fehrenbacher, L.; Pan, M.; Venook, A.P.; Fisher, G.A.; Tempero, M.A.; Ko, A.H.; Korn, W.M.; Hwang, J.; et al. Oxaliplatin-Fluoropyrimidine Chemotherapy Plus Bevacizumab in Advanced Neuroendocrine Tumors: An Analysis of 2 Phase II Trials. Pancreas 2016, 45, 1394–1400. [Google Scholar] [CrossRef]
- Siperstein, A.E.; Rogers, S.J.; Hansen, P.D.; Gitomirsky, A. Laparoscopic thermal ablation of hepatic neuroendocrine tumor metastases. Surgery 1997, 122, 1147–1154; Discussion 1154–1145. [Google Scholar] [CrossRef]
- Mazzaglia, P.J.; Berber, E.; Milas, M.; Siperstein, A.E. Laparoscopic radiofrequency ablation of neuroendocrine liver metastases: A 10-year experience evaluating predictors of survival. Surgery 2007, 142, 10–19. [Google Scholar] [CrossRef]
- Perrodin, S.F.; Renzulli, M.M.; Maurer, M.H.; Kim-Fuchs, C.; Candinas, D.; Beldi, G.; Lachenmayer, A. Can Microwave Ablation Be an Alternative to Resection for the Treatment of Neuroendocrine Liver Metastases? Endocr. Pract. 2020, 26, 378–387. [Google Scholar] [CrossRef]
- Mohan, H.; Nicholson, P.; Winter, D.C.; O’Shea, D.; O’Toole, D.; Geoghegan, J.; Maguire, D.; Hoti, E.; Traynor, O.; Cantwell, C.P. Radiofrequency ablation for neuroendocrine liver metastases: A systematic review. J. Vasc. Interv. Radiol. 2015, 26, 935–942.e1. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, B.; Lin, M.; Zhang, X.; Zheng, Y.; Xie, X.; Xu, M.; Xie, X. Ultrasound-guided percutaneous radiofrequency ablation in treatment of neuroendocrine tumor liver metastases: AS single-center experience. Int. J. Hyperth. 2022, 39, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Taner, T.; Atwell, T.D.; Zhang, L.; Oberg, T.N.; Harmsen, W.S.; Slettedahl, S.W.; Kendrick, M.L.; Nagorney, D.M.; Que, F.G. Adjunctive radiofrequency ablation of metastatic neuroendocrine cancer to the liver complements surgical resection. HPB (Oxford). 2013, 15, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, M.; Fiore, F.; Modica, R.; Marotta, V.; Marciello, F.; Ramundo, V.; Di Sarno, A.; Carratu, A.; di Roseto Cde, L.; Tafuto, S.; et al. Hepatic arterial embolization in patients with neuroendocrine tumors. J. Exp. Clin. Cancer Res. 2014, 33, 43. [Google Scholar] [CrossRef]
- Loewe, C.; Schindl, M.; Cejna, M.; Niederle, B.; Lammer, J.; Thurnher, S. Permanent transarterial embolization of neuroendocrine metastases of the liver using cyanoacrylate and lipiodol: Assessment of mid- and long-term results. AJR Am. J. Roentgenol. 2003, 180, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Choi, J.; Cantor, A.B.; Kvols, L.K. Selective hepatic artery embolization for treatment of patients with metastatic carcinoid and pancreatic endocrine tumors. Cancer Control 2006, 13, 72–78. [Google Scholar] [CrossRef]
- Sward, C.; Johanson, V.; Nieveen van Dijkum, E.; Jansson, S.; Nilsson, O.; Wangberg, B.; Ahlman, H.; Kolby, L. Prolonged survival after hepatic artery embolization in patients with midgut carcinoid syndrome. Br. J. Surg. 2009, 96, 517–521. [Google Scholar] [CrossRef]
- Madoff, D.C.; Gupta, S.; Ahrar, K.; Murthy, R.; Yao, J.C. Update on the management of neuroendocrine hepatic metastases. J. Vasc. Interv. Radiol. 2006, 17, 1235–1250. [Google Scholar] [CrossRef]
- Touloupas, C.; Faron, M.; Hadoux, J.; Deschamps, F.; Roux, C.; Ronot, M.; Yevich, S.; Joskin, J.; Gelli, M.; Barbe, R.; et al. Long Term Efficacy and Assessment of Tumor Response of Transarterial Chemoembolization in Neuroendocrine Liver Metastases: A 15-Year Monocentric Experience. Cancers 2021, 13, 5366. [Google Scholar] [CrossRef]
- Kitano, M.; Davidson, G.W.; Shirley, L.A.; Schmidt, C.R.; Guy, G.E.; Khabiri, H.; Dowell, J.D.; Shah, M.H.; Bloomston, M. Transarterial Chemoembolization for Metastatic Neuroendocrine Tumors With Massive Hepatic Tumor Burden: Is the Benefit Worth the Risk? Ann. Surg. Oncol. 2016, 23, 4008–4015. [Google Scholar] [CrossRef]
- Rhee, T.K.; Lewandowski, R.J.; Liu, D.M.; Mulcahy, M.F.; Takahashi, G.; Hansen, P.D.; Benson, A.B., 3rd; Kennedy, A.S.; Omary, R.A.; Salem, R. 90Y Radioembolization for metastatic neuroendocrine liver tumors: Preliminary results from a multi-institutional experience. Ann. Surg. 2008, 247, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Wang, W. Yttrium-90 radioembolization for unresectable metastatic neuroendocrine liver tumor: A systematic review. Eur. J. Radiol. 2018, 100, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Tsang, E.S.; Loree, J.M.; Davies, J.M.; Gill, S.; Liu, D.; Ho, S.; Renouf, D.J.; Lim, H.J.; Kennecke, H.F. Efficacy and Prognostic Factors for Y-90 Radioembolization (Y-90) in Metastatic Neuroendocrine Tumors with Liver Metastases. Can. J. Gastroenterol. Hepatol. 2020, 2020, 5104082. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.L.; Reidy-Lagunes, D.; Anthony, L.B.; Bertino, E.M.; Brendtro, K.; Chan, J.A.; Chen, H.; Jensen, R.T.; Kim, M.K.; Klimstra, D.S.; et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas 2013, 42, 557–577. [Google Scholar] [CrossRef]
- Howe, J.R.; Merchant, N.B.; Conrad, C.; Keutgen, X.M.; Hallet, J.; Drebin, J.A.; Minter, R.M.; Lairmore, T.C.; Tseng, J.F.; Zeh, H.J.; et al. The North American Neuroendocrine Tumor Society Consensus Paper on the Surgical Management of Pancreatic Neuroendocrine Tumors. Pancreas 2020, 49, 1–33. [Google Scholar] [CrossRef]
- Mayo, S.C.; Herman, J.M.; Cosgrove, D.; Bhagat, N.; Kamel, I.; Geschwind, J.F.; Pawlik, T.M. Emerging approaches in the management of patients with neuroendocrine liver metastasis: Role of liver-directed and systemic therapies. J. Am. Coll. Surg. 2013, 216, 123–134. [Google Scholar] [CrossRef]
- Mayo, S.C.; de Jong, M.C.; Pulitano, C.; Clary, B.M.; Reddy, S.K.; Gamblin, T.C.; Celinksi, S.A.; Kooby, D.A.; Staley, C.A.; Stokes, J.B.; et al. Surgical management of hepatic neuroendocrine tumor metastasis: Results from an international multi-institutional analysis. Ann. Surg. Oncol. 2010, 17, 3129–3136. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Li, S.; Zhu, W.H.; Zhang, D.F. Microwave ablation combined with hepatectomy for treatment of neuroendocrine tumor liver metastases. World J. Clin. Cases 2021, 9, 5064–5072. [Google Scholar] [CrossRef]
- Martin, R.C.; Scoggins, C.R.; McMasters, K.M. Safety and efficacy of microwave ablation of hepatic tumors: A prospective review of a 5-year experience. Ann. Surg. Oncol. 2010, 17, 171–178. [Google Scholar] [CrossRef]
- Jensen, E.H.; Kvols, L.; McLoughlin, J.M.; Lewis, J.M.; Alvarado, M.D.; Yeatman, T.; Malafa, M.; Shibata, D. Biomarkers predict outcomes following cytoreductive surgery for hepatic metastases from functional carcinoid tumors. Ann. Surg. Oncol. 2007, 14, 780–785. [Google Scholar] [CrossRef]
- Xie, H.; Liu, J.; Yadav, S.; Keutgen, X.M.; Hobday, T.J.; Strosberg, J.R.; Halfdanarson, T.R. The Role of Perioperative Systemic Therapy in Localized Pancreatic Neuroendocrine Neoplasms. Neuroendocrinology 2020, 110, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Alese, O.B.; Jiang, R.; Shaib, W.; Wu, C.; Akce, M.; Behera, M.; El-Rayes, B.F. High-Grade Gastrointestinal Neuroendocrine Carcinoma Management and Outcomes: A National Cancer Database Study. Oncologist 2019, 24, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Cloyd, J.M.; Omichi, K.; Mizuno, T.; Kawaguchi, Y.; Tzeng, C.D.; Conrad, C.; Chun, Y.S.; Aloia, T.A.; Katz, M.H.G.; Lee, J.E.; et al. Preoperative Fluorouracil, Doxorubicin, and Streptozocin for the Treatment of Pancreatic Neuroendocrine Liver Metastases. Ann. Surg. Oncol. 2018, 25, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Ambe, C.M.; Nguyen, P.; Centeno, B.A.; Choi, J.; Strosberg, J.; Kvols, L.; Hodul, P.; Hoffe, S.; Malafa, M.P. Multimodality Management of “Borderline Resectable” Pancreatic Neuroendocrine Tumors: Report of a Single-Institution Experience. Cancer Control 2017, 24, 1073274817729076. [Google Scholar] [CrossRef] [PubMed]
- Voong, K.R.; Rashid, A.; Crane, C.H.; Minsky, B.D.; Krishnan, S.; Yao, J.C.; Wolff, R.A.; Skibber, J.M.; Feig, B.W.; Chang, G.J.; et al. Chemoradiation for High-grade Neuroendocrine Carcinoma of the Rectum and Anal Canal. Am. J. Clin. Oncol. 2017, 40, 555–560. [Google Scholar] [CrossRef]
- DePietro, D.M.; Gatmaytan, I.; Hunt, S.; Nadolski, G.; Woodard, A.; Soulen, M.; Gade, T.; Ackerman, D. Optical Genome Mapping: A Novel Approach to Identifying Structural Variants in Metastatic Neuroendocrine Tumors. In Endocrine Abstracts; North American Neuroendocrine Tumor Society: Washington, DC, USA, 2022. [Google Scholar]
- Tsunokake, J.; Fujishima, F.; Watanabe, H.; Sato, I.; Miura, K.; Sakamoto, K.; Suzuki, H.; Sawai, T.; Itakura, Y.; Hoshi, T.; et al. Tumor Microenvironment in Mixed Neuroendocrine Non-Neuroendocrine Neoplasms: Interaction between Tumors and Immune Cells, and Potential Effects of Neuroendocrine Differentiation on the Tumor Microenvironment. Cancers 2022, 14, 2152. [Google Scholar] [CrossRef]
- Crabtree, J.S. Epigenetic Regulation in Gastroenteropancreatic Neuroendocrine Tumors. Front. Oncol. 2022, 12, 901435. [Google Scholar] [CrossRef]
- Colao, A.; de Nigris, F.; Modica, R.; Napoli, C. Clinical Epigenetics of Neuroendocrine Tumors: The Road Ahead. Front. Endocrinol. 2020, 11, 604341. [Google Scholar] [CrossRef]


{kind=link}
| Neuroendocrine Neoplasm | Classification | Mitotic Rate (Mitoses/2 mm2) | Ki-67 Index |
|---|---|---|---|
| Gastroenteropancreatic Well-differentiated NET | NET, grade 1 | <2 | <3% |
| NET, grade 2 | 2–20 | 3–20% | |
| NET, grade 3 | >20 | >20% | |
| Upper aerodigestive tract and salivary glands Well-differentiated NET | NET, grade 1 | <2 | <20% |
| NET, grade 2 | 2–10 | <20% | |
| NET, grade 3 | >10 | >20% | |
| Lung and thymus Well-differentiated NET | Typical carcinoid, NET, grade 1 | <2 | |
| Atypical carcinoid, NET, grade 2 | 2–10 | ||
| Carcinoids/NETs with elevated mitotic counts and/or Ki-67 index | >10 | >30% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexander, E.S.; Ziv, E. Neuroendocrine Tumors: Genomics and Molecular Biomarkers with a Focus on Metastatic Disease. Cancers 2023, 15, 2249. https://doi.org/10.3390/cancers15082249
Alexander ES, Ziv E. Neuroendocrine Tumors: Genomics and Molecular Biomarkers with a Focus on Metastatic Disease. Cancers. 2023; 15(8):2249. https://doi.org/10.3390/cancers15082249
Chicago/Turabian StyleAlexander, Erica S., and Etay Ziv. 2023. "Neuroendocrine Tumors: Genomics and Molecular Biomarkers with a Focus on Metastatic Disease" Cancers 15, no. 8: 2249. https://doi.org/10.3390/cancers15082249
APA StyleAlexander, E. S., & Ziv, E. (2023). Neuroendocrine Tumors: Genomics and Molecular Biomarkers with a Focus on Metastatic Disease. Cancers, 15(8), 2249. https://doi.org/10.3390/cancers15082249