
A Quick Guide to CAF Subtypes in Pancreatic Cancer

 ,
,
Abstract
Simple Summary
Abstract
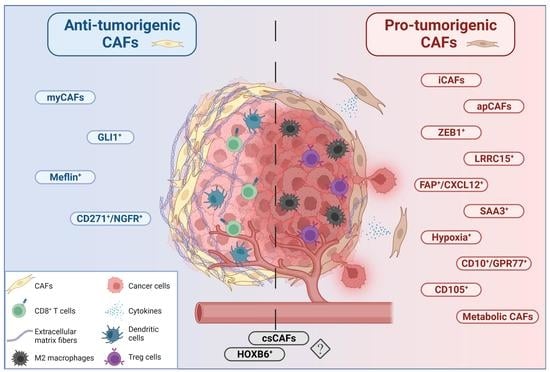
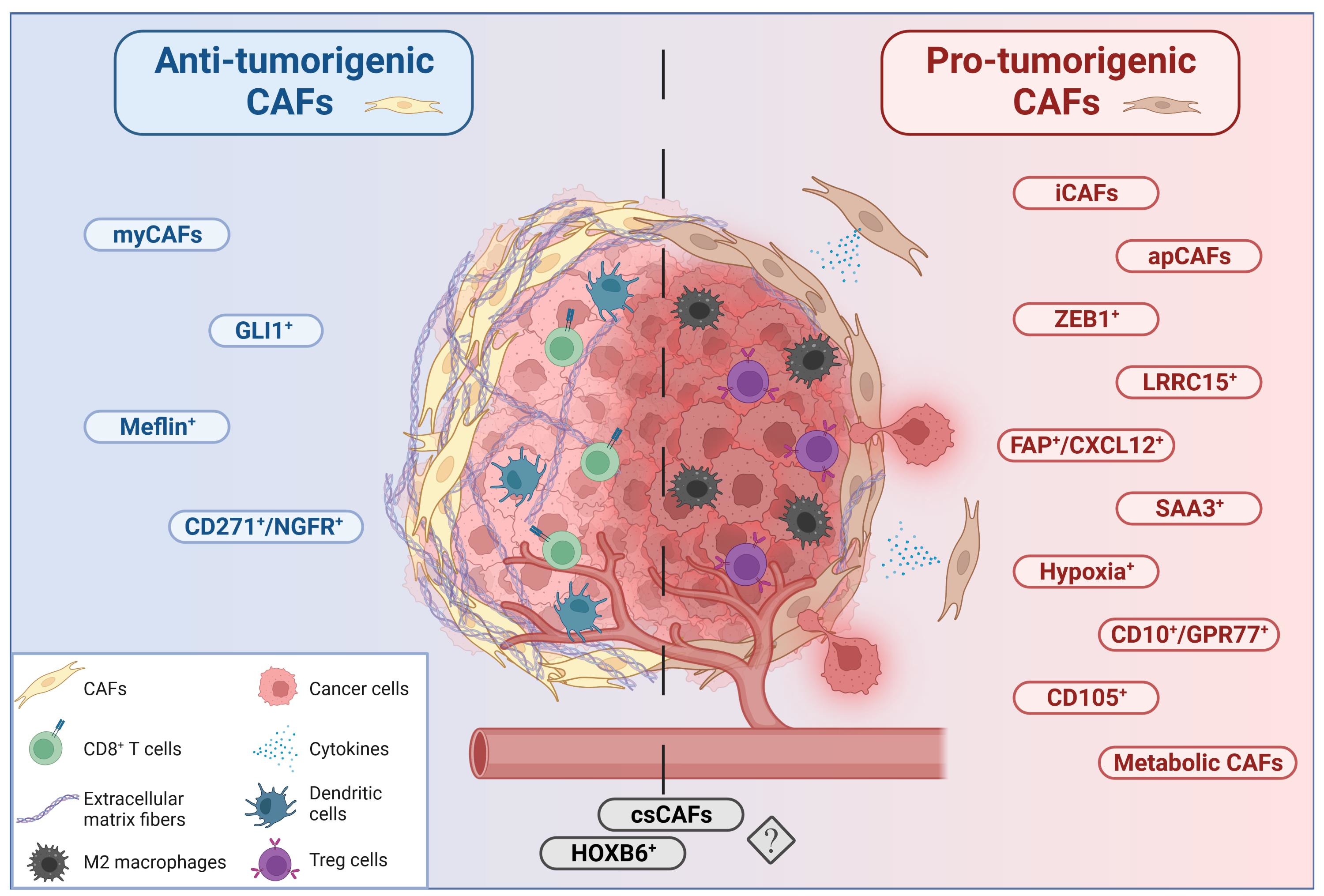{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. A Large Signaling Network Activates Fibroblasts
3. The Origin of CAFs
4. Heterogeneity of Cancer-Associated Fibroblasts in PDAC
5. Myofibroblastic CAFs (myCAFs)
6. myCAFs: Complexities around Collagen
7. ZEB1+ CAFs
8. LRRC15+ CAFs
9. GLI1+ and HOXB6+ CAFs
10. Inflammatory CAFs (iCAFs)
11. SAA3+ CAFs
12. Complement-Secreting CAFs (CsCAFs)
13. FAP+/CXCL12+ CAFs
14. CD10+/GPR77+ CAFs
15. Hypoxia+ CAFs
16. Antigen-Presenting CAFs (apCAFs)
17. CD105+ CAFs
18. Meflin+ CAFs
19. CD271/NGFR+ CAFs
20. Metabolic CAFs
21. Stromal Targeting: Where Do We Stand?
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stoker, M.G.; Shearer, M.; O'Neill, C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J. Cell Sci. 1966, 1, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Paland, N.; Kamer, I.; Kogan-Sakin, I.; Madar, S.; Goldfinger, N.; Rotter, V. Differential influence of normal and cancer-associated fibroblasts on the growth of human epithelial cells in an in vitro cocultivation model of prostate cancer. Mol. Cancer Res. 2009, 7, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; Denardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.L.; Sholl, L.M.; Ben-Aharon, I.; Beck, A.H.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.M.; Whitesell, L.; et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef]
- Shaashua, L.; Ben-Shmuel, A.; Pevsner-Fischer, M.; Friedman, G.; Levi-Galibov, O.; Nandakumar, S.; Barki, D.; Nevo, R.; Brown, L.E.; Zhang, W.; et al. BRCA mutational status shapes the stromal microenvironment of pancreatic cancer linking clusterin expression in cancer associated fibroblasts with HSF1 signaling. Nat. Commun. 2022, 13, 6513. [Google Scholar] [CrossRef]
- Levi-Galibov, O.; Lavon, H.; Wassermann-Dozorets, R.; Pevsner-Fischer, M.; Mayer, S.; Wershof, E.; Stein, Y.; Brown, L.E.; Zhang, W.; Friedman, G.; et al. Heat Shock Factor 1-dependent extracellular matrix remodeling mediates the transition from chronic intestinal inflammation to colon cancer. Nat. Commun. 2020, 11, 6245. [Google Scholar] [CrossRef]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.C.W.; Tchou, J.; et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat. Commun. 2019, 10, 130. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, H.; Ma, Q.; Huang, R.; Lu, J.; Liang, X.; Liu, X.; Zhang, Z.; Yu, L.; Pang, J.; et al. YAP1-mediated pancreatic stellate cell activation inhibits pancreatic cancer cell proliferation. Cancer Lett. 2019, 462, 51–60. [Google Scholar] [CrossRef]
- Wohlfahrt, T.; Rauber, S.; Uebe, S.; Luber, M.; Soare, A.; Ekici, A.; Weber, S.; Matei, A.-E.; Chen, C.-W.; Maier, C.; et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 2019, 566, 344–349. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Jana, S.; Aujla, P.; Hu, M.; Kilic, T.; Zhabyeyev, P.; McCulloch, C.A.; Oudit, G.Y.; Kassiri, Z. Gelsolin is an important mediator of Angiotensin II-induced activation of cardiac fibroblasts and fibrosis. FASEB J. 2021, 35, e21932. [Google Scholar] [CrossRef]
- Zaghdoudi, S.; Decaup, E.; Belhabib, I.; Samain, R.; Cassant-Sourdy, S.; Rochotte, J.; Brunel, A.; Schlaepfer, D.; Cros, J.; Neuzillet, C.; et al. FAK activity in cancer-associated fibroblasts is a prognostic marker and a druggable key metastatic player in pancreatic cancer. EMBO Mol. Med. 2020, 12, e12010. [Google Scholar] [CrossRef]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Procopio, M.-G.; Laszlo, C.; Al Labban, D.; Kim, D.E.; Bordignon, P.; Jo, S.-H.; Goruppi, S.; Menietti, E.; Ostano, P.; Ala, U.; et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat. Cell Biol. 2015, 17, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Thoen, L.F.R.; Guimarães, E.L.M.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.I.; Kim, D.H.; Sung, K.W.; Shim, S.M.; Cha-Molstad, H.; Soung, N.K.; Lee, K.H.; Hwang, J.; Lee, H.G.; Kwon, Y.T.; et al. p62-Induced Cancer-Associated Fibroblast Activation via the Nrf2-ATF6 Pathway Promotes Lung Tumorigenesis. Cancers 2021, 13, 864. [Google Scholar] [CrossRef]
- Linares, J.F.; Cid-Diaz, T.; Duran, A.; Osrodek, M.; Martinez-Ordoñez, A.; Reina-Campos, M.; Kuo, H.-H.; Elemento, O.; Martin, M.L.; Cordes, T.; et al. The lactate-NAD+ axis activates cancer-associated fibroblasts by downregulating p62. Cell Rep. 2022, 39, 110792. [Google Scholar] [CrossRef]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.-L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer–Associated Fibroblasts. Cancer Discov. 2022, 12, 484–501. [Google Scholar] [CrossRef]
- Iwamoto, C.; Ohuchida, K.; Shinkawa, T.; Okuda, S.; Otsubo, Y.; Okumura, T.; Sagara, A.; Koikawa, K.; Ando, Y.; Shindo, K.; et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 2021, 512, 15–27. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Oda, T.; Mori, N.; Kida, Y.S. Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts in vitro. FEBS Open Bio 2020, 10, 2268–2281. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Rønnov-Jessen, L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 2003, 162, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-tissue organization of the fibroblast lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Grippo, P.J.; Munshi, H.G.; Rasheed, Z.A.; Matsui, W.; Maitra, A. (Eds.) Pancreatic Cancer and Tumor Microenvironment: Pathology of Pancreatic Stroma in PDAC; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology 2010, 139, 1041–1051.e1-8. [Google Scholar] [CrossRef]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, e129212. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef]
- Hupfer, A.; Brichkina, A.; Koeniger, A.; Keber, C.; Denkert, C.; Pfefferle, P.; Helmprobst, F.; Pagenstecher, A.; Visekruna, A.; Lauth, M. Matrix stiffness drives stromal autophagy and promotes formation of a protumorigenic niche. Proc. Natl. Acad. Sci. USA 2021, 118, e2105367118. [Google Scholar] [CrossRef]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; Del Monte-Nieto, G.; Conway, J.R.W.; et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017, 9, eaai8504. [Google Scholar] [CrossRef]
- Murray, E.R.; Menezes, S.; Henry, J.C.; Williams, J.L.; Alba-Castellón, L.; Baskaran, P.; Quétier, I.; Desai, A.; Marshall, J.J.T.; Rosewell, I.; et al. Disruption of pancreatic stellate cell myofibroblast phenotype promotes pancreatic tumor invasion. Cell Rep. 2022, 38, 110227. [Google Scholar] [CrossRef]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Lovisa, S.; Ambrose, C.G.; McAndrews, K.M.; Sugimoto, H.; Kalluri, R. Type-I collagen produced by distinct fibroblast lineages reveals specific function during embryogenesis and Osteogenesis Imperfecta. Nat. Commun. 2021, 12, 7199. [Google Scholar] [CrossRef]
- Torphy, R.J.; Wang, Z.; True-Yasaki, A.; Volmar, K.E.; Rashid, N.; Yeh, B.; Anderson, J.M.; Johansen, J.S.; Hollingsworth, M.A.; Yeh, J.J.; et al. Stromal Content Is Correlated with Tissue Site, Contrast Retention, and Survival in Pancreatic Adenocarcinoma. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Jiang, H.; Torphy, R.J.; Steiger, K.; Hongo, H.; Ritchie, A.J.; Kriegsmann, M.; Horst, D.; Umetsu, S.E.; Joseph, N.M.; McGregor, K.; et al. Pancreatic ductal adenocarcinoma progression is restrained by stromal matrix. J. Clin. Investig. 2020, 130, 4704–4709. [Google Scholar] [CrossRef]
- Bever, K.M.; Sugar, E.A.; Bigelow, E.; Sharma, R.; Laheru, D.; Wolfgang, C.L.; Jaffee, E.M.; Anders, R.A.; de Jesus-Acosta, A.; Zheng, L. The prognostic value of stroma in pancreatic cancer in patients receiving adjuvant therapy. HPB 2015, 17, 292–298. [Google Scholar] [CrossRef]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser-Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef]
- Leitinger, B. Transmembrane collagen receptors. Annu. Rev. Cell Dev. Biol. 2011, 27, 265–290. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Hamberger, F.; Ravichandra, A.; Miller, M.; Nair, A.; Affo, S.; Filliol, A.; Chin, L.; Savage, T.M.; Yin, D.; et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Investig. 2021, 131, e146987. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, S.; Tavormina, J.; Tampe, D.; Zeisberg, M.; Wang, H.; Mahadevan, K.K.; Wu, C.-J.; Sugimoto, H.; Chang, C.-C.; et al. Oncogenic collagen I homotrimers from cancer cells bind to α3β1 integrin and impact tumor microbiome and immunity to promote pancreatic cancer. Cancer Cell 2022, 40, 818–834.e9. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Huang, Y.; Clauser, K.R.; Rickelt, S.; Lau, A.N.; Carr, S.A.; Vander Heiden, M.G.; Hynes, R.O. Suppression of pancreatic ductal adenocarcinoma growth and metastasis by fibrillar collagens produced selectively by tumor cells. Nat. Commun. 2021, 12, 2328. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Kohler, I.; Timme, S.; Kiefer, S.; Werner, M.; Schilling, O.; Vashist, Y.; Makowiec, F.; Brabletz, T.; Hopt, U.T.; et al. Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery 2014, 156, 97–108. [Google Scholar] [CrossRef]
- Galván, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, X.; Huang, L.; Wang, W.; Jiang, G.; Dean, K.C.; Clem, B.; Telang, S.; Jenson, A.B.; Cuatrecasas, M.; et al. Different thresholds of ZEB1 are required for Ras-mediated tumour initiation and metastasis. Nat. Commun. 2014, 5, 5660. [Google Scholar] [CrossRef]
- Sangrador, I.; Molero, X.; Campbell, F.; Franch-Expósito, S.; Rovira-Rigau, M.; Samper, E.; Domínguez-Fraile, M.; Fillat, C.; Castells, A.; Vaquero, E.C. Zeb1 in Stromal Myofibroblasts Promotes Kras-Driven Development of Pancreatic Cancer. Cancer Res. 2018, 78, 2624–2637. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Tsai, C.-H.; Lai, Y.-L.; Yu, C.-C.; Chi, W.-Y.; Li, J.J.; Chang, W.-W. Arecoline-induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated by ZEB1. J. Cell. Mol. Med. 2014, 18, 698–708. [Google Scholar] [CrossRef]
- Satoh, K.; Hata, M.; Yokota, H. A novel member of the leucine-rich repeat superfamily induced in rat astrocytes by beta-amyloid. Biochem. Biophys. Res. Commun. 2002, 290, 756–762. [Google Scholar] [CrossRef]
- Purcell, J.W.; Tanlimco, S.G.; Hickson, J.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; McGonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody-Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, H.; Fan, S.; Bi, Y.; Hao, M.; Shang, J. Cancer-associated fibroblast-derived LRRC15 promotes the migration and invasion of triple-negative breast cancer cells via Wnt/β-catenin signalling pathway regulation. Mol. Med. Rep. 2022, 25, 2, 1–8. [Google Scholar] [CrossRef]
- Krishnamurty, A.T.; Shyer, J.A.; Thai, M.; Gandham, V.; Buechler, M.B.; Yang, Y.A.; Pradhan, R.N.; Wang, A.W.; Sanchez, P.L.; Qu, Y.; et al. LRRC15+ myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 2022, 611, 148–154. [Google Scholar] [CrossRef]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.; Scales, M.K.; El-Tawil, Y.S.; Shaikh, A.Z.; Wen, H.-J.; Bednar, F.; Allen, B.L.; et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 581–599. [Google Scholar] [CrossRef]
- Galperin, I.; Dempwolff, L.; Diederich, W.E.; Lauth, M. Inhibiting Hedgehog: An Update on Pharmacological Compounds and Targeting Strategies. J. Med. Chem. 2019, 62, 8392–8411. [Google Scholar] [CrossRef]
- Lauth, M. RAS and Hedgehog--partners in crime. Front. Biosci. 2011, 17, 2259–2270. [Google Scholar] [CrossRef]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgard, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef]
- Fendrich, V.; Oh, E.; Bang, S.; Karikari, C.; Ottenhof, N.; Bisht, S.; Lauth, M.; Brossart, P.; Katsanis, N.; Maitra, A.; et al. Ectopic overexpression of Sonic Hedgehog (Shh) induces stromal expansion and metaplasia in the adult murine pancreas. Neoplasia 2011, 13, 923–930. [Google Scholar] [CrossRef]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef]
- Javelaud, D.; Alexaki, V.I.; Dennler, S.; Mohammad, K.S.; Guise, T.A.; Mauviel, A. TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011, 71, 5606–5610. [Google Scholar] [CrossRef]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Boyd, L.N.C.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and plasticity of cancer-associated fibroblasts in the pancreatic tumor microenvironment. Semin. Cancer Biol. 2022, 82, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Castellanos, J.A.; VanSaun, M.N.; Dai, X.; Ambrose, M.; Guo, Q.; Xiong, Y.; Merchant, N.B. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016, 7, 65982–65992. [Google Scholar] [CrossRef]
- Kim, H.W.; Lee, J.-C.; Paik, K.-H.; Kang, J.; Kim, J.; Hwang, J.-H. Serum interleukin-6 is associated with pancreatic ductal adenocarcinoma progression pattern. Medicine 2017, 96, e5926. [Google Scholar] [CrossRef]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGFβR2-mutant tumors exposes vulnerabilities to stromal TGFβ blockade in pancreatic cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Kesh, K.; Garrido, V.T.; Dosch, A.; Durden, B.; Gupta, V.K.; Sharma, N.S.; Lyle, M.; Nagathihalli, N.; Merchant, N.; Saluja, A.; et al. Stroma secreted IL6 selects for "stem-like" population and alters pancreatic tumor microenvironment by reprogramming metabolic pathways. Cell Death Dis. 2020, 11, 967. [Google Scholar] [CrossRef]
- Lee, J.W.; Stone, M.L.; Porrett, P.M.; Thomas, S.K.; Komar, C.A.; Li, J.H.; Delman, D.; Graham, K.; Gladney, W.L.; Hua, X.; et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019, 567, 249–252. [Google Scholar] [CrossRef]
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.W.; Pan, X.; Beatty, G.L. IL6 Receptor Blockade Enhances Chemotherapy Efficacy in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; Correa de Sampaio, P.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022, 12, 1580–1597. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Wang, M.-T.; Fer, N.; Galeas, J.; Collisson, E.A.; Kim, S.E.; Sharib, J.; McCormick, F. Blockade of leukemia inhibitory factor as a therapeutic approach to KRAS driven pancreatic cancer. Nat. Commun. 2019, 10, 3055. [Google Scholar] [CrossRef]
- Picard, F.S.R.; Lutz, V.; Brichkina, A.; Neuhaus, F.; Ruckenbrod, T.; Hupfer, A.; Raifer, H.; Klein, M.; Bopp, T.; Pfefferle, P.I.; et al. IL-17A-producing CD8+ T cells promote PDAC via induction of inflammatory cancer-associated fibroblasts. Gut 2023, 1–13. [Google Scholar] [CrossRef]
- Bressy, C.; Lac, S.; Nigri, J.; Leca, J.; Roques, J.; Lavaut, M.-N.; Secq, V.; Guillaumond, F.; Bui, T.-T.; Pietrasz, D.; et al. LIF Drives Neural Remodeling in Pancreatic Cancer and Offers a New Candidate Biomarker. Cancer Res. 2018, 78, 909–921. [Google Scholar] [CrossRef]
- Djurec, M.; Graña, O.; Lee, A.; Troulé, K.; Espinet, E.; Cabras, L.; Navas, C.; Blasco, M.T.; Martín-Díaz, L.; Burdiel, M.; et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E1147–E1156. [Google Scholar] [CrossRef]
- Yao, L.; Rathnakar, B.H.; Kwon, H.R.; Sakashita, H.; Kim, J.H.; Rackley, A.; Tomasek, J.J.; Berry, W.L.; Olson, L.E. Temporal control of PDGFRα regulates the fibroblast-to-myofibroblast transition in wound healing. Cell Rep. 2022, 40, 111192. [Google Scholar] [CrossRef]
- Cui Zhou, D.; Jayasinghe, R.G.; Chen, S.; Herndon, J.M.; Iglesia, M.D.; Navale, P.; Wendl, M.C.; Caravan, W.; Sato, K.; Storrs, E.; et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 2022, 54, 1390–1405. [Google Scholar] [CrossRef]
- Lefler, J.E.; MarElia-Bennett, C.B.; Thies, K.A.; Hildreth, B.E.; Sharma, S.M.; Pitarresi, J.R.; Han, L.; Everett, C.; Koivisto, C.; Cuitino, M.C.; et al. STAT3 in tumor fibroblasts promotes an immunosuppressive microenvironment in pancreatic cancer. Life Sci. Alliance 2022, 5, e202201460. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.-H. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef]
- Malle, E.; Sodin-Semrl, S.; Kovacevic, A. Serum amyloid A: An acute-phase protein involved in tumour pathogenesis. Cell. Mol. Life Sci. 2009, 66, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef] [PubMed]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef]
- Kawase, T.; Yasui, Y.; Nishina, S.; Hara, Y.; Yanatori, I.; Tomiyama, Y.; Nakashima, Y.; Yoshida, K.; Kishi, F.; Nakamura, M.; et al. Fibroblast activation protein-α-expressing fibroblasts promote the progression of pancreatic ductal adenocarcinoma. BMC Gastroenterol. 2015, 15, 109. [Google Scholar] [CrossRef]
- Santos, A.M.; Jung, J.; Aziz, N.; Kissil, J.L.; Puré, E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Investig. 2009, 119, 3613–3625. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef]
- Mazur, A.; Holthoff, E.; Vadali, S.; Kelly, T.; Post, S.R. Cleavage of Type I Collagen by Fibroblast Activation Protein-α Enhances Class A Scavenger Receptor Mediated Macrophage Adhesion. PLoS ONE 2016, 11, e0150287. [Google Scholar] [CrossRef]
- Lee, H.-O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 245. [Google Scholar] [CrossRef]
- Lo, A.; Li, C.-P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Puré, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017, 2, e92232. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Typing of pancreatic cancer-associated fibroblasts identifies different subpopulations. World J. Gastroenterol. 2018, 24, 4663–4678. [Google Scholar] [CrossRef]
- Shindo, K.; Aishima, S.; Ohuchida, K.; Fujiwara, K.; Fujino, M.; Mizuuchi, Y.; Hattori, M.; Mizumoto, K.; Tanaka, M.; Oda, Y. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol. Cancer 2013, 12, 168. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genom. 2010, 11, 22. [Google Scholar] [CrossRef]
- Yu, S.; Lu, Y.; Su, A.; Chen, J.; Li, J.; Zhou, B.; Liu, X.; Xia, Q.; Li, Y.; Li, J.; et al. A CD10-OGP Membrane Peptolytic Signaling Axis in Fibroblasts Regulates Lipid Metabolism of Cancer Stem Cells via SCD1. Adv. Sci. 2021, 8, e2101848. [Google Scholar] [CrossRef]
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013, 4, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhao, Z.; Zhang, J.; Wang, W.; Zhu, Y. High expressions of CD10, FAP and GPR77 in CAFs are associated with chemoresistance and worse prognosis in gastric cancer. Front. Oncol. 2022, 12, 984817. [Google Scholar] [CrossRef] [PubMed]
- Ammirante, M.; Shalapour, S.; Kang, Y.; Jamieson, C.A.M.; Karin, M. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl. Acad. Sci. USA 2014, 111, 14776–14781. [Google Scholar] [CrossRef] [PubMed]
- Ziani, L.; Buart, S.; Chouaib, S.; Thiery, J. Hypoxia increases melanoma-associated fibroblasts immunosuppressive potential and inhibitory effect on T cell-mediated cytotoxicity. OncoImmunology 2021, 10, 1950953. [Google Scholar] [CrossRef] [PubMed]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef]
- Du, Y.; Long, Q.; Zhang, L.; Shi, Y.; Liu, X.; Li, X.; Guan, B.; Tian, Y.; Wang, X.; Li, L.; et al. Curcumin inhibits cancer-associated fibroblast-driven prostate cancer invasion through MAOA/mTOR/HIF-1α signaling. Int. J. Oncol. 2015, 47, 2064–2072. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef]
- Mello, A.M.; Ngodup, T.; Lee, Y.; Donahue, K.L.; Li, J.; Rao, A.; Carpenter, E.S.; Crawford, H.C.; Di Pasca Magliano, M.; Lee, K.E. Hypoxia promotes an inflammatory phenotype of fibroblasts in pancreatic cancer. Oncogenesis 2022, 11, 56. [Google Scholar] [CrossRef]
- Schwörer, S.; Ros, M.; Tsanov, K.M.; Cimino, F.V.; Lowe, S.W.; Carmona-Fontaine, C.; Thompson, C.B. Hypoxia potentiates the inflammatory fibroblast phenotype promoted by pancreatic cancer cell-derived cytokines. Cancer Res. 2023, CAN-22-2316, Online ahead of print. [Google Scholar] [CrossRef]
- Garcia Garcia, C.J.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology 2022, 162, 2018–2031. [Google Scholar] [CrossRef]
- Kim, J.-W.; Evans, C.; Weidemann, A.; Takeda, N.; Lee, Y.S.; Stockmann, C.; Branco-Price, C.; Brandberg, F.; Leone, G.; Ostrowski, M.C.; et al. Loss of fibroblast HIF-1α accelerates tumorigenesis. Cancer Res. 2012, 72, 3187–3195. [Google Scholar] [CrossRef]
- Madsen, C.D.; Pedersen, J.T.; Venning, F.A.; Singh, L.B.; Moeendarbary, E.; Charras, G.; Cox, T.R.; Sahai, E.; Erler, J.T. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 2015, 16, 1394–1408. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673.e7. [Google Scholar] [CrossRef]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244.e20. [Google Scholar] [CrossRef]
- Paauwe, M.; Schoonderwoerd, M.J.A.; Helderman, R.F.C.P.; Harryvan, T.J.; Groenewoud, A.; van Pelt, G.W.; Bor, R.; Hemmer, D.M.; Versteeg, H.H.; Snaar-Jagalska, B.E.; et al. Endoglin Expression on Cancer-Associated Fibroblasts Regulates Invasion and Stimulates Colorectal Cancer Metastasis. Clin. Cancer Res. 2018, 24, 6331–6344. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Zhong, W.; Culp, S.; Shaffer, K.; Hoover, J.; Wickramasinghe, D.; Ruefli-Brasse, A. Endoglin requirement for BMP9 signaling in endothelial cells reveals new mechanism of action for selective anti-endoglin antibodies. PLoS ONE 2012, 7, e50920. [Google Scholar] [CrossRef]
- Rosen, L.S.; Hurwitz, H.I.; Wong, M.K.; Goldman, J.; Mendelson, D.S.; Figg, W.D.; Spencer, S.; Adams, B.J.; Alvarez, D.; Seon, B.K.; et al. A phase I first-in-human study of TRC105 (Anti-Endoglin Antibody) in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 4820–4829. [Google Scholar] [CrossRef]
- Schoonderwoerd, M.J.A.; Hakuno, S.K.; Sassen, M.; Kuhlemaijer, E.B.; Paauwe, M.; Slingerland, M.; Fransen, M.F.; Hawinkels, L.J.A.C. Targeting Endoglin Expressing Cells in the Tumor Microenvironment Does Not Inhibit Tumor Growth in a Pancreatic Cancer Mouse Model. Onco. Targets. Ther. 2021, 14, 5205–5220. [Google Scholar] [CrossRef]
- Maeda, K.; Enomoto, A.; Hara, A.; Asai, N.; Kobayashi, T.; Horinouchi, A.; Maruyama, S.; Ishikawa, Y.; Nishiyama, T.; Kiyoi, H.; et al. Identification of Meflin as a Potential Marker for Mesenchymal Stromal Cells. Sci. Rep. 2016, 6, 22288. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef]
- Miyai, Y.; Sugiyama, D.; Hase, T.; Asai, N.; Taki, T.; Nishida, K.; Fukui, T.; Chen-Yoshikawa, T.F.; Kobayashi, H.; Mii, S.; et al. Meflin-positive cancer-associated fibroblasts enhance tumor response to immune checkpoint blockade. Life Sci. Alliance 2022, 5, e202101230. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Mizutani, Y.; Esaki, N.; Ponik, S.M.; Burkel, B.M.; Weng, L.; Kuwata, K.; Masamune, A.; Ishihara, S.; Haga, H.; et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene 2022, 41, 2764. [Google Scholar] [CrossRef] [PubMed]
- Trim, N.; Morgan, S.; Evans, M.; Issa, R.; Fine, D.; Afford, S.; Wilkins, B.; Iredale, J. Hepatic stellate cells express the low affinity nerve growth factor receptor p75 and undergo apoptosis in response to nerve growth factor stimulation. Am. J. Pathol. 2000, 156, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Ohuchida, K.; Mizumoto, K.; Shindo, K.; Eguchi, D.; Kozono, S.; Ikenaga, N.; Ohtsuka, T.; Takahata, S.; Aishima, S.; et al. CD271+ subpopulation of pancreatic stellate cells correlates with prognosis of pancreatic cancer and is regulated by interaction with cancer cells. PLoS ONE 2012, 7, e52682. [Google Scholar] [CrossRef]
- Kasashima, H.; Yashiro, M.; Nakamae, H.; Masuda, G.; Kinoshita, H.; Morisaki, T.; Fukuoka, T.; Hasegawa, T.; Sakurai, K.; Toyokawa, T.; et al. Bone marrow-derived stromal cells are associated with gastric cancer progression. Br. J. Cancer 2015, 113, 443–452. [Google Scholar] [CrossRef]
- Minami, T.; Aoyagi, K.; Kawahara, A.; Murakami, N.; Isobe, T.; Tanaka, Y.; Kaku, H.; Fujita, F.; Akagi, Y. Evaluation of the expression of bone marrow-derived mesenchymal stem cells and cancer-associated fibroblasts in the stroma of gastric cancer tissue. Ann. Gastroenterol. Surg. 2020, 4, 464–474. [Google Scholar] [CrossRef]
- Bose, S.; Zhang, C.; Le, A. Glucose Metabolism in Cancer: The Warburg Effect and Beyond. In The Heterogeneity of Cancer Metabolism; Springer: Cham, Switzerland, 2021; pp. 3–15. [Google Scholar]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.-L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Yang, Y.; Gomez, M.; Marsh, T.; Poillet-Perez, L.; Sawant, A.; Chen, L.; Park, N.R.; Jackson, S.R.; Hu, Z.; Alon, N.; et al. Autophagy in PDGFRα+ mesenchymal cells is essential for intestinal stem cell survival. Proc. Natl. Acad. Sci. USA 2022, 119, e2202016119. [Google Scholar] [CrossRef]
- Goruppi, S.; Jo, S.-H.; Laszlo, C.; Clocchiatti, A.; Neel, V.; Dotto, G.P. Autophagy Controls CSL/RBPJκ Stability through a p62/SQSTM1-Dependent Mechanism. Cell Rep. 2018, 24, 3108–3114.e4. [Google Scholar] [CrossRef]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef]
- Linares, J.F.; Cordes, T.; Duran, A.; Reina-Campos, M.; Valencia, T.; Ahn, C.S.; Castilla, E.A.; Moscat, J.; Metallo, C.M.; Diaz-Meco, M.T. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab. 2017, 26, 817–829.e6. [Google Scholar] [CrossRef]
- Mestre-Farrera, A.; Bruch-Oms, M.; Peña, R.; Rodríguez-Morató, J.; Alba-Castellón, L.; Comerma, L.; Quintela-Fandino, M.; Duñach, M.; Baulida, J.; Pozo, Ó.J.; et al. Glutamine-Directed Migration of Cancer-Activated Fibroblasts Facilitates Epithelial Tumor Invasion. Cancer Res. 2021, 81, 438–451. [Google Scholar] [CrossRef]
- Neesse, A.; Bauer, C.A.; Ohlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Harris, W.P.; Beck, J.T.; Berdov, B.A.; Wagner, S.A.; Pshevlotsky, E.M.; Tjulandin, S.A.; Gladkov, O.A.; Holcombe, R.F.; Korn, R.; et al. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Cyphert, J.M.; Trempus, C.S.; Garantziotis, S. Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int. J. Cell Biol. 2015, 2015, 563818. [Google Scholar] [CrossRef]
- Takasugi, M.; Firsanov, D.; Tombline, G.; Ning, H.; Ablaeva, J.; Seluanov, A.; Gorbunova, V. Naked mole-rat very-high-molecular-mass hyaluronan exhibits superior cytoprotective properties. Nat. Commun. 2020, 11, 2376. [Google Scholar] [CrossRef]
- Tian, X.; Azpurua, J.; Hine, C.; Vaidya, A.; Myakishev-Rempel, M.; Ablaeva, J.; Mao, Z.; Nevo, E.; Gorbunova, V.; Seluanov, A. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 2013, 499, 346–349. [Google Scholar] [CrossRef]
- Yates, T.J.; Lopez, L.E.; Lokeshwar, S.D.; Ortiz, N.; Kallifatidis, G.; Jordan, A.; Hoye, K.; Altman, N.; Lokeshwar, V.B. Dietary supplement 4-methylumbelliferone: An effective chemopreventive and therapeutic agent for prostate cancer. J. Natl. Cancer Inst. 2015, 107, djv085. [Google Scholar] [CrossRef]
- Brichkina, A.; Bertero, T.; Loh, H.M.; Nguyen, N.T.M.; Emelyanov, A.; Rigade, S.; Ilie, M.; Hofman, P.; Gaggioli, C.; Bulavin, D.V. p38MAPK builds a hyaluronan cancer niche to drive lung tumorigenesis. Genes Dev. 2016, 30, 2623–2636. [Google Scholar] [CrossRef]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e1-14. [Google Scholar] [CrossRef]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; de Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; del Río Hernández, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Meng, K.P.; Majedi, F.S.; Thauland, T.J.; Butte, M.J. Mechanosensing through YAP controls T cell activation and metabolism. J. Exp. Med. 2020, 217, e20200053. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Aung, K.L.; McWhirter, E.; Welch, S.; Wang, L.; Lovell, S.; Stayner, L.-A.; Ali, S.; Malpage, A.; Makepeace, B.; Ramachandran, M.; et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2022, 13, 3216–3226. [Google Scholar] [CrossRef]
- Mouratidis, P.X.E.; Costa, M.; Rivens, I.; Repasky, E.E.; Ter Haar, G. Pulsed focused ultrasound can improve the anti-cancer effects of immune checkpoint inhibitors in murine pancreatic cancer. J. R. Soc. Interface 2021, 18, 20210266. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.A.; Rivera, L.B.; Carbon, J.G.; Toombs, J.E.; Chang, C.-L.; Bradshaw, A.D.; Brekken, R.A. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFβ activation. PLoS ONE 2012, 7, e31384. [Google Scholar] [CrossRef] [PubMed]
- Kasi, A.; Allen, J.; Mehta, K.; Dandawate, P.; Saha, S.; Bossmann, S.; Anant, S.; Sun, W. Association of losartan with outcomes in metastatic pancreatic cancer patients treated with chemotherapy. J. Clin. Transl. Res. 2021, 7, 257–262. [Google Scholar] [PubMed]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy with FOLFIRINOX in Combination with Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Cortes, E.; Lachowski, D.; Robinson, B.; Sarper, M.; Teppo, J.S.; Thorpe, S.D.; Lieberthal, T.J.; Iwamoto, K.; Lee, D.A.; Okada-Hatakeyama, M.; et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 2019, 20, e46557. [Google Scholar] [CrossRef]
- Cortes, E.; Sarper, M.; Robinson, B.; Lachowski, D.; Chronopoulos, A.; Thorpe, S.D.; Lee, D.A.; Del Río Hernández, A.E. GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. 2019, 20, e46556. [Google Scholar] [CrossRef]
- Brennan, E.P.; Nolan, K.A.; Börgeson, E.; Gough, O.S.; McEvoy, C.M.; Docherty, N.G.; Higgins, D.F.; Murphy, M.; Sadlier, D.M.; Ali-Shah, S.T.; et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFβR1. J. Am. Soc. Nephrol. 2013, 24, 627–637. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Wu, S.-H.; Luo, Y.-Y.; Li, B.-J.; Li, S.-J.; Lu, H.-Y.; Jin, R.; Sun, Z.-Y. Lipoxin A4 Attenuates Bronchopulmonary Dysplasia via Upregulation of Let-7c and Downregulation of TGF-β1 Signaling Pathway. Inflammation 2017, 40, 2094–2108. [Google Scholar] [CrossRef]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature's way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar] [CrossRef]
- Schnittert, J.; Heinrich, M.A.; Kuninty, P.R.; Storm, G.; Prakash, J. Reprogramming tumor stroma using an endogenous lipid lipoxin A4 to treat pancreatic cancer. Cancer Lett. 2018, 420, 247–258. [Google Scholar] [CrossRef]
- Zong, L.; Chen, K.; Jiang, Z.; Chen, X.; Sun, L.; Ma, J.; Zhou, C.; Xu, Q.; Duan, W.; Han, L.; et al. Lipoxin A4 reverses mesenchymal phenotypes to attenuate invasion and metastasis via the inhibition of autocrine TGF-β1 signaling in pancreatic cancer. J. Exp. Clin. Cancer Res. 2017, 36, 181. [Google Scholar] [CrossRef]
- Yuan, J.; Lin, F.; Chen, L.; Chen, W.; Pan, X.; Bai, Y.; Cai, Y.; Lu, H. Lipoxin A4 regulates M1/M2 macrophage polarization via FPR2-IRF pathway. Inflammopharmacology 2022, 30, 487–498. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brichkina, A.; Polo, P.; Sharma, S.D.; Visestamkul, N.; Lauth, M. A Quick Guide to CAF Subtypes in Pancreatic Cancer. Cancers 2023, 15, 2614. https://doi.org/10.3390/cancers15092614
Brichkina A, Polo P, Sharma SD, Visestamkul N, Lauth M. A Quick Guide to CAF Subtypes in Pancreatic Cancer. Cancers. 2023; 15(9):2614. https://doi.org/10.3390/cancers15092614
Chicago/Turabian StyleBrichkina, Anna, Pierfrancesco Polo, Shrey Dharamvir Sharma, Nico Visestamkul, and Matthias Lauth. 2023. "A Quick Guide to CAF Subtypes in Pancreatic Cancer" Cancers 15, no. 9: 2614. https://doi.org/10.3390/cancers15092614
APA StyleBrichkina, A., Polo, P., Sharma, S. D., Visestamkul, N., & Lauth, M. (2023). A Quick Guide to CAF Subtypes in Pancreatic Cancer. Cancers, 15(9), 2614. https://doi.org/10.3390/cancers15092614