

The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma

Abstract
Simple Summary
Abstract
1. Introduction
2. GSC and Hypoxia-Related Signatures
3. GSC and Hypoxia-Related Genes
4. GSC and Hypoxia-Related Pathways
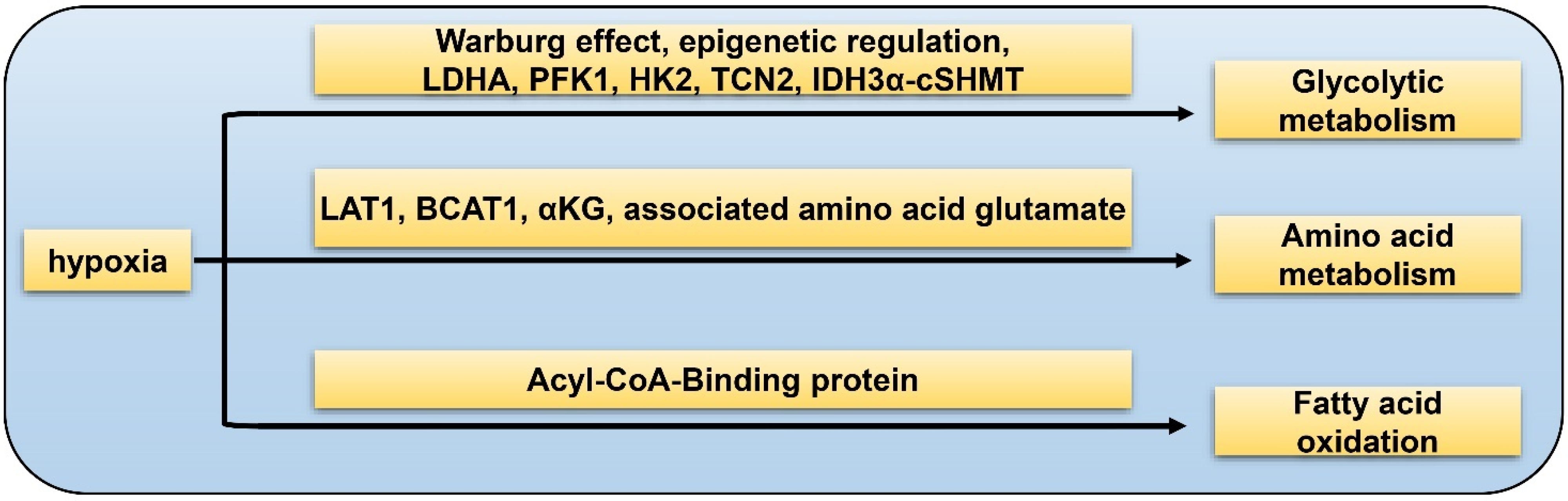
5. GSC and Hypoxia-Related Metabolism
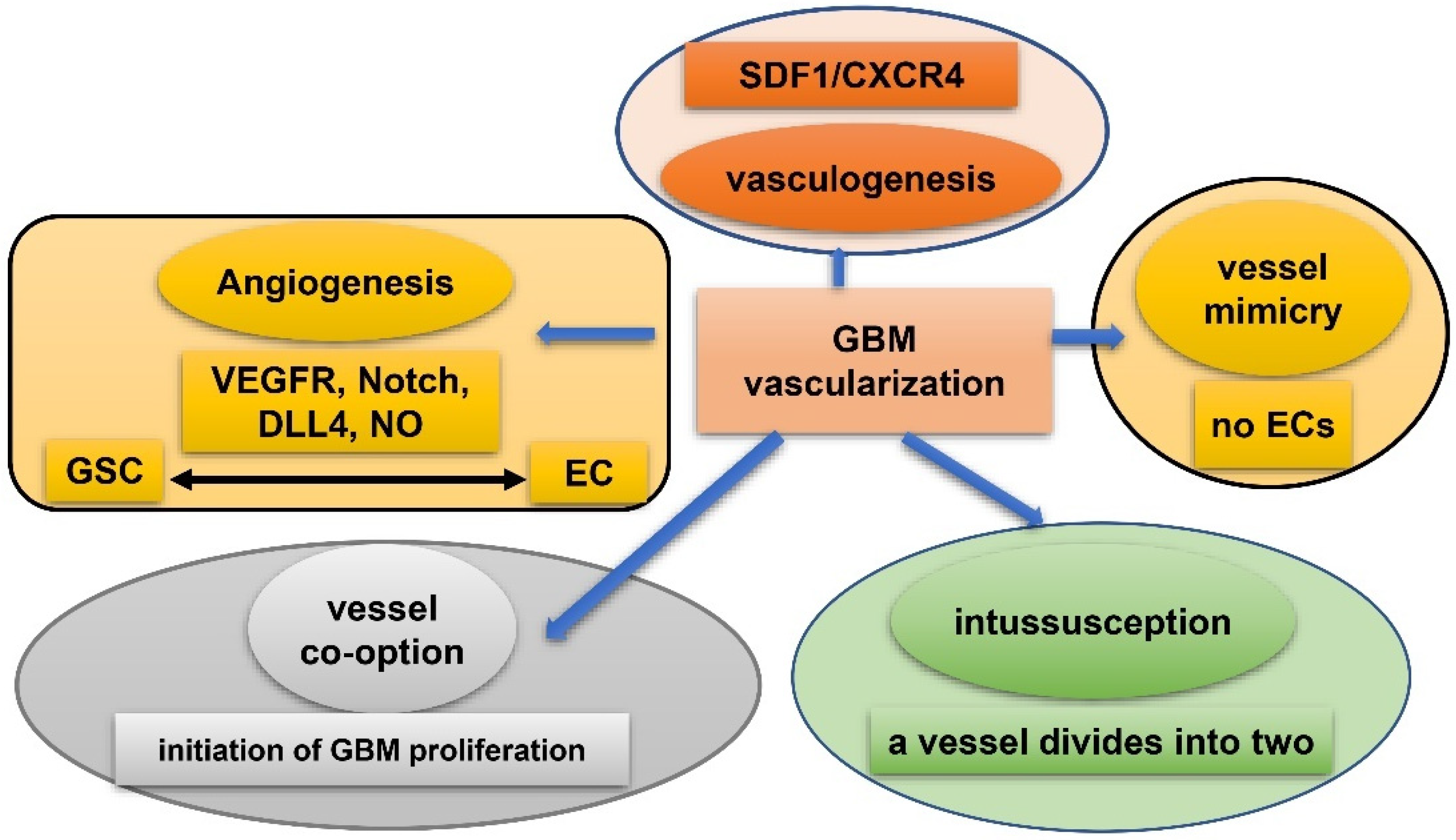
6. GSC and Hypoxia-Related Vasculature
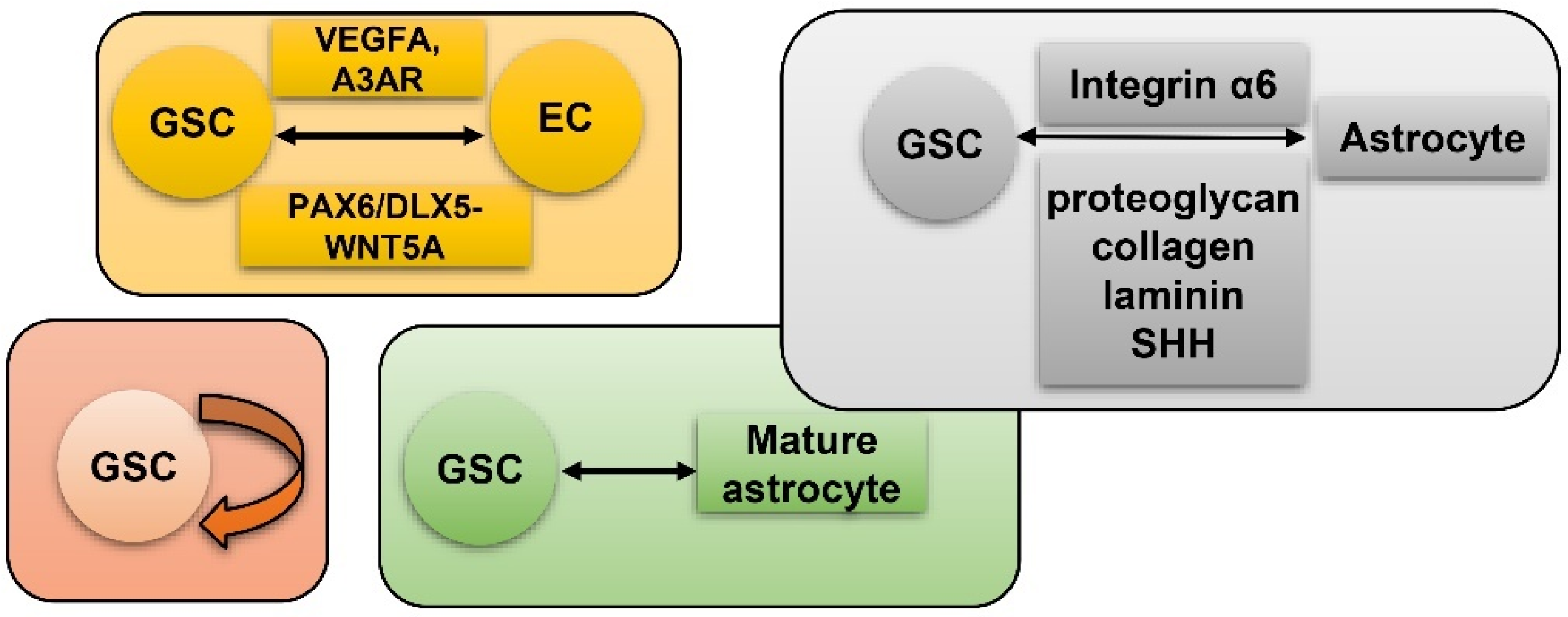
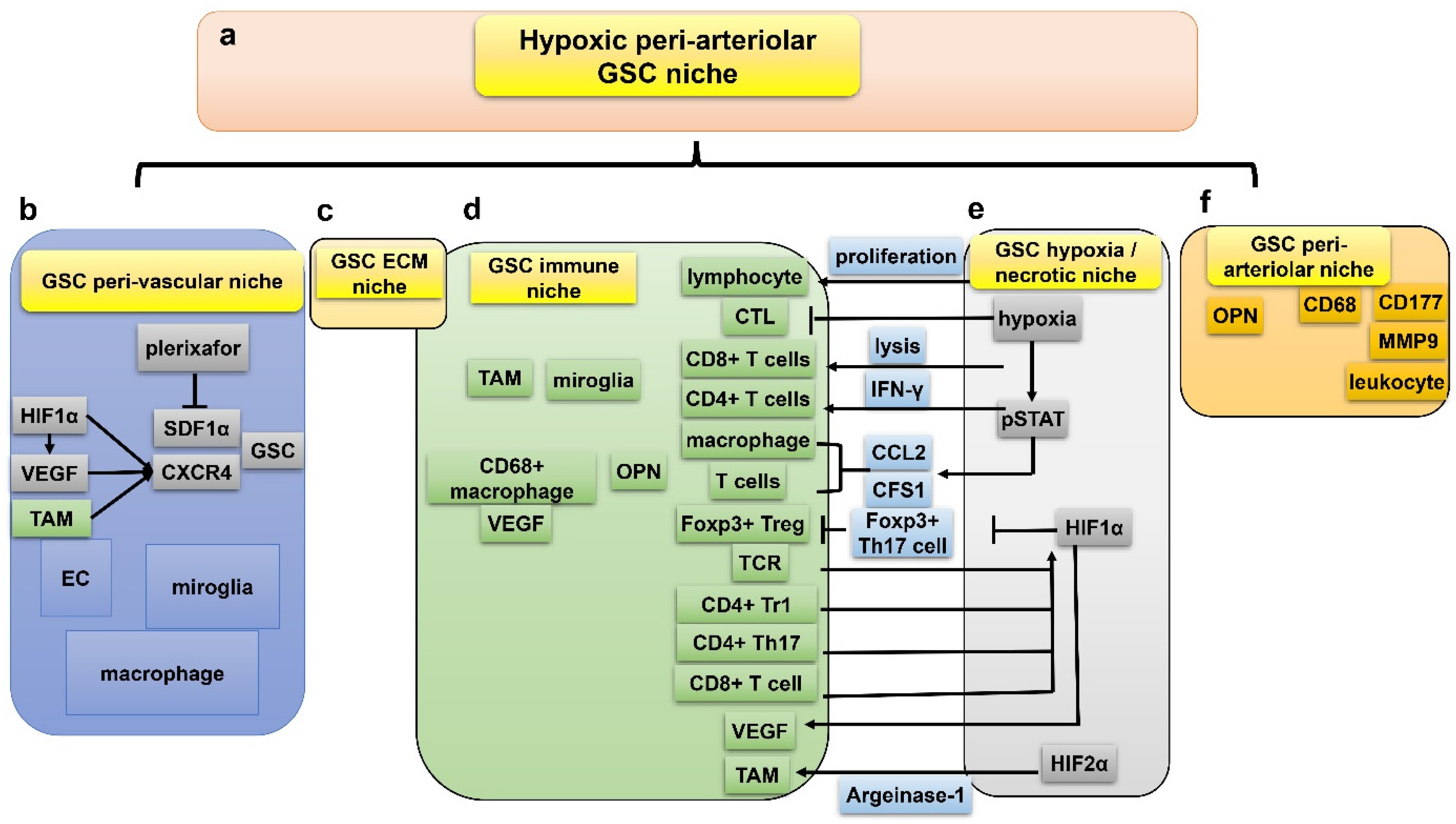
7. GSC and Hypoxia-Related Niches
7.1. Peri-Vascular Niche of GSC
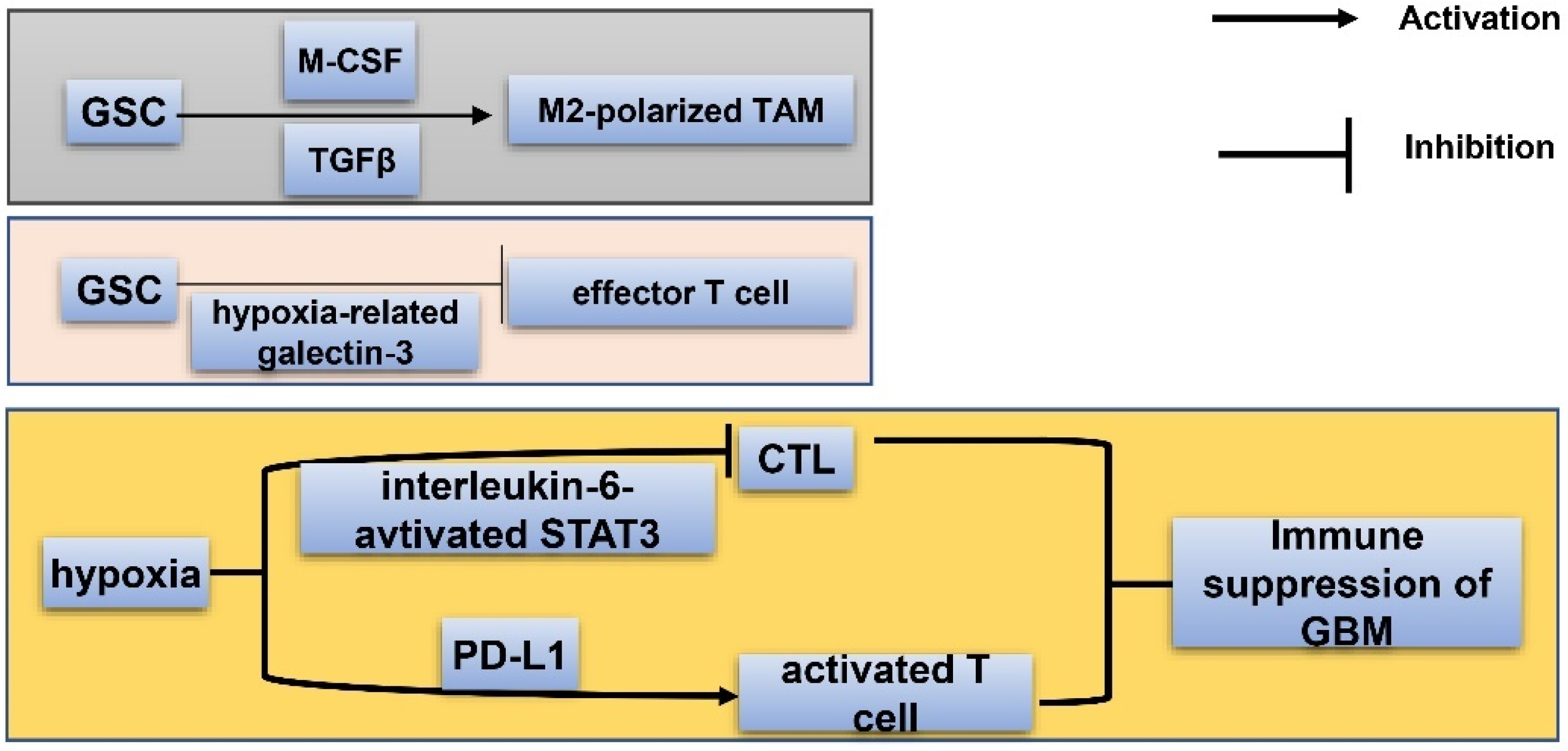
7.2. Immune Niche of GSC
7.3. Hypoxia/Necrotic Niche of GSC
7.4. ECM Niche of GSC
7.5. Peri-Arteriolar Niche of GSC
7.6. Interactions between the Five GSC Niches
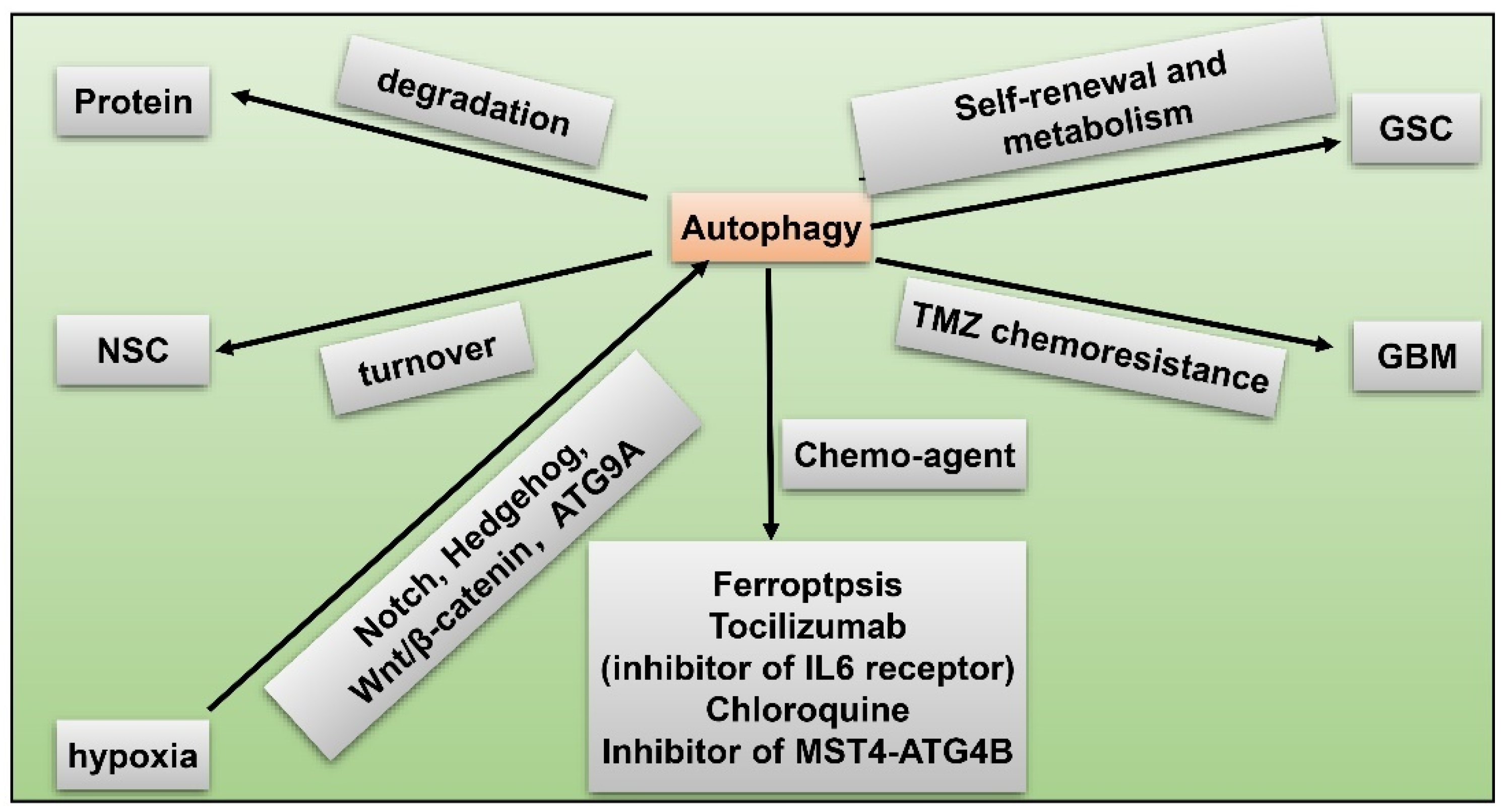
8. GSC and Hypoxia-Related Autophagy
9. GSC and Hypoxia-Related Therapeutic Resistance
10. GSC and Hypoxia-Related Chemotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemo Agent | Function | Reference |
|---|---|---|
| TMZ | associates with HIF-1α and prolong survival span of GBM patients | (Lo Dico et al., 2018 [120]; Struve et al., 2020 [117]) |
| TMZ plus EGFRvIII | EGFRvIII enhances hypoxia-induced death and cooperates with TMZ to prolong survival of patients with MGMT promoter methylated GBM | (Struve et al., 2020 [117]; Luger et al., 2020 [121]) |
| TMZ plus SNAP | SNAP induces HIF-1α and cooperates with TMZ to benefit survival span of GBM patients with MGMT promoter methylated | (Tsai et al., 2019 [122]) |
| TMZ plus metformin | reverts chemoresistance of GBM during hypoxia via inhibition of PI3K/mTOR pathway | (Lo Dico et al., 2019 [120]) |
| Biweekly TMZ plus bevacizumab | well tolerated by refractory GBM patients but increases regional hypoxia | (Badruddoja et al., 2017 [123]; Gerstner et al., 2020 [124]) |
| TMZ plus Decitabine | increases cytotoxicity of HIF-1α-related chemo-agent | (Gallitto et al., 2020 [127]) |
| TMZ plus imipramine | reduces cytotoxic effect of TMZ under hypoxia | (Bielecka and Obuchowicz, 2017 [126]) |
| TMZ plus tranylcypromine | reduces cytotoxic effect of TMZ under hypoxia | (Bielecka and Obuchowicz, 2017 [126]) |
| N45 | inhibits proliferation through hypoxia-associated ROS/PI3K/Akt pathway in TMZ-resistant GBM | (Zhang et al., 2020 [128]) |
| Tacrolimus (FK506) | reduce GBM tumor volume and hypoxia-induce surface markers (ki67, GFAP and nestin) in GSC | (Torres et al., 2018 [119]) |
| UDCA bortezomib plus BTZ | stabilizes expression of HIF-1α and a promising therapy for GBM patients | (Yao et al., 2020 [130]) |
| BAL101553 | targets hypoxia-mediated angiogenesis of GBM | (Bergès et al., 2020 [131]) |
| Bevacizumab plus carmustine | not enhance incidence of hematologic toxicity but attributes to regional hypoxia in recurrent GBM | (Yerram et al., 2019 [132]) |
| nimotuzumab | an anti-EGFR antibody that upregulates survival span of GBM patients | (Ronellenfitsch et al., 2018 [135]) |
| Evofosfamide plus bevacizumab | activated during hypoxia and well tolerated by bevacizumab-regressive GBM patients | (Brenner et al., 2018 [133]; Takakusagi et al., 2018 [134]) |
| amitriptyline | stimulates phenotypical switch from GSCs to non-GSCs | (Bielecka-Wajdman et al., 2017 [125]) |
| digoxin | inhibits HIF-1α and HIF-2α to target GBM | (Patocka et al., 2020 [136]) |
| digitoxin | suppresses HIF-1α to target GSCs | (Lee et al., 2017 [137]) |
| Cetuximab | reduces translation of HIF-1α to target GBM | (Ferreira et al., 2020 [138]) |
| Topotecan | reduces translation of HIF-1α to target GBM | (Bernstock et al., 2017 [139]) |
11. GSC and Hypoxia-Related Radiotherapy
12. GSC and Hypoxia-Related Radio-, Immunotherapy
13. GSC and Hyperbaric Oxygen Therapy
14. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.; Mao, X.G.; Cao, W.D.; Zhen, H.N.; Hu, S.J. Malignant Intracranial High Grade Glioma and Current Treatment Strategy. Curr. Cancer Drug Targets 2019, 19, 101–108. [Google Scholar] [CrossRef]
- Park, J.E.; Kim, H.S.; Park, S.Y.; Jung, S.C.; Kim, J.H.; Heo, H.Y. Identification of Early Response to Anti-Angiogenic Therapy in Recurrent Glioblastoma: Amide Proton Transfer-weighted and Perfusion-weighted MRI compared with Diffusion-weighted MRI. Radiology 2020, 295, 397–406. [Google Scholar] [CrossRef]
- Henderson, F., Jr.; Brem, S.; O’Rourke, D.M.; Nasrallah, M.; Buch, V.P.; Young, A.J.; Doot, R.K.; Pantel, A.; Desai, A.; Bagley, S.J.; et al. (18)F-Fluciclovine PET to distinguish treatment-related effects from disease progression in recurrent glioblastoma: PET fusion with MRI guides neurosurgical sampling. Neuro Oncol. Pract. 2020, 7, 152–157. [Google Scholar] [CrossRef]
- Mao, X.G.; Zhang, X.; Zhen, H.N. Progress on potential strategies to target brain tumor stem cells. Cell. Mol. Neurobiol. 2009, 29, 141–155. [Google Scholar] [CrossRef]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and Reprogramming the Tumor-Propagating Potential of Glioblastoma Stem-like Cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef]
- Lee, G.H.; Hong, K.T.; Choi, J.Y.; Shin, H.Y.; Lee, W.W.; Kang, H.J. Immunosenescent characteristics of T cells in young patients following haploidentical haematopoietic stem cell transplantation from parental donors. Clin. Transl. Immunol. 2020, 9, e1124. [Google Scholar] [CrossRef]
- Ho, N.T.T.; Rahane, C.S.; Pramanik, S.; Kim, P.S.; Kutzner, A.; Heese, K. FAM72, Glioblastoma Multiforme (GBM) and Beyond. Cancers 2021, 13, 1025. [Google Scholar] [CrossRef]
- Valiyaveettil, D.; Malik, M.; Akram, K.S.; Ahmed, S.F.; Joseph, D.M. Prospective study to assess the survival outcomes of planned irradiation of ipsilateral subventricular and periventricular zones in glioblastoma. Ecancermedicalscience 2020, 14, 1021. [Google Scholar] [CrossRef]
- Kumar, U. Immunolocalization of Gas7 in the Subgranular Zone of Mice Hippocampus. Prague Med. Rep. 2019, 120, 117–123. [Google Scholar] [CrossRef]
- Niklasson, C.U.; Fredlund, E.; Monni, E.; Lindvall, J.M.; Kokaia, Z.; Hammarlund, E.U.; Bronner, M.E.; Mohlin, S. Hypoxia inducible factor-2alpha importance for migration, proliferation, and self-renewal of trunk neural crest cells. Dev. Dyn. 2021, 250, 191–236. [Google Scholar] [CrossRef]
- Peng, S.; Zhang, J.; Tan, X.; Huang, Y.; Xu, J.; Silk, N.; Zhang, D.; Liu, Q.; Jiang, J. The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front. Endocrinol. 2020, 11, 586857. [Google Scholar] [CrossRef]
- Večeřa, J.; Procházková, J.; Šumberová, V.; Pánská, V.; Paculová, H.; Lánová, M.K.; Mašek, J.; Bohačiaková, D.; Andersson, E.R.; Pacherník, J. Hypoxia/Hif1α prevents premature neuronal differentiation of neural stem cells through the activation of Hes1. Stem Cell Res. 2020, 45, 101770. [Google Scholar] [CrossRef]
- Zhou, Y.; Dong, X.; Xiu, P.; Wang, X.; Yang, J.; Li, L.; Li, Z.; Sun, P.; Shi, X.; Zhong, J. Meloxicam, a Selective COX-2 Inhibitor, Mediates Hypoxia-Inducible Factor- (HIF-) 1α Signaling in Hepatocellular Carcinoma. Oxidative Med. Cell. Longev. 2020, 2020, 7079308. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell. Mol. Life Sci. CMLS 2021, 78, 195–206. [Google Scholar] [CrossRef]
- Satija, S.; Kaur, H.; Tambuwala, M.M.; Sharma, P.; Vyas, M.; Khurana, N.; Sharma, N.; Bakshi, H.A.; Charbe, N.B.; Zacconi, F.C.; et al. Hypoxia-inducible factor (HIF): Fuel for cancer progression. Curr. Mol. Pharmacol. 2021, 14, 321–332. [Google Scholar] [CrossRef]
- Kung-Chun Chiu, D.; Pui-Wah Tse, A.; Law, C.T.; Ming-Jing Xu, I.; Lee, D.; Chen, M.; Kit-Ho Lai, R.; Wai-Hin Yuen, V.; Wing-Sum Cheu, J.; Wai-Hung Ho, D.; et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell. Death Dis. 2019, 10, 934. [Google Scholar] [CrossRef]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System; WHO: Lyon, France, 2010. [Google Scholar]
- Li, G.; Li, Y.; Liu, X.; Wang, Z.; Zhang, C.; Wu, F.; Jiang, H.; Zhang, W.; Bao, Z.; Wang, Y.; et al. ALDH1A3 induces mesenchymal differentiation and serves as a predictor for survival in glioblastoma. Cell. Death Dis. 2018, 9, 1190. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Z.; Chen, C.; Fu, X.; Wang, J.; Fei, X.; Yan, X.; Xu, R. A low MW inhibitor of CD44 dimerization for the treatment of glioblastoma. Br. J. Pharmacol. 2020, 177, 3009–3023. [Google Scholar] [CrossRef]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Saito, N.; Miyazawa, K.; Miyazono, K. Glioma-initiating cells retain their tumorigenicity through integration of the Sox axis and Oct4 protein. J. Biol. Chem. 2011, 286, 41434–41441. [Google Scholar] [CrossRef]
- Wang, F.; Wang, A.Y.; Chesnelong, C.; Yang, Y.; Nabbi, A.; Thalappilly, S.; Alekseev, V.; Riabowol, K. ING5 activity in self-renewal of glioblastoma stem cells via calcium and follicle stimulating hormone pathways. Oncogene 2018, 37, 286–301. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schanzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nister, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef]
- Herting, C.J.; Chen, Z.; Pitter, K.L.; Szulzewsky, F.; Kaffes, I.; Kaluzova, M.; Park, J.C.; Cimino, P.J.; Brennan, C.; Wang, B.; et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017, 65, 1914–1926. [Google Scholar] [CrossRef]
- Armocida, D.; Pesce, A.; Di Giammarco, F.; Frati, A.; Santoro, A.; Salvati, M. Long Term Survival in Patients Suffering from Glio-blastoma Multiforme: A Single-Center Observational Cohort Study. Diagnostics 2019, 9, 209. [Google Scholar] [CrossRef]
- Orzan, F.; De Bacco, F.; Crisafulli, G.; Pellegatta, S.; Mussolin, B.; Siravegna, G.; D’Ambrosio, A.; Comoglio, P.M.; Finocchiaro, G.; Boccaccio, C. Genetic Evolution of Glioblastoma Stem-Like Cells From Primary to Recurrent Tumor. Stem Cells 2017, 35, 2218–2228. [Google Scholar] [CrossRef]
- Van Noorden, C.J.F.; Hira, V.V.V.; van Dijck, A.J.; Novak, M.; Breznik, B.; Molenaar, R.J. Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy? Cells 2021, 10, 705. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef]
- Voss, D.M.; Spina, R.; Carter, D.L.; Lim, K.S.; Jeffery, C.J.; Bar, E.E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292. [Google Scholar] [CrossRef]
- Gao, D.; Nyalali, A.M.K.; Hou, Y.; Xu, Y.; Zhou, J.; Zhao, W.; Huang, B.; Li, F. 2,5-Dimethyl Celecoxib Inhibits Proliferation and Cell Cycle and Induces Apoptosis in Glioblastoma by Suppressing CIP2A/PP2A/Akt Signaling Axis. J. Mol. Neurosci. 2021, 71, 1703–1713. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, L.; Gong, S.; Xiong, S.; Wang, J.; Zou, D.; Pan, J.; Deng, Y.; Yan, Q.; Wu, N.; et al. HIF1α/HIF2α-Sox2/Klf4 promotes the malignant progression of glioblastoma via the EGFR-PI3K/AKT signalling pathway with positive feedback under hypoxia. Cell. Death Dis. 2021, 12, 312. [Google Scholar] [CrossRef]
- Malmström, A.; Łysiak, M.; Åkesson, L.; Jakobsen, I.; Mudaisi, M.; Milos, P.; Hallbeck, M.; Fomichov, V.; Broholm, H.; Grunnet, K.; et al. ABCB1 single-nucleotide variants and survival in patients with glioblastoma treated with radiotherapy concomitant with temozolomide. Pharm. J. 2020, 20, 213–219. [Google Scholar] [CrossRef]
- Huang, W.; Ding, X.; Ye, H.; Wang, J.; Shao, J.; Huang, T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway. Neuroreport 2018, 29, 1578–1585. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Iwanami, A.; Gini, B.; Zanca, C.; Matsutani, T.; Assuncao, A.; Nael, A.; Dang, J.; Yang, H.; Zhu, S.; Kohyama, J.; et al. PML mediates glioblastoma resistance to mammalian target of rapamycin (mTOR)-targeted therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 4339–4344. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef]
- Li, H.; Chen, L.; Li, J.J.; Zhou, Q.; Huang, A.; Liu, W.W.; Wang, K.; Gao, L.; Qi, S.T.; Lu, Y.T. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018, 11, 70. [Google Scholar] [CrossRef]
- Nicolas, S.; Abdellatef, S.; Haddad, M.A.; Fakhoury, I.; El-Sibai, M. Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells 2019, 8, 1397. [Google Scholar] [CrossRef]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef]
- Grassi, E.S.; Pantazopoulou, V.; Pietras, A. Hypoxia-induced release, nuclear translocation, and signaling activity of a DLK1 intracellular fragment in glioma. Oncogene 2020, 39, 4028–4044. [Google Scholar] [CrossRef]
- Maciaczyk, D.; Picard, D.; Zhao, L.; Koch, K.; Herrera-Rios, D.; Li, G.; Marquardt, V.; Pauck, D.; Hoerbelt, T.; Zhang, W.; et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br. J. Cancer 2017, 117, 102–112. [Google Scholar] [CrossRef]
- Kreuger, J.; Claesson-Welsh, L.; Olsson, A.-K.; Dimberg, A. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar]
- Almiron Bonnin, D.A.; Havrda, M.C.; Lee, M.C.; Liu, H.; Zhang, Z.; Nguyen, L.N.; Harrington, L.X.; Hassanpour, S.; Cheng, C.; Israel, M.A. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene 2018, 37, 1107–1118. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J. Bevacizumab Plus Irinotecan in Recurrent Glioblastoma Multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef]
- Lu, F.I.; Wang, Y.T.; Wang, Y.S.; Wu, C.Y.; Li, C.C. Involvement of BIG1 and BIG2 in regulating VEGF expression and angiogenesis. FASEB J. Off Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 9959–9973. [Google Scholar] [CrossRef]
- Long, Y.; Tao, H.; Karachi, A.; Grippin, A.J.; Jin, L.; Chang, Y.E.; Zhang, W.; Dyson, K.A.; Hou, A.Y.; Na, M.; et al. Dysregulation of Glutamate Transport Enhances Treg Function That Promotes VEGF Blockade Resistance in Glioblastoma. Cancer Res. 2020, 80, 499–509. [Google Scholar] [CrossRef]
- Boso, D.; Rampazzo, E.; Zanon, C.; Bresolin, S.; Maule, F.; Porcù, E.; Cani, A.; Della Puppa, A.; Trentin, L.; Basso, G.; et al. HIF-1α/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics 2019, 9, 4860–4877. [Google Scholar] [CrossRef]
- Lin, Y.T.; Wu, K.J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 39. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef]
- Guerrero, P.A.; Tchaicha, J.H.; Chen, Z.; Morales, J.E.; Mccarty, N.; Wang, Q.; Sulman, E.P.; Fuller, G.; Lang, F.F.; Rao, G. Glioblastoma stem cells exploit the αvβ8 integrin-TGFβ1 signaling axis to drive tumor initiation and progression. Oncogene 2017, 36, 6568–6580. [Google Scholar] [CrossRef]
- Mangani, D.; Weller, M.; Seyed Sadr, E.; Willscher, E.; Seystahl, K.; Reifenberger, G.; Tabatabai, G.; Binder, H.; Schneider, H. Limited role for transforming growth factor-β pathway activation-mediated escape from VEGF inhibition in murine glioma models. Neuro Oncol. 2016, 18, 1610–1621. [Google Scholar] [CrossRef]
- Wu, W.; Hu, Q.; Nie, E.; Yu, T.; Wu, Y.; Zhi, T.; Jiang, K.; Shen, F.; Wang, Y.; Zhang, J.; et al. Hypoxia induces H19 expression through direct and indirect Hif-1α activity, promoting oncogenic effects in glioblastoma. Sci. Rep. 2017, 7, 45029. [Google Scholar] [CrossRef]
- Mao, X.G.; Wang, C.; Liu, D.Y.; Zhang, X.; Wang, L.; Yan, M.; Zhang, W.; Zhu, J.; Li, Z.C.; Mi, C.; et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget 2016, 7, 47808–47820. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell. Proteom. MCP 2017, 16, 1906–1921. [Google Scholar] [CrossRef]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, A.; Chandel, N.S.; et al. IDH3α regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019, 5, eaat0456. [Google Scholar] [CrossRef]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2019, 57, 45–51. [Google Scholar] [CrossRef]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef]
- Maus, A.; Peters, G.J. Erratum to: Glutamate and α-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 1143. [Google Scholar] [CrossRef]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgöz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.K.; Alfonso, J. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab. 2019, 30, 274–289.e275. [Google Scholar] [CrossRef]
- Javelot, H.; Michel, B.; Marquis, A.; Didelot, N.; Socha, M.; Javelot, T.; Petitpain, N. Acute withdrawal syndrome after discontinuation of a short analgesic treatment with tramadol. Therapie 2016, 71, 347–348. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Liu, M.; Burow, M.E.; Wang, G. Targeting CXCL12/CXCR4 Axis in Tumor Immunotherapy. Curr. Med. Chem. 2019, 26, 3026–3041. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef]
- Hu, B.; Wang, Q.; Wang, Y.A.; Hua, S.; Sauvé, C.G.; Ong, D.; Lan, Z.D.; Chang, Q.; Ho, Y.W.; Monasterio, M.M.; et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 2016, 167, 1281–1295.e18. [Google Scholar] [CrossRef]
- Rocha, R.; Torres, Á.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martín, R.; Quezada, C. The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef]
- Lathia, J.D.; Li, M.; Hall, P.E.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.R.; Huang, P.; et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Corsini, N.S.; Martin-Villalba, A. Integrin alpha 6: Anchors away for glioma stem cells. Cell Stem Cell 2010, 6, 403–404. [Google Scholar] [CrossRef]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Jeng, K.S.; Chang, C.F.; Lin, S.S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M. Endothelial Cells Create a Stem Cell Niche in Glioblastoma by Providing NOTCH Ligands That Nurture Self-Renewal of Cancer Stem-Like Cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Mei, X.; Chen, Y.S.; Chen, F.R.; Xi, S.Y.; Chen, Z.P. Glioblastoma stem cell differentiation into endothelial cells evidenced through live-cell imaging. Neuro Oncol. 2017, 19, 1109–1118. [Google Scholar] [CrossRef]
- Hsu, S.P.C.; Chen, Y.C.; Chiang, H.C.; Huang, Y.C.; Huang, C.C.; Wang, H.E.; Wang, Y.S.; Chi, K.H. Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J. Neuro Oncol. 2020, 146, 417–426. [Google Scholar] [CrossRef]
- Arcuri, C.; Fioretti, B.; Bianchi, R.; Mecca, C.; Tubaro, C.; Beccari, T.; Franciolini, F.; Giambanco, I.; Donato, R. Microglia-glioma cross-talk: A two way approach to new strategies against glioma. Front. Biosci. 2017, 22, 268–309. [Google Scholar] [CrossRef]
- Singh, R.; Mishra, M.K.; Aggarwal, H. Inflammation, Immunity, and Cancer. Mediat. Inflamm. 2017, 2017, 6027305. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Q.; Li, L.; Chen, K.; Yang, J.; Dixit, D.; Gimple, R.C.; Ci, S.; Lu, C.; Hu, L.; et al. beta2-Microglobulin Maintains Glioblastoma Stem Cells and Induces M2-like Polarization of Tumor-Associated Macrophages. Cancer Res. 2022, 82, 3321–3334. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Kwok, D.; Okada, H. T-Cell based therapies for overcoming neuroanatomical and immunosuppressive challenges within the glioma microenvironment. J. Neuro Oncol. 2020, 147, 281–295. [Google Scholar] [CrossRef]
- Peng, W.; Wang, H.Y.; Miyahara, Y.; Peng, G.; Wang, R.-F. Tumor-Associated Galectin-3 Modulates the Function of Tumor-Reactive T Cells. Cancer Res. 2008, 68, 7228–7236. [Google Scholar] [CrossRef]
- Weenink, B.; French, P.J.; Sillevis Smitt, P.A.E.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef]
- Tong, L.; Li, J.; Li, Q.; Wang, X.; Medikonda, R.; Zhao, T.; Li, T.; Ma, H.; Yi, L.; Liu, P.; et al. ACT001 reduces the expression of PD-L1 by inhibiting the phosphorylation of STAT3 in glioblastoma. Theranostics 2020, 10, 5943–5956. [Google Scholar] [CrossRef]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef]
- Boyd, N.H.; Walker, K.; Ayokanmbi, A.; Gordon, E.R.; Whetsel, J.; Smith, C.M.; Sanchez, R.G.; Lubin, F.D.; Chakraborty, A.; Tran, A.N.; et al. Chromodomain Helicase DNA-Binding Protein 7 Is Suppressed in the Perinecrotic/Ischemic Microenvironment and Is a Novel Regulator of Glioblastoma Angiogenesis. Stem Cells 2019, 37, 453–462. [Google Scholar] [CrossRef]
- Filatova, A.; Seidel, S.; Böğürcü, N.; Gräf, S.; Garvalov, B.K.; Acker, T. Acidosis Acts through HSP90 in a PHD/VHL-Independent Manner to Promote HIF Function and Stem Cell Maintenance in Glioma. Cancer Res. 2016, 76, 5845–5856. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, J.; Zhang, Z.; Gao, Z.; Qi, Y.; Qiu, W.; Pan, Z.; Guo, Q.; Li, B.; Zhao, S.; et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 2021, 12, 373. [Google Scholar] [CrossRef]
- Dong, F.; Qin, X.; Wang, B.; Li, Q.; Hu, J.; Cheng, X.; Guo, D.; Cheng, F.; Fang, C.; Tan, Y.; et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 5876–5888. [Google Scholar] [CrossRef]
- Mondal, A.; Kumari Singh, D.; Panda, S.; Shiras, A. Extracellular Vesicles As Modulators of Tumor Microenvironment and Disease Progression in Glioma. Front. Oncol. 2017, 7, 144. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef]
- Reinhard, J.; Brösicke, N.; Theocharidis, U.; Faissner, A. The extracellular matrix niche microenvironment of neural and cancer stem cells in the brain. Int. J. Biochem. Cell Biol. 2016, 81, 174–183. [Google Scholar] [CrossRef]
- Faissner, A.; Roll, L.; Theocharidis, U. Tenascin-C in the matrisome of neural stem and progenitor cells. Mol. Cell. Neurosci. 2017, 81, 22–31. [Google Scholar] [CrossRef]
- Belter, A.; Gudanis, D.; Rolle, K.; Piwecka, M.; Gdaniec, Z.; Naskręt-Barciszewska, M.Z.; Barciszewski, J. Mature miRNAs form secondary structure, which suggests their function beyond RISC. PLoS ONE 2014, 9, e113848. [Google Scholar] [CrossRef]
- Mao, X.G.; Xue, X.Y.; Lv, R.; Ji, A.; Shi, T.Y.; Chen, X.Y.; Jiang, X.F.; Zhang, X. CEBPD is a master transcriptional factor for hypoxia regulated proteins in glioblastoma and augments hypoxia induced invasion through extracellular matrix-integrin mediated EGFR/PI3K pathway. Cell Death Dis. 2023, 14, 269. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Van Noorden, C.J.F.; Carraway, H.E.; Maciejewski, J.P.; Molenaar, R.J. Novel therapeutic strategies to target leukemic cells that hijack compartmentalized continuous hematopoietic stem cell niches. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 183–198. [Google Scholar] [CrossRef]
- Aderetti, D.A.; Hira, V.V.V.; Molenaar, R.J.; van Noorden, C.J.F. The hypoxic peri-arteriolar glioma stem cell niche, an integrated concept of five types of niches in human glioblastoma. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 346–354. [Google Scholar] [CrossRef]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef]
- An, Z.; Knobbe-Thomsen, C.B.; Wan, X.; Fan, Q.W.; Reifenberger, G.; Weiss, W.A. EGFR Cooperates with EGFRvIII to Recruit Macrophages in Glioblastoma. Cancer Res. 2018, 78, 6785–6794. [Google Scholar] [CrossRef]
- Guo, X.Y.; Zhang, G.H.; Wang, Z.N.; Duan, H.; Xie, T.; Liang, L.; Cui, R.; Hu, H.R.; Wu, Y.; Dong, J.J.; et al. A novel Foxp3-related immune prognostic signature for glioblastoma multiforme based on immunogenomic profiling. Aging 2021, 13, 3501–3517. [Google Scholar] [CrossRef]
- Kiyokawa, J.; Kawamura, Y.; Ghouse, S.M.; Acar, S.; Barçın, E.; Martínez-Quintanilla, J.; Martuza, R.L.; Alemany, R.; Rabkin, S.D.; Shah, K.; et al. Modification of Extracellular Matrix Enhances Oncolytic Adenovirus Immunotherapy in Glioblastoma. Clin. Cancer Res. Off J. Am. Assoc. Cancer Res. 2021, 27, 889–902. [Google Scholar] [CrossRef]
- Brat, D.J.; Meir, E.G.V. Glomeruloid Microvascular Proliferation Orchestrated by VPF/VEGF: A New World of Angiogenesis Research. Am. J. Pathol. 2001, 158, 789–796. [Google Scholar] [CrossRef]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Peng, P.; Zhu, H.; Liu, D.; Chen, Z.; Zhang, X.; Guo, Z.; Dong, M.; Wan, L.; Zhang, P.; Liu, G.; et al. TGFBI secreted by tumor-associated macrophages promotes glioblastoma stem cell-driven tumor growth via integrin alphavbeta5-Src-Stat3 signaling. Theranostics 2022, 12, 4221–4236. [Google Scholar] [CrossRef]
- Ho, S.Y.; Ling, T.Y.; Lin, H.Y.; Liou, J.T.J.; Liu, F.C.; Chen, I.; Lee, S.W.; Hsu, Y.; Lai, D.M.; Liou, H.H. SDF-1/CXCR4 Signaling Maintains Stemness Signature in Mouse Neural Stem/Progenitor Cells. Stem Cells Int. 2017, 2017, 2493752. [Google Scholar] [CrossRef]
- Lee, E.Q.; Duda, D.G.; Muzikansky, A.; Gerstner, E.R.; Kuhn, J.G.; Reardon, D.A.; Nayak, L.; Norden, A.D.; Doherty, L.; LaFrankie, D.; et al. Phase I and Biomarker Study of Plerixafor and Bevacizumab in Recurrent High-Grade Glioma. Clin. Cancer Res. Off J. Am. Assoc. Cancer Res. 2018, 24, 4643–4649. [Google Scholar] [CrossRef]
- Jawhari, S.; Ratinaud, M.H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘menage-a-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef]
- Abdul Rahim, S.A.; Dirkse, A.; Oudin, A.; Schuster, A.; Bohler, J.; Barthelemy, V.; Muller, A.; Vallar, L.; Janji, B.; Golebiewska, A.; et al. Regulation of hypoxia-induced autophagy in glioblastoma involves ATG9A. Br. J. Cancer 2017, 117, 813–825. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Xue, H.; Yuan, G.; Guo, X.; Liu, Q.; Zhang, J.; Gao, X.; Guo, X.; Xu, S.; Li, T.; Shao, Q.; et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: Hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy 2016, 12, 1129–1152. [Google Scholar] [CrossRef]
- Julio Sotelo, E.B.; López-González, M.A. Adding Chloroquine to Conventional Treatment for Glioblastoma Multiforme: A Randomized, Double-Blind, Placebo-Controlled Trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855.e8. [Google Scholar] [CrossRef]
- Pottoo, F.H.; Javed, M.N.; Rahman, J.U.; Abu-Izneid, T.; Khan, F.A. Targeted delivery of miRNA based therapeuticals in the clinical management of Glioblastoma Multiforme. Semin. Cancer Biol. 2020, 69, 391–398. [Google Scholar] [CrossRef]
- Dixit, D.; Prager, B.C.; Gimple, R.C.; Poh, H.X.; Wang, Y.; Wu, Q.; Qiu, Z.; Kidwell, R.L.; Kim, L.J.Y.; Xie, Q.; et al. The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer Discov. 2021, 11, 480–499. [Google Scholar] [CrossRef]
- Struve, N.; Binder, Z.A.; Stead, L.F.; Brend, T.; Bagley, S.J.; Faulkner, C.; Ott, L.; Müller-Goebel, J.; Weik, A.S.; Hoffer, K.; et al. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene 2020, 39, 3041–3055. [Google Scholar] [CrossRef]
- Jalota, A.; Kumar, M.; Das, B.C.; Yadav, A.K.; Chosdol, K.; Sinha, S. A drug combination targeting hypoxia induced chemoresistance and stemness in glioma cells. Oncotarget 2018, 9, 18351–18366. [Google Scholar] [CrossRef]
- Torres, Á.; Arriagada, V.; Erices, J.I.; Toro, M.; Rocha, J.D.; Niechi, I.; Carrasco, C.; Oyarzún, C.; Quezada, C. FK506 Attenuates the MRP1-Mediated Chemoresistant Phenotype in Glioblastoma Stem-Like Cells. Int. J. Mol. Sci. 2018, 19, 2697. [Google Scholar] [CrossRef]
- Lo Dico, A.; Martelli, C.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Hypoxia-Inducible Factor-1α Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front. Oncol. 2018, 8, 249. [Google Scholar] [CrossRef]
- Luger, A.L.; Lorenz, N.I.; Urban, H.; Divé, I.; Engel, A.L.; Strassheimer, F.; Dettmer, K.; Zeiner, P.S.; Shaid, S.; Struve, N.; et al. Activation of Epidermal Growth Factor Receptor Sensitizes Glioblastoma Cells to Hypoxia-Induced Cell Death. Cancers 2020, 12, 2144. [Google Scholar] [CrossRef]
- Tsai, C.K.; Huang, L.C.; Wu, Y.P.; Kan, I.Y.; Hueng, D.Y. SNAP reverses temozolomide resistance in human glioblastoma multiforme cells through down-regulation of MGMT. FASEB J. Off Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 14171–14184. [Google Scholar] [CrossRef]
- Badruddoja, M.A.; Pazzi, M.; Sanan, A.; Schroeder, K.; Kuzma, K.; Norton, T.; Scully, T.; Mahadevan, D.; Ahmadi, M.M. Phase II study of bi-weekly temozolomide plus bevacizumab for adult patients with recurrent glioblastoma. Cancer Chemother. Pharmacol. 2017, 80, 715–721. [Google Scholar] [CrossRef]
- Gerstner, E.R.; Emblem, K.E.; Yen, Y.F.; Dietrich, J.; Jordan, J.T.; Catana, C.; Wenchin, K.L.; Hooker, J.M.; Duda, D.G.; Rosen, B.R.; et al. Vascular dysfunction promotes regional hypoxia after bevacizumab therapy in recurrent glioblastoma patients. Neuro Oncol. Adv. 2020, 2, vdaa157. [Google Scholar] [CrossRef]
- Bielecka-Wajdman, A.M.; Lesiak, M.; Ludyga, T.; Sieroń, A.; Obuchowicz, E. Reversing glioma malignancy: A new look at the role of antidepressant drugs as adjuvant therapy for glioblastoma multiforme. Cancer Chemother. Pharmacol. 2017, 79, 1249–1256. [Google Scholar] [CrossRef]
- Bielecka, A.M.; Obuchowicz, E. Antidepressant drugs can modify cytotoxic action of temozolomide. Eur. J. Cancer Care 2017, 26, e12551. [Google Scholar] [CrossRef]
- Gallitto, M.; Cheng He, R.; Inocencio, J.F.; Wang, H.; Zhang, Y.; Deikus, G.; Wasserman, I.; Strahl, M.; Smith, M.; Sebra, R.; et al. Epigenetic preconditioning with decitabine sensitizes glioblastoma to temozolomide via induction of MLH1. J. Neuro Oncol. 2020, 147, 557–566. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Y.; Li, H.; Ji, Y.; Fang, F.; Tang, H.; Qiu, P. A steroidal saponin form Paris vietnamensis (Takht.) reverses temozolomide resistance in glioblastoma cells via inducing apoptosis through ROS/PI3K/Akt pathway. Biosci. Trends 2020, 14, 123–133. [Google Scholar] [CrossRef]
- Ludman, T.; Melemedjian, O.K. Bortezomib and metformin opposingly regulate the expression of hypoxia-inducible factor alpha and the consequent development of chemotherapy-induced painful peripheral neuropathy. Mol. Pain 2019, 15, 1744806919850043. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, X.; Zhao, F.; Wang, S.; Chen, A.; Huang, B.; Wang, J.; Li, X. Ursodeoxycholic Acid Inhibits Glioblastoma Progression via Endoplasmic Reticulum Stress Related Apoptosis and Synergizes with the Proteasome Inhibitor Bortezomib. ACS Chem. Neurosci. 2020, 11, 1337–1346. [Google Scholar] [CrossRef]
- Bergès, R.; Tchoghandjian, A.; Sergé, A.; Honoré, S.; Figarella-Branger, D.; Bachmann, F.; Lane, H.A.; Braguer, D. EB1-dependent long survival of glioblastoma-grafted mice with the oral tubulin-binder BAL101553 is associated with inhibition of tumor angiogenesis. Oncotarget 2020, 11, 759–774. [Google Scholar] [CrossRef]
- Yerram, P.; Reiss, S.N.; Modelevsky, L.; Gavrilovic, I.T.; Kaley, T. Evaluation of toxicity of carmustine with or without bevacizumab in patients with recurrent or progressive high grade gliomas. J. Neuro Oncol. 2019, 145, 57–63. [Google Scholar] [CrossRef]
- Brenner, A.; Zuniga, R.; Sun, J.D.; Floyd, J.; Hart, C.P.; Kroll, S.; Fichtel, L.; Cavazos, D.; Caflisch, L.; Gruslova, A.; et al. Hypoxia-activated evofosfamide for treatment of recurrent bevacizumab-refractory glioblastoma: A phase I surgical study. Neuro Oncol. 2018, 20, 1231–1239. [Google Scholar] [CrossRef]
- Takakusagi, Y.; Kishimoto, S.; Naz, S.; Matsumoto, S.; Saito, K.; Hart, C.P.; Mitchell, J.B.; Krishna, M.C. Radiotherapy Synergizes with the Hypoxia-Activated Prodrug Evofosfamide: In Vitro and In Vivo Studies. Antioxid. Redox Signal. 2018, 28, 131–140. [Google Scholar] [CrossRef]
- Ronellenfitsch, M.W.; Zeiner, P.S.; Mittelbronn, M.; Urban, H.; Pietsch, T.; Reuter, D.; Senft, C.; Steinbach, J.P.; Westphal, M.; Harter, P.N. Akt and mTORC1 signaling as predictive biomarkers for the EGFR antibody nimotuzumab in glioblastoma. Acta Neuropathol. Commun. 2018, 6, 81. [Google Scholar] [CrossRef]
- Patocka, J.; Nepovimova, E.; Wu, W.; Kuca, K. Digoxin: Pharmacology and toxicology-A review. Environ. Toxicol. Pharmacol. 2020, 79, 103400. [Google Scholar] [CrossRef]
- Lee, D.H.; Cheul Oh, S.; Giles, A.J.; Jung, J.; Gilbert, M.R.; Park, D.M. Cardiac glycosides suppress the maintenance of stemness and malignancy via inhibiting HIF-1α in human glioma stem cells. Oncotarget 2017, 8, 40233–40245. [Google Scholar] [CrossRef]
- Ferreira, N.N.; Granja, S.; Boni, F.I.; Ferreira, L.M.B.; Reis, R.M.; Baltazar, F.; Gremião, M.P.D. A novel strategy for glioblastoma treatment combining alpha-cyano-4-hydroxycinnamic acid with cetuximab using nanotechnology-based delivery systems. Drug. Deliv. Transl. Res. 2020, 10, 594–609. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Ye, D.; Gessler, F.A.; Lee, Y.J.; Peruzzotti-Jametti, L.; Baumgarten, P.; Johnson, K.R.; Maric, D.; Yang, W.; Kögel, D.; et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci. Rep. 2017, 7, 7425. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, X.; Li, S.; Yang, S. The total flavonoid of Eucommia ulmoides sensitizes human glioblastoma cells to radiotherapy via HIF-α/MMP-2 pathway and activates intrinsic apoptosis pathway. OncoTargets Ther. 2019, 12, 5515–5524. [Google Scholar] [CrossRef]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugué, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Mao, X.G.; Xue, X.Y.; Wang, L.; Wang, L.; Li, L.; Zhang, X. Hypoxia Regulated Gene Network in Glioblastoma Has Special Algebraic Topology Structures and Revealed Communications Involving Warburg Effect and Immune Regulation. Cell. Mol. Neurobiol. 2019, 39, 1093–1114. [Google Scholar] [CrossRef]
- Huang, L.; Boling, W.; Zhang, J.H. Hyperbaric oxygen therapy as adjunctive strategy in treatment of glioblastoma multiforme. Med. Gas Res. 2018, 8, 24–28. [Google Scholar] [CrossRef]
- Alpuim Costa, D.; Sampaio-Alves, M.; Netto, E.; Fernandez, G.; Oliveira, E.; Teixeira, A.; Daniel, P.M.; Bernardo, G.S.; Amaro, C. Hyperbaric Oxygen Therapy as a Complementary Treatment in Glioblastoma-A Scoping Review. Front. Neurol. 2022, 13, 886603. [Google Scholar] [CrossRef]
- Stępień, K.; Ostrowski, R.P.; Matyja, E. Hyperbaric oxygen as an adjunctive therapy in treatment of malignancies, including brain tumours. Med. Oncol. 2016, 33, 101. [Google Scholar] [CrossRef]
- Chang, C.H. Hyperbaric oxygen and radiation therapy in the management of glioblastoma. Natl. Cancer Inst. Monogr. 1977, 46, 163–169. [Google Scholar]
- Ogawa, K.; Kohshi, K.; Ishiuchi, S.; Matsushita, M.; Yoshimi, N.; Murayama, S. Old but new methods in radiation oncology: Hyperbaric oxygen therapy. Int. J. Clin. Oncol. 2013, 18, 364–370. [Google Scholar] [CrossRef]
- Ogawa, K.; Ishiuchi, S.; Inoue, O.; Yoshii, Y.; Saito, A.; Watanabe, T.; Iraha, S.; Toita, T.; Kakinohana, Y.; Ariga, T.; et al. Phase II Trial of Radiotherapy After Hyperbaric Oxygenation With Multiagent Chemotherapy (Procarbazine, Nimustine, and Vincristine) for High-Grade Gliomas: Long-Term Results. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 732–738. [Google Scholar] [CrossRef]
- Yahara, K.; Ohguri, T.; Udono, H.; Yamamoto, J.; Tomura, K.; Onoda, T.; Imada, H.; Nishizawa, S.; Korogi, Y. Radiotherapy using IMRT boosts after hyperbaric oxygen therapy with chemotherapy for glioblastoma. J. Radiat. Res. 2017, 58, 351–356. [Google Scholar] [CrossRef]







| Signature | Gene | Pathways | lncRNA | Protein |
|---|---|---|---|---|
| CD9 | EGFR | DLK1 | lncRNA H19 | HILPDA (HIG2) |
| SP | TP53 mutation | Notch (CBF1) | ||
| CD133 | IDH mutation | VEGF | ||
| Olig2 | MCT4 | JAK1//2-STAT3 | ||
| integrin αβ | PP2A | Wnt (TCF-1, LEF-1) | ||
| ALDH | Klf4 | avβ8-integrin-TGF-β1 | ||
| CD44 | ABCB1 | |||
| Sox2 | PTEN | |||
| Oct4 | PML | |||
| nestin |
| Post-Surgical Recurrence | Radio Resistance | Chemo Resistance |
|---|---|---|
| GSC infiltrate proximate normal tissues | cell cycles alter | DNA double-strand break upregulates |
| tumor vascularization upregulates | cell cycle-related proteins alter | p38-ERK1/2 axis increases |
| diffusion around proximate tissues | expression of Notch increases | COX2 elevates |
| GSC produces insulin-like growth factor 1 (IGF1) | multidrug resistance-associated protein 1 (MRP1) | |
| DNA-damage response activates via musashi-1 |
| Agent | Mechanism | Function | Reference |
|---|---|---|---|
| total flavonoid of Eucommia ulmoides | downregulates HIF-a/MMP-2 pathway and upregulates apoptosis | increase effect of GBM radiotherapy | (Wang et al., 2019 [140]) |
| Olaparib | a promising radiosensitizer | improves prognosis of GBM patients | (Lesueur et al., 2019 [141]) |
| Olaparib plus temozolomide | combined with intensity modulated radiotherapy | spares healthy tissues and preserves neurocognitive functions to improve prognosis of GBM patients | (Lesueur et al., 2019 [141]) |
| nivolumab | a PD-1 inhibitor associates with PTEN mutation and MAPK enrichment | displays therapeutic efficacy of GBM | (Zhao et al., 2019 [35]) |
| pembrolizumab | a PD-1 inhibitor associates with PTEN mutation and MAPK enrichment | displays therapeutic efficacy of GBM | (Hsu et al., 2020 [75]) |
| nanoparticles | penetrates GBM niche and combines with chemo-, radio- and photodynamic therapies | displays therapeutic efficacy of GBM | (Yang et al., 2021 [142]) |
| Definition | Category | Benefit | Side-Effect | Difficulty | Reference |
|---|---|---|---|---|---|
| An adjuvant therapy to chemo-and-radio therapy in post-surgical GBM patients | radiotherapy during HBOT | prolong survival span of GBM patients | conclusive seizure and radiation-correlated necrosis | radiation establishment and underlying damage to normal tissues surrounded | (Chang, 1977 [148]; Ogawa et al., 2013 [149]) |
| radiation within 15 min after HBOT | improve prognoses of GBM patients, with progression-free survival rate reaching 46.5% | cause no late toxicities in GBM | requires more clinical validation | (Ogawa et al., 2012 [150]; Yahara et al., 2017 [151]) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, T.; Zhu, J.; Zhang, X.; Mao, X. The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma. Cancers 2023, 15, 2613. https://doi.org/10.3390/cancers15092613
Shi T, Zhu J, Zhang X, Mao X. The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma. Cancers. 2023; 15(9):2613. https://doi.org/10.3390/cancers15092613
Chicago/Turabian StyleShi, Tingyu, Jun Zhu, Xiang Zhang, and Xinggang Mao. 2023. "The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma" Cancers 15, no. 9: 2613. https://doi.org/10.3390/cancers15092613
APA StyleShi, T., Zhu, J., Zhang, X., & Mao, X. (2023). The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma. Cancers, 15(9), 2613. https://doi.org/10.3390/cancers15092613