
Understanding the Tumor Immune Microenvironment in Renal Cell Carcinoma

 ,
,  , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
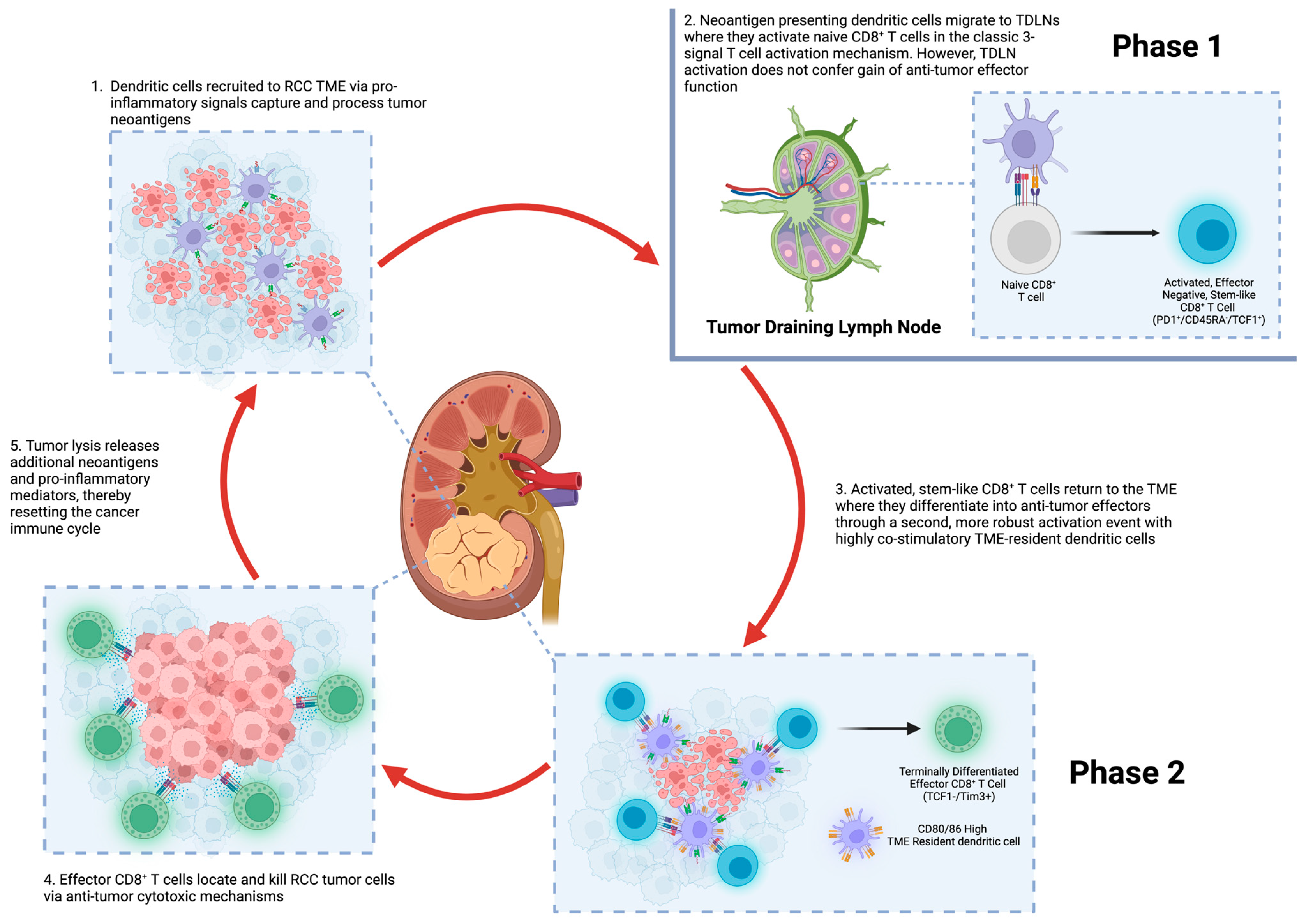
2. The Immune Response to Cancer
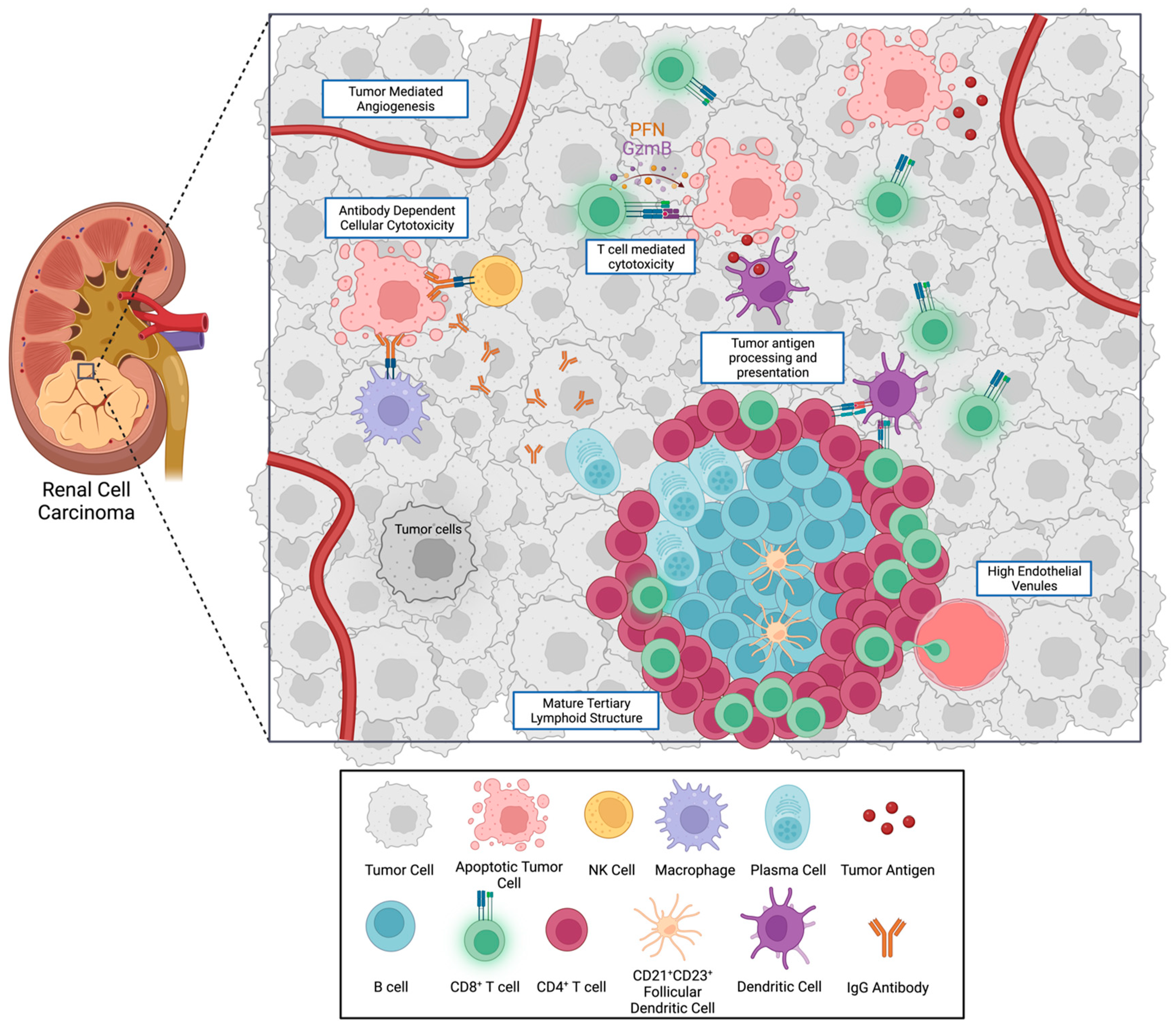
3. Cell Types of the Tumor Immune Microenvironment
4. Characteristics of the Renal Cell Carcinoma Tumor Immune Microenvironment
4.1. Immune Microenvironment Changes during Tumor Development and Treatment
4.2. Renal Cell Carcinoma Tumor Antigens and Genomic Correlations with Immune Response
4.3. The Predictive and Prognostic Capability of the Tumor Immune Microenvironment in RCC
5. Future Directions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seer Cancer Stat Facts: Kidney and Renal Pelvis Cancer. National Cancer Institute: Bethesda, MD, USA, 2022. Available online: https://seer.Cancer.Gov/statfacts/html/kidrp.Html (accessed on 5 April 2023).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Mattila, K.E.; Laajala, T.D.; Tornberg, S.V.; Kilpeläinen, T.P.; Vainio, P. A three-feature prediction model for metastasis-free survival after surgery of localized clear cell renal cell carcinoma. Sci. Rep. 2021, 11, 8650. [Google Scholar] [CrossRef] [PubMed]
- Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Lohse, C.M.; Frank, I.; Kwon, E.D.; Weaver, A.L.; Parker, A.S.; Zincke, H. Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003, 97, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall Survival and Updated Results for Sunitinib Compared with Interferon Alfa in Patients With Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Aren Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F.; et al. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 checkmate 025 trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Chang, Y.-H.; Hajek, J.; Symeonides, S.N.; Lee, J.L.; Sarwar, N.; et al. Adjuvant Pembrolizumab after Nephrectomy in Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 385, 683–694. [Google Scholar] [CrossRef]
- Powles, T.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Symeonides, S.N.; Hajek, J.; Gurney, H.; Chang, Y.-H.; Lee, J.L.; et al. Pembrolizumab versus placebo as post-nephrectomy adjuvant therapy for clear cell renal cell carcinoma (KEYNOTE-564): 30-month follow-up analysis of a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022, 23, 1133–1144. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Germain, C.; de Reyniès, A.; Laurent-Puig, P.; Zucman-Rossi, J.; Dieu-Nosjean, M.-C.; Sautès-Fridman, C.; Fridman, W.H. Immune Contexture, Immunoscore, and Malignant Cell Molecular Subgroups for Prognostic and Theranostic Classifications of Cancers. Adv. Immunol. 2016, 130, 95–190. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, R.G.; Hakimi, A.A.; Chan, T.A. Genomics-based immuno-oncology: Bridging the gap between immunology and tumor biology. Hum. Mol. Genet. 2020, 29, R214–R225. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Prokhnevska, N.; Cardenas, M.A.; Valanparambil, R.M.; Sobierajska, E.; Barwick, B.G.; Jansen, C.; Moon, A.R.; Gregorova, P.; Delbalzo, L.; Greenwald, R.; et al. CD8+ T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity 2022, 56, 107–124. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Braun, D.A.; Street, K.; Burke, K.P.; Cookmeyer, D.L.; Denize, T.; Pedersen, C.B.; Gohil, S.H.; Schindler, N.; Pomerance, L.; Hirsch, L.; et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 2021, 39, 632–648.e8. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics 2023, 13, 1607–1631. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Vano, Y.; Giraldo, N.A.; Fridman, W.H.; Sautès-Fridman, C. Oncoimmunology, A Practical Guide for Cancer Immunotherapy; Springer: Berlin/Heidelberg, Germany, 2017; pp. 5–21. [Google Scholar]
- Nixon, B.G.; Kuo, F.; Ji, L.; Liu, M.; Capistrano, K.; Do, M.; Franklin, R.A.; Wu, X.; Kansler, E.R.; Srivastava, R.M.; et al. Tumor-associated macrophages expressing the transcription factor IRF8 promote T cell exhaustion in cancer. Immunity 2022, 55, 2044–2058.e5. [Google Scholar] [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e6. [Google Scholar] [CrossRef]
- Shen, H.; Liu, J.; Chen, S.; Ma, X.; Ying, Y.; Li, J.; Wang, W.; Wang, X.; Xie, L. Prognostic Value of Tumor-Associated Macrophages in Clear Cell Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 657318. [Google Scholar] [CrossRef]
- Conejo-Garcia, J.R.; Rutkowski, M.R.; Cubillos-Ruiz, J.R. State-of-the-art of regulatory dendritic cells in cancer. Pharmacol. Ther. 2016, 164, 97–104. [Google Scholar] [CrossRef]
- Verneau, J.; Sautés-Fridman, C.; Sun, C.-M. Dendritic cells in the tumor microenvironment: Prognostic and theranostic impact. Semin. Immunol. 2020, 48, 101410. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Varn, F.S.; Wang, Y.; Mullins, D.W.; Fiering, S.; Cheng, C. Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer Res. 2017, 77, 1271–1282. [Google Scholar] [CrossRef]
- Wang, T.; Lu, R.; Kapur, P.; Jaiswal, B.S.; Hannan, R.; Zhang, Z.; Pedrosa, I.; Luke, J.J.; Zhang, H.; Goldstein, L.D.; et al. An Empirical Approach Leveraging Tumorgrafts to Dissect the Tumor Microenvironment in Renal Cell Carcinoma Identifies Missing Link to Prognostic Inflammatory Factors. Cancer Discov. 2018, 8, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition—Novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 2021, 18, 199–214. [Google Scholar] [CrossRef] [PubMed]
- McRitchie, B.R.; Akkaya, B. Exhaust the exhausters: Targeting regulatory t cells in the tumor microenvironment. Front. Immunol. 2022, 13, 940052. [Google Scholar] [CrossRef] [PubMed]
- Ahrends, T.; Borst, J. The opposing roles of cd4+ t cells in anti-tumour immunity. Immunology 2018, 154, 582–592. [Google Scholar] [CrossRef]
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A. Interleukin-35 limits anti-tumor immunity. Immunity 2016, 44, 316–329. [Google Scholar] [CrossRef]
- Van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. Cd8+ t cell states in human cancer: Insights from single-cell analysis. Nature reviews. Cancer 2020, 20, 218–232. [Google Scholar]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.-M.; Italiano, A.; Sautès-Fridman, C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef]
- Downs-Canner, S.M.; Meier, J.; Vincent, B.G.; Serody, J.S. B Cell Function in the Tumor Microenvironment. Annu. Rev. Immunol. 2022, 40, 169–193. [Google Scholar] [CrossRef]
- Lauss, M.; Donia, M.; Svane, I.M.; Jönsson, G. B cells and tertiary lymphoid structures: Friends or foes in cancer immunotherapy? Clin. Cancer Res. 2022, 28, 1751–1758. [Google Scholar] [CrossRef]
- Iglesia, M.D.; Parker, J.S.; Hoadley, K.; Serody, J.S.; Perou, C.; Vincent, B.G. Genomic Analysis of Immune Cell Infiltrates Across 11 Tumor Types. J. Natl. Cancer Inst. 2016, 108, djw144. [Google Scholar] [CrossRef]
- Murakami, Y.; Saito, H.; Shimizu, S.; Kono, Y.; Shishido, Y.; Miyatani, K.; Matsunaga, T.; Fukumoto, Y.; Ashida, K.; Sakabe, T.; et al. Increased regulatory B cells are involved in immune evasion in patients with gastric cancer. Sci. Rep. 2019, 9, 13083. [Google Scholar] [CrossRef]
- Meylan, M.; Petitprez, F.; Becht, E.; Bougoüin, A.; Pupier, G.; Calvez, A.; Giglioli, I.; Verkarre, V.; Lacroix, G.; Verneau, J.; et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 2022, 55, 527–541.e5. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Şenbabaoğlu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; Van Allen, E.M.; de Velasco, G.; Miao, D.; Ostrovnaya, I.; Drill, E.; Luna, A.; et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Cubas, A.A.D.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef]
- Li, R.; Ferdinand, J.R.; Loudon, K.W.; Bowyer, G.S.; Laidlaw, S.; Muyas, F.; Mamanova, L.; Neves, J.B.; Bolt, L.; Fasouli, E.S.; et al. Mapping single-cell transcriptomes in the intra-tumoral and associated territories of kidney cancer. Cancer Cell 2022, 40, 1583–1599.e10. [Google Scholar] [CrossRef]
- Calderaro, J.; Petitprez, F.; Becht, E.; Laurent, A.; Hirsch, T.Z.; Rousseau, B.; Luciani, A.; Amaddeo, G.; Derman, J.; Charpy, C.; et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J. Hepatol. 2019, 70, 58–65. [Google Scholar] [CrossRef]
- Moussion, C.; Girard, J.-P. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature 2011, 479, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.-C.; Goc, J.; Giraldo, N.A.; Sautès-Fridman, C.; Fridman, W.H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014, 35, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Goc, J.; Fridman, W.-H.; Sautès-Fridman, C.; Dieu-Nosjean, M.-C. Characteristics of tertiary lymphoid structures in primary cancers. Oncoimmunology 2013, 2, e26836. [Google Scholar] [CrossRef] [PubMed]
- Johansson-Percival, A.; He, B.; Li, Z.-J.; Kjellén, A.; Russell, K.; Li, J.; Larma, I.; Ganss, R. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat. Immunol. 2017, 18, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Johansson-Percival, A.; Ganss, R. Therapeutic Induction of Tertiary Lymphoid Structures in Cancer Through Stromal Remodeling. Front. Immunol. 2021, 12, 674375. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G.; Durando, M.; Cooke, M. CXCL13 and Its Receptor CXCR5 in Cancer: Inflammation, Immune Response, and Beyond. Front. Endocrinol. 2019, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Dimberg, A.; Verma, V. Editorial: Tertiary Lymphoid Structures: From Basic Biology to Translational Impact in Cancer. Front. Immunol. 2022, 13, 870862. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, eabf9419. [Google Scholar] [CrossRef]
- Vaghjiani, R.G.; Skitzki, J.J. Tertiary Lymphoid Structures as Mediators of Immunotherapy Response. Cancers 2022, 14, 3748. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.J.; Dhanasekaran, S.M.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.-S.M.; Chang, H.-Y.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179, 964–983.e31. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Voss, M.H.; Kuo, F.; Sanchez, A.; Liu, M.; Nixon, B.G.; Vuong, L.; Ostrovnaya, I.; Chen, Y.-B.; Reuter, V.; et al. Transcriptomic Profiling of the Tumor Microenvironment Reveals Distinct Subgroups of Clear Cell Renal Cell Cancer: Data from a Randomized Phase III Trial. Cancer Discov. 2019, 9, 510–525. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Angelo, M.S.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef]
- Remark, R.; Alifano, M.; Cremer, I.; Lupo, A.; Dieu-Nosjean, M.-C.; Riquet, M.; Crozet, L.; Ouakrim, H.; Goc, J.; Cazes, A.; et al. Characteristics and Clinical Impacts of the Immune Environments in Colorectal and Renal Cell Carcinoma Lung Metastases: Influence of Tumor Origin. Clin. Cancer Res. 2013, 19, 4079–4091. [Google Scholar] [CrossRef]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A.; et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 2021, 39, 649–661.e5. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bossé, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor Microenvironment Dynamics in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.D.; Virumbrales-Muñoz, M.; Beebe, D.J.; Abel, E.J. Models of Renal Cell Carcinoma Used to Investigate Molecular Mechanisms and Develop New Therapeutics. Front. Oncol. 2022, 12, 871252. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef]
- Hoefflin, R.; Harlander, S.; Schäfer, S.; Metzger, P.; Kuo, F.; Schönenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.-Y.; et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, L.; Xia, Y.; Qi, Y.; Chen, Y.; Chen, L.; Zhang, P.; Kong, Y.; Qu, Y.; Wang, Z.; et al. Tumor infiltrating mast cells determine oncogenic HIF-2α-conferred immune evasion in clear cell renal cell carcinoma. Cancer Immunol. Immunother. 2019, 68, 731–741. [Google Scholar] [CrossRef]
- Liu, X.-D.; Kong, W.; Peterson, C.B.; McGrail, D.J.; Hoang, A.; Zhang, X.; Lam, T.; Pilie, P.G.; Zhu, H.; Beckermann, K.E.; et al. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat. Commun. 2020, 11, 2135. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Carlisle, J.W.; Jansen, C.S.; Cardenas, M.A.; Sobierajska, E.; Reyes, A.M.; Greenwald, R.; Del Balzo, L.; Prokhnevska, N.; Kucuk, O.; Carthon, B.C.; et al. Clinical outcome following checkpoint therapy in renal cell carcinoma is associated with a burst of activated CD8 T cells in blood. J. Immunother. Cancer 2022, 10, e004803. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Pagès, F.; Skliris, G.P.; Verkarre, V.; Vano, Y.; Mejean, A.; Saint-Aubert, N.; Lacroix, L.; Natario, I.; et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin. Cancer Res. 2015, 21, 3031–3040. [Google Scholar] [CrossRef]
- Ghatalia, P.; Gordetsky, J.; Kuo, F.; Dulaimi, E.; Cai, K.Q.; Devarajan, K.; Bae, S.; Naik, G.; Chan, T.A.; Uzzo, R.; et al. Prognostic impact of immune gene expression signature and tumor infiltrating immune cells in localized clear cell renal cell carcinoma. J. Immunother. Cancer 2019, 7, 139. [Google Scholar] [CrossRef]
- Cotta, B.H.; Choueiri, T.K.; Cieslik, M.; Ghatalia, P.; Mehra, R.; Morgan, T.M.; Palapattu, G.S.; Shuch, B.; Vaishampayan, U.; Van Allen, E.; et al. Current Landscape of Genomic Biomarkers in Clear Cell Renal Cell Carcinoma. Eur. Urol. 2023, in press. [CrossRef]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.-F.; Leng, N.; Abbas, A.R.; et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 2020, 38, 803–817.e4. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef]
- Xu, Y.; Miller, C.P.; Warren, E.H.; Tykodi, S.S. Current status of antigen-specific T-cell immunotherapy for advanced renal-cell carcinoma. Hum. Vaccines Immunother. 2021, 17, 1882–1896. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Routy, B.; le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Park, E.M.; Chelvanambi, M.; Bhutiani, N.; Kroemer, G.; Zitvogel, L.; Wargo, J.A. Targeting the gut and tumor microbiota in cancer. Nat. Med. 2022, 28, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Dizman, N.; Meza, L.; Bergerot, P.; Alcantara, M.; Dorff, T.; Lyou, Y.; Frankel, P.; Cui, Y.; Mira, V.; Llamas, M.; et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: A randomized phase 1 trial. Nat. Med. 2022, 28, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Molina-Cerrillo, J.; Santoni, G.; Lam, E.T.; Massari, F.; Mollica, V.; Mazzaschi, G.; Rapoport, B.L.; Grande, E.; Buti, S. Role of Clock Genes and Circadian Rhythm in Renal Cell Carcinoma: Recent Evidence and Therapeutic Consequences. Cancers 2023, 15, 408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | RCC Subtype | Stages Studied | Method of Categorization | Tumor Microenvironment Categorization | Category Characteristics | Category Prognosis |
|---|---|---|---|---|---|---|
| Senbabaoglu et al., 2016 [48] | Clear cell | All Stages | TCGA based gene expression signatures | Non-infiltrated | Low T cell infiltration, low stromal score, increased metabolism and mitochondrial related genes | Best |
| Heterogeneously Infiltrated | Increased angiogenesis-related gene expression, c-KIT and Smad1 | Intermediate | ||||
| T cell Enriched | High T cell infiltration, high granzyme B and IFNγ expression | Worst | ||||
| Giraldo et al., 2017 [66] | Clear cell | Localized RCC | Multiparametric flow cytometric immunophenotypic analysis | Immune activated | Increased CD8+ clonality, increased cytotoxic genes | Best |
| Immune silent | Low levels tumor infiltrating lymphocytes | Intermediate | ||||
| Immune regulated | M2-rich, poorly cytotoxic, increased Treg | Worst | ||||
| Clark et al., 2019 [65] | Clear cell | All Stages | Transcriptomic and proteomic microenvironment signatures | VEGF immune desert | Elevated stromal score, endothelial enrichment | Best |
| CD8- Inflamed | Innate immune signature, fibroblast signature | Intermediate | ||||
| Metabolic immune desert | Low immune and stromal scores, elevated mitochondrial, OXPHOS, glycolysis protein expression | Intermediate | ||||
| CD8+ inflamed | High CD8+ infiltration, BAP1 mutations, CD38 expression, IFNγ signaling | Worst | ||||
| Hakimi et al., 2019 [67] | Clear cell | Metastatic | Somatic mutation analysis, RNA and protein expression | Cluster 3 | High PBRM1 mutations, high angiogenesis score, moderate immune infiltration | Best |
| Cluster 2 | Moderate angiogenesis score, moderate immune infiltration | Intermediate | ||||
| Cluster 1 | Lowest angiogenesis score, lowest immune infiltration | Intermediate | ||||
| Cluster 4 | High BAP1 mutation, moderate angiogenesis score, high immune infiltration, higher PD-L1 expression | Worst | ||||
| Braun et al., 2020 [68] | Clear cell | Metastatic | Integrated genetic, transcriptomic and immunopathologic analysis | Immune Excluded | 5-fold more CD8+ T cells at the tumor margin than in tumor center | No difference in prognosis |
| Immune Desert | Not excluded and below 25th percentile for CD8+ T cells, 50 cells/mm2 in the tumor center | |||||
| Immune Infiltrated | Not excluded and ≥25th percentile for CD8+ T cells in the tumor center. Enriched for M1 macrophages, CD4+ memory T cells, NK cells |
| Biomarker | Description | Investigational Use | Limitations |
|---|---|---|---|
| PD-L1 [6,10,84] | Cell surface protein |
|
|
| Tumor Mutational Burden [80] | Number of non-synonymous somatic mutations in tumor DNA |
|
|
| Tumor Neoantigen Burden [80] | Number of immunogenic tumor-specific proteins (neoantigens) predicted by quantity of somatic mutations |
|
|
| Renal 101 Immuno signature [85] | 26-gene immune based signature |
|
|
| T-effector score [80] | Five gene signature (CD8A, EOMES, PRF1, IFNG, CD274) |
|
|
| Myeloid inflammation [80] | Six gene signature (IL-6, CXCL1, CXCL2, CXCL3, CXCL8, PTGS2) |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shapiro, D.D.; Dolan, B.; Laklouk, I.A.; Rassi, S.; Lozar, T.; Emamekhoo, H.; Wentland, A.L.; Lubner, M.G.; Abel, E.J. Understanding the Tumor Immune Microenvironment in Renal Cell Carcinoma. Cancers 2023, 15, 2500. https://doi.org/10.3390/cancers15092500
Shapiro DD, Dolan B, Laklouk IA, Rassi S, Lozar T, Emamekhoo H, Wentland AL, Lubner MG, Abel EJ. Understanding the Tumor Immune Microenvironment in Renal Cell Carcinoma. Cancers. 2023; 15(9):2500. https://doi.org/10.3390/cancers15092500
Chicago/Turabian StyleShapiro, Daniel D., Brendan Dolan, Israa A. Laklouk, Sahar Rassi, Taja Lozar, Hamid Emamekhoo, Andrew L. Wentland, Meghan G. Lubner, and Edwin Jason Abel. 2023. "Understanding the Tumor Immune Microenvironment in Renal Cell Carcinoma" Cancers 15, no. 9: 2500. https://doi.org/10.3390/cancers15092500
APA StyleShapiro, D. D., Dolan, B., Laklouk, I. A., Rassi, S., Lozar, T., Emamekhoo, H., Wentland, A. L., Lubner, M. G., & Abel, E. J. (2023). Understanding the Tumor Immune Microenvironment in Renal Cell Carcinoma. Cancers, 15(9), 2500. https://doi.org/10.3390/cancers15092500