
Obesity Is Associated with Immunometabolic Changes in Adipose Tissue That May Drive Treatment Resistance in Breast Cancer: Immune-Metabolic Reprogramming and Novel Therapeutic Strategies

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Immunometabolic Changes in Adipose Tissue of Patients with Obesity
3. Innate Immunity in Adipose Tissue of Patients with Obesity
4. Adaptive Immunity in Adipose Tissue of Patients with Obesity
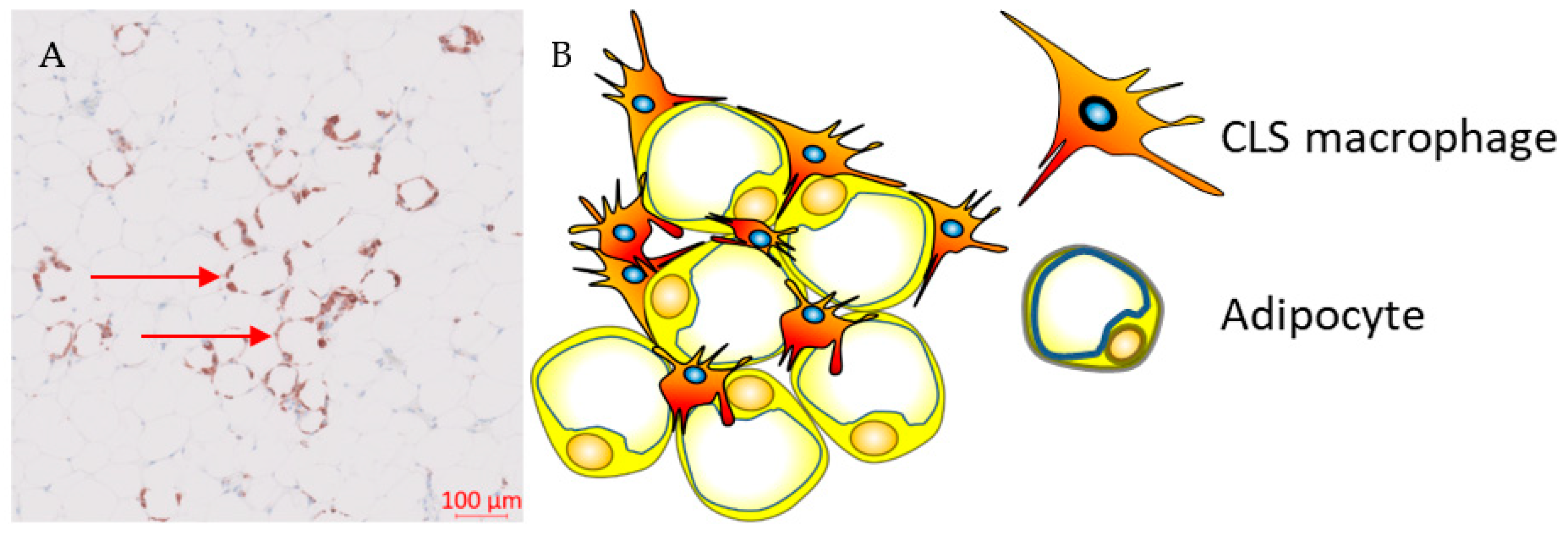
5. Adipose Tissue Macrophages and Breast Cancer
6. BMI and Clinical Outcomes in Different Breast Cancer Subtypes and Responses to Treatment
7. HER2+ Breast Cancer
8. Oestrogen Receptor Negative Breast Cancer
9. Oestrogen Receptor Positive Breast Cancer
10. Adiposity and Response to Cancer Immunotherapy in Breast Cancer
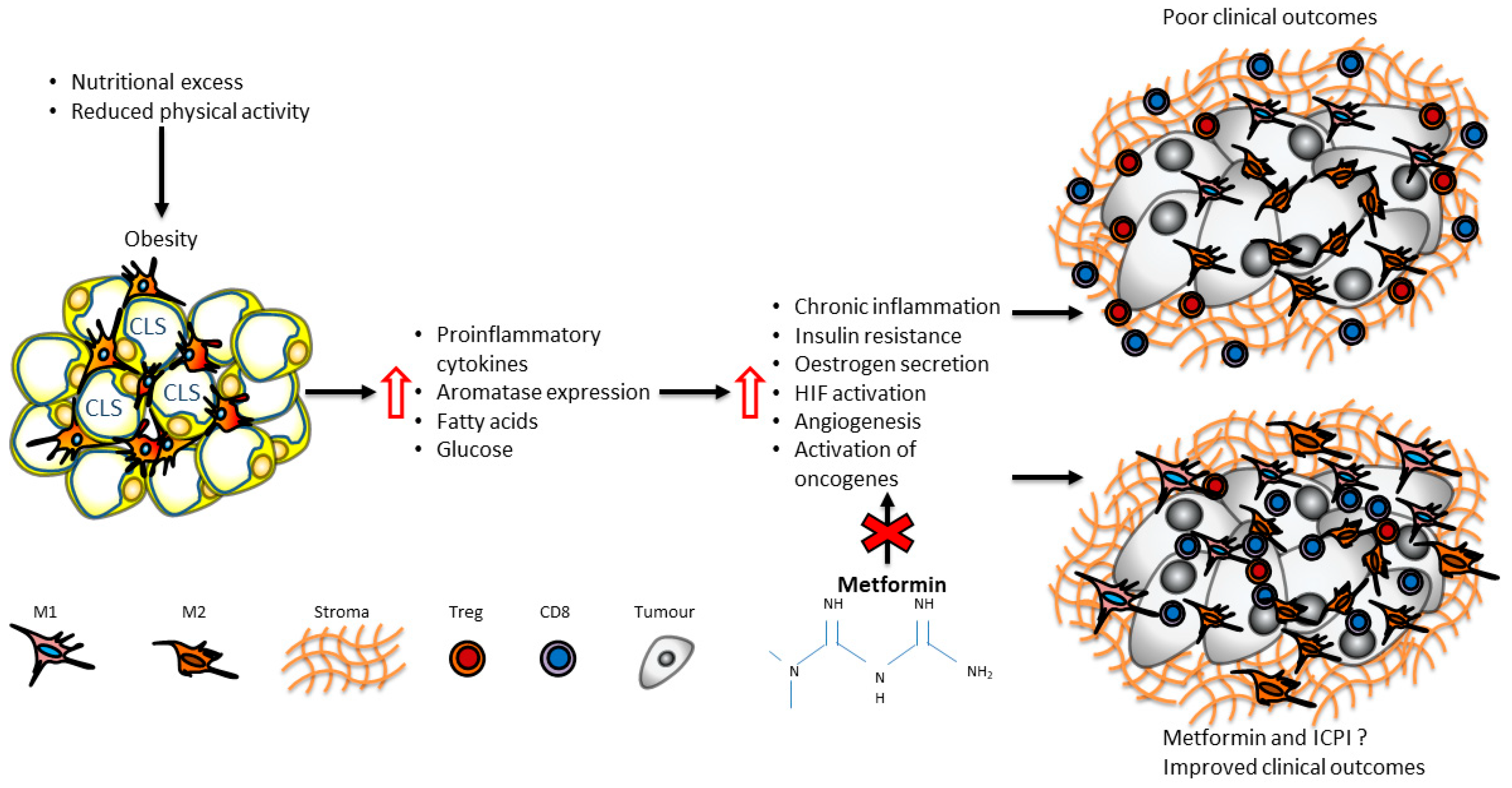
11. Metabolic Interventions in Immunometabolic Reprograming in Breast Cancer: The Paradigm of Metformin
12. Future Directions
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Jeibouei, S.; Akbari, M.E.; Kalbasi, A.; Aref, A.R.; Ajoudanian, M.; Rezvani, A.; Zali, H. Personalized medicine in breast cancer: Pharmacogenomics approaches. Pharm. Pers. Med. 2019, 12, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [PubMed]
- James, F.R.; Wootton, S.; Jackson, A.; Wiseman, M.; Copson, E.R.; Cutress, R.I. Obesity in breast cancer—What is the risk factor? Eur. J. Cancer 2015, 51, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Vieira, A.; Aune, D.; Bandera, E.; Greenwood, D.; McTiernan, A.; Rosenblatt, D.N.; Thune, I.; Norat, T. Body mass index and survival in women with breast cancer—Systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 2014, 25, 1901–1914. [Google Scholar] [CrossRef]
- Lebiedowska, A.; Hartman-Petrycka, M.; Błońska-Fajfrowska, B. How reliable is BMI? Bioimpedance analysis of body composition in underweight, normal weight, overweight, and obese women. Ir. J. Med. Sci. 2020, 190, 993–998. [Google Scholar] [CrossRef]
- Christ, A.; Latz, E. The Western lifestyle has lasting effects on metaflammation. Nat. Rev. Immunol. 2019, 19, 267–268. [Google Scholar] [CrossRef]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef]
- Qu, L.; Matz, A.J.; Karlinsey, K.; Cao, Z.; Vella, A.T.; Zhou, B. Macrophages at the Crossroad of Meta-Inflammation and Inflammaging. Genes 2022, 13, 2074. [Google Scholar] [CrossRef]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef]
- Schmidt, V.; Hogan, A.E.; Fallon, P.G.; Schwartz, C. Obesity-Mediated Immune Modulation: One Step Forward, (Th)2 Steps Back. Front. Immunol. 2022, 13, 932893. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Martín-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. Oncoimmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Tapia, E. Reduction of Obesity Associated Breast Cancer Risk in a Phase II Clinical Trial of Metformin; The University of Arizona: Tucson, AZ, USA, 2020. [Google Scholar]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef]
- Torres, N.; Vargas-Castillo, A.E.; Tovar, A.R. Adipose Tissue: White Adipose Tissue Structure and Function. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 35–42. [Google Scholar]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Musiyenko, S.I.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA Damage and Dysfunction, and Oxidative Stress Are Associated with Endoplasmic Reticulum Stress, Protein Degradation and Apoptosis in High Fat Diet-Induced Insulin Resistance Mice. PLoS ONE 2013, 8, e54059. [Google Scholar] [CrossRef]
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed]
- Lolmède, K.; Front, V.D.D.S.; Galitzky, J.; Lafontan, M.; Bouloumié, A. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int. J. Obes. 2003, 27, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Hosoya, Y.; Yamashita, H.; Fujita, H.; Ohsugi, M.; Tobe, K.; Kadowaki, T.; Nagai, R.; et al. Adipogenesis in Obesity Requires Close Interplay Between Differentiating Adipocytes, Stromal Cells, and Blood Vessels. Diabetes 2007, 56, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Nocon, A.; Fry, J.; Sherban, A.; Rui, X.; Jiang, B.; Xu, X.J.; Han, J.; Yan, Y.; Yang, Q.; et al. AMPK Activation by Metformin Suppresses Abnormal Extracellular Matrix Remodeling in Adipose Tissue and Ameliorates Insulin Resistance in Obesity. Diabetes 2016, 65, 2295–2310. [Google Scholar] [CrossRef]
- Kubo, H.; Sawada, S.; Satoh, M.; Asai, Y.; Kodama, S.; Sato, T.; Tomiyama, S.; Seike, J.; Takahashi, K.; Kaneko, K.; et al. Insulin-like growth factor-1 levels are associated with high comorbidity of metabolic disorders in obese subjects; a Japanese single-center, retrospective-study. Sci. Rep. 2022, 12, 20130. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Bagchi, D.P.; Nishii, A.; Li, Z.; DelProposto, J.B.; Corsa, C.A.; Mori, H.; Hardij, J.; Learman, B.S.; Lumeng, C.N.; MacDougald, O.A. Wnt/β-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Mol. Metab. 2020, 42, 101078. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2018, 25, 141–151. [Google Scholar] [CrossRef]
- Ouldamer, L.; Jourdan, M.-L.; Pinault, M.; Arbion, F.; Goupille, C. Accumulation of Arachidonic Acid, Precursor of Pro-Inflammatory Eicosanoids, in Adipose Tissue of Obese Women: Association with Breast Cancer Aggressiveness Indicators. Biomedicines 2022, 10, 995. [Google Scholar] [CrossRef]
- Marchand, G.B.; Carreau, A.-M.; Weisnagel, S.J.; Bergeron, J.; Labrie, F.; Lemieux, S.; Tchernof, A. Increased body fat mass explains the positive association between circulating estradiol and insulin resistance in postmenopausal women. Am. J. Physiol. Metab. 2018, 314, E448–E456. [Google Scholar] [CrossRef]
- Pasquali, R. Obesity and androgens: Facts and perspectives. Fertil. Steril. 2006, 85, 1319–1340. [Google Scholar] [CrossRef]
- Richard, C.; Wadowski, M.; Goruk, S.; Cameron, L.; Sharma, A.M.; Field, C.J. Individuals with obesity and type 2 diabetes have additional immune dysfunction compared with obese individuals who are metabolically healthy. BMJ Open Diabetes Res. Care 2017, 5, e000379. [Google Scholar] [CrossRef]
- Sheridan, P.A.; Paich, H.A.; Handy, J.; Karlsson, E.A.; Hudgens, M.G.; Sammon, A.B.; Holland, L.A.; Weir, S.; Noah, T.L.; Beck, M.A. Obesity is associated with impaired immune response to influenza vaccination in humans. Int. J. Obes. 2012, 36, 1072–1077. [Google Scholar] [CrossRef]
- Harris, B.H.L.; Macaulay, V.M.; Harris, D.A.; Klenerman, P.; Karpe, F.; Lord, S.R.; Harris, A.L.; Buffa, F.M. Obesity: A perfect storm for carcinogenesis. Cancer Metastasis Rev. 2022, 41, 491–515. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Xu, X.; Grijalva, A.; Skowronski, A.; van Eijk, M.; Serlie, M.J.; Ferrante, A.W. Obesity Activates a Program of Lysosomal-Dependent Lipid Metabolism in Adipose Tissue Macrophages Independently of Classic Activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef]
- Zuany-Amorim, C.; Hastewell, J.; Walker, C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat. Rev. Drug Discov. 2002, 1, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2018, 15, 139–154. [Google Scholar] [CrossRef]
- Birts, C.N.; Savva, C.; Laversin, S.A.; Lefas, A.; Krishnan, J.; Schapira, A.; Ashton-Key, M.; Crispin, M.; Johnson, P.W.M.; Blaydes, J.P.; et al. Prognostic significance of crown-like structures to trastuzumab response in patients with primary invasive HER2 + breast carcinoma. Sci. Rep. 2022, 12, 7802. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Perrard, X.D.; Wang, Q.; Perrard, J.L.; Polsani, V.R.; Jones, P.H.; Smith, C.W.; Ballantyne, C.M. CD11c Expression in Adipose Tissue and Blood and Its Role in Diet-Induced Obesity. Arter. Thromb. Vasc. Biol. 2010, 30, 186–192. [Google Scholar] [CrossRef]
- Nakajima, S.; Koh, V.; Kua, L.F.; So, J.; Davide, L.; Lim, K.S.; Petersen, S.H.; Yong, W.-P.; Shabbir, A.; Kono, K. Accumulation of CD11c+CD163+ Adipose Tissue Macrophages through Upregulation of Intracellular 11beta-HSD1 in Human Obesity. J. Immunol. 2016, 197, 3735–3745. [Google Scholar] [CrossRef]
- Wentworth, J.M.; Naselli, G.; Brown, W.A.; Doyle, L.; Phipson, B.; Smyth, G.K.; Wabitsch, M.; O’Brien, P.E.; Harrison, L.C. Pro-Inflammatory CD11c+CD206+ Adipose Tissue Macrophages Are Associated with Insulin Resistance in Human Obesity. Diabetes 2010, 59, 1648–1656. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Morris, P.G.; Howe, L.R.; Giri, D.D.; Morrow, M.; Wang, H.; Pollak, M.; Jones, L.W.; et al. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin. Cancer Res. 2016, 22, 2283–2289. [Google Scholar] [CrossRef]
- Vaysse, C.; Lømo, J.; Garred, Ø.; Fjeldheim, F.; Lofteroed, T.; Schlichting, E.; McTiernan, A.; Frydenberg, H.; Husøy, A.; Lundgren, S.; et al. Inflammation of mammary adipose tissue occurs in overweight and obese patients exhibiting early-stage breast cancer. NPJ Breast Cancer 2017, 3, 19. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Chen, I.-C.; Zhou, X.K.; Giri, D.D.; Falcone, D.J.; Winston, L.A.; Wang, H.; Williams, S.; Lu, Y.-S.; Hsueh, T.-H.; et al. Adiposity, Inflammation, and Breast Cancer Pathogenesis in Asian Women. Cancer Prev. Res. 2018, 11, 227–236. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Brown, K.A.; Zhou, X.K.; Gucalp, A.; Subbaramaiah, K.; Giri, D.D.; Zahid, H.; Bhardwaj, P.; Wendel, N.K.; Falcone, D.J.; et al. Metabolic Obesity, Adipose Inflammation and Elevated Breast Aromatase in Women with Normal Body Mass Index. Cancer Prev. Res. 2017, 10, 235–243. [Google Scholar] [CrossRef]
- Cha, Y.J.; Kim, E.-S.; Koo, J.S. Tumor-associated macrophages and crown-like structures in adipose tissue in breast cancer. Breast Cancer Res. Treat. 2018, 170, 15–25. [Google Scholar] [CrossRef]
- Koru-Sengul, T.; Santander, A.M.; Miao, F.; Sanchez, L.G.; Jorda, M.; Glück, S.; Ince, T.A.; Nadji, M.; Chen, Z.; Penichet, M.L.; et al. Breast cancers from black women exhibit higher numbers of immunosuppressive macrophages with proliferative activity and of crown-like structures associated with lower survival compared to non-black Latinas and Caucasians. Breast Cancer Res. Treat. 2016, 158, 113–126. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Ghossein, R.A.; Morris, L.G.; Zhou, X.K.; Kochhar, A.; Morris, P.G.; Pfister, D.G.; Patel, S.G.; Boyle, J.O.; Hudis, C.A.; et al. White adipose tissue inflammation and cancer-specific survival in patients with squamous cell carcinoma of the oral tongue. Cancer 2016, 122, 3794–3802. [Google Scholar] [CrossRef]
- Maliniak, M.L.; Cheriyan, A.M.; Sherman, M.E.; Liu, Y.; Gogineni, K.; Liu, J.; He, J.; Krishnamurti, U.; Miller-Kleinhenz, J.; Ashiqueali, R.; et al. Detection of crown-like structures in breast adipose tissue and clinical outcomes among African-American and White women with breast cancer. Breast Cancer Res. 2020, 22, 65. [Google Scholar] [CrossRef]
- Griner, S.E.; Wang, K.J.; Joshi, J.P.; Nahta, R. Mechanisms of Adipocytokine-Mediated Trastuzumab Resistance in HER2-Positive Breast Cancer Cell Lines. Curr. Pharm. Pers. Med. 2013, 11, 31–41. [Google Scholar] [CrossRef]
- Giordano, C.; Vizza, D.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Sisci, D.; De Amicis, F.; Fuqua, S.A.; Catalano, S.; et al. Leptin increases HER2 protein levels through a STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol. Oncol. 2012, 7, 379–391. [Google Scholar] [CrossRef]
- Soma, D.; Kitayama, J.; Yamashita, H.; Miyato, H.; Ishikawa, M.; Nagawa, H. Leptin Augments Proliferation of Breast Cancer Cells via Transactivation of HER2. J. Surg. Res. 2008, 149, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Fiorio, E.; Mercanti, A.; Terrasi, M.; Micciolo, R.; Remo, A.; Auriemma, A.; Molino, A.; Parolin, V.; Di Stefano, B.; Bonetti, F.; et al. Leptin/HER2 crosstalk in breast cancer: In vitro study and preliminary in vivo analysis. BMC Cancer 2008, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Subbaramaiah, K.; Wang, H.; Giri, D.D.; Morrow, M.; Falcone, D.J.; Wendel, N.K.; et al. Menopause Is a Determinant of Breast Aromatase Expression and Its Associations With BMI, Inflammation, and Systemic Markers. J. Clin. Endocrinol. Metab. 2017, 102, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Mullooly, M.; Yang, H.P.; Falk, R.T.; Nyante, S.J.; Cora, R.; Pfeiffer, R.M.; Radisky, D.C.; Visscher, D.W.; Hartmann, L.C.; Carter, J.M.; et al. Relationship between crown-like structures and sex-steroid hormones in breast adipose tissue and serum among postmenopausal breast cancer patients. Breast Cancer Res. 2017, 19, 8. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Du, B.; Zhou, X.K.; Sue, E.; Giri, D.; Harbus, M.D.; Falcone, D.J.; Hudis, C.A.; Subbaramaiah, K.; Dannenberg, A.J. Estrogen Protects against Obesity-Induced Mammary Gland Inflammation in Mice. Cancer Prev. Res. 2015, 8, 751–759. [Google Scholar] [CrossRef]
- Foley, J.F. Obesity and antitumor immunity. Sci. Signal. 2022, 15, eabq0080. [Google Scholar] [CrossRef]
- Krasniqi, E.; Pizzuti, L.; Barchiesi, G.; Sergi, D.; Carpano, S.; Botti, C.; Kayal, R.; Sanguineti, G.; Marchetti, P.; Botticelli, A.; et al. Impact of BMI on HER2+ metastatic breast cancer patients treated with pertuzumab and/or trastuzumab emtansine. Real-world evidence. J. Cell. Physiol. 2020, 235, 7900–7910. [Google Scholar] [CrossRef]
- Martel, S.; Poletto, E.; Ferreira, A.R.; Lambertini, M.; Sottotetti, F.; Bertolini, I.; Montemurro, F.; Bernardo, A.; Risi, E.; Zanardi, E.; et al. Impact of body mass index on the clinical outcomes of patients with HER2-positive metastatic breast cancer. Breast 2018, 37, 142–147. [Google Scholar] [CrossRef]
- Agresti, R.; Meneghini, E.; Baili, P.; Minicozzi, P.; Turco, A.; Cavallo, I.; Funaro, F.; Amash, H.; Berrino, F.; Tagliabue, E.; et al. Association of adiposity, dysmetabolisms, and inflammation with aggressive breast cancer subtypes: A cross-sectional study. Breast Cancer Res. Treat. 2016, 157, 179–189. [Google Scholar] [CrossRef]
- Alkhateeb, A.A.; Leitzel, K.; Ali, S.M.; Campbell-Baird, C.; Evans, M.; Fuchs, E.-M.; Köstler, W.J.; Lipton, A.; Connor, J. Elevation in Inflammatory Serum Biomarkers Predicts Response to Trastuzumab-Containing Therapy. PLoS ONE 2012, 7, e51379. [Google Scholar] [CrossRef]
- Korkaya, H.; Kim, G.-I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 Inflammatory Loop Mediates Trastuzumab Resistance in HER2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef]
- Liu, S.; Lee, J.S.; Jie, C.; Park, M.H.; Iwakura, Y.; Patel, Y.; Soni, M.; Reisman, D.; Chen, H. HER2 Overexpression Triggers an IL1α Proinflammatory Circuit to Drive Tumorigenesis and Promote Chemotherapy Resistance. Cancer Res. 2018, 78, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Harborg, S.; Zachariae, R.; Olsen, J.; Johannsen, M.; Cronin-Fenton, D.; Bøggild, H.; Borgquist, S. Overweight and prognosis in triple-negative breast cancer patients: A systematic review and meta-analysis. NPJ Breast Cancer 2021, 7, 119. [Google Scholar] [CrossRef]
- Heng, Y.J.; Wang, J.; Ahearn, T.U.; Brown, S.B.; Zhang, X.; Ambrosone, C.B.; de Andrade, V.P.; Brufsky, A.M.; Couch, F.J.; King, T.A.; et al. Molecular mechanisms linking high body mass index to breast cancer etiology in post-menopausal breast tumor and tumor-adjacent tissues. Breast Cancer Res. Treat. 2018, 173, 667–677. [Google Scholar] [CrossRef]
- Li, H.; Meng, Y.; He, S.; Tan, X.; Zhang, Y.; Zhang, X.; Wang, L.; Zheng, W. Macrophages, Chronic Inflammation, and Insulin Resistance. Cells 2022, 11, 2001. [Google Scholar] [CrossRef]
- Massihnia, D.; Galvano, A.; Fanale, D.; Perez, A.; Castiglia, M.; Incorvaia, L.; Listì, A.; Rizzo, S.; Cicero, G.; Bazan, V.; et al. Triple negative breast cancer: Shedding light onto the role of pi3k/akt/mtor pathway. Oncotarget 2016, 7, 60712–60722. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, L.; Zheng, H.; Zeng, Y. Cytokines secreted from adipose tissues mediate tumor proliferation and metastasis in triple negative breast cancer. BMC Cancer 2022, 22, 886. [Google Scholar] [CrossRef]
- Evangelista, G.C.M.; Salvador, P.A.; Soares, S.M.A.; Barros, L.R.C.; Xavier, F.H.D.C.; Abdo, L.M.; Gualberto, A.C.M.; Macedo, G.C.; Clavijo-Salomon, M.A.; Gameiro, J. 4T1 Mammary Carcinoma Colonization of Metastatic Niches Is Accelerated by Obesity. Front. Oncol. 2019, 9, 685. [Google Scholar] [CrossRef]
- Fuentes-Mattei, E.; Velazquez-Torres, G.; Phan, L.; Zhang, F.; Chou, P.-C.; Shin, J.-H.; Choi, H.H.; Chen, J.-S.; Zhao, R.; Chen, J.; et al. Effects of Obesity on Transcriptomic Changes and Cancer Hallmarks in Estrogen Receptor–Positive Breast Cancer. Gynecol. Oncol. 2014, 106, dju158. [Google Scholar] [CrossRef]
- Madeddu, C.; Gramignano, G.; Floris, C.; Murenu, G.; Sollai, G.; Macciò, A. Role of inflammation and oxidative stress in post-menopausal oestrogen-dependent breast cancer. J. Cell. Mol. Med. 2014, 18, 2519–2529. [Google Scholar] [CrossRef]
- Quigley, D.A.; Tahiri, A.; Lüders, T.; Riis, M.H.; Balmain, A.; Børresen-Dale, A.-L.; Bukholm, I.; Kristensen, V. Age, estrogen, and immune response in breast adenocarcinoma and adjacent normal tissue. Oncoimmunology 2017, 6, e1356142. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Picon-Ruiz, M.; Aurrekoetxea-Rodriguez, I.; de Paiva, V.N.; D’amico, M.; Yoon, H.; Radhakrishnan, R.; Morata-Tarifa, C.; Ince, T.; Lippman, M.E.; et al. The Major Pre- and Postmenopausal Estrogens Play Opposing Roles in Obesity-Driven Mammary Inflammation and Breast Cancer Development. Cell Metab. 2020, 31, 1154–1172.e9. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546.e9. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Aren Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Blank Christian, U.; Haanen John, B.; Ribas, A.; Schumacher Ton, N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Adams, S.; Gatti-Mays, M.E.; Kalinsky, K.; Korde, L.A.; Sharon, E.; Amiri-Kordestani, L.; Bear, H.; McArthur, H.L.; Frank, E.; Perlmutter, J. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol. 2019, 5, 1205–1214. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Park, J.H.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.-B.; et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. Ann. Oncol. 2019, 30, 1941–1949. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]
- An, Y.; Wu, Z.; Wang, N.; Yang, Z.; Li, Y.; Xu, B.; Sun, M. Association between body mass index and survival outcomes for cancer patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. J. Transl. Med. 2020, 18, 235. [Google Scholar] [CrossRef]
- Maslov, D.; Tawagi, K.; Simenson, V.; Yuan, H.; Parent, C.; Bamnolker, A.; Goel, R.; Blake, Z.; Kc, M.; Matrana, M.R.; et al. Impact of body mass index on survival rates in patients receiving immune checkpoint inhibitors. J. Clin. Oncol. 2020, 38, e15108. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Procaccini, C.; Jirillo, E.; Matarese, G. Leptin as an immunomodulator. Mol. Asp. Med. 2012, 33, 35–45. [Google Scholar] [CrossRef]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2019, 31, 148–161.e5. [Google Scholar] [CrossRef]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef]
- Dyck, L.; Prendeville, H.; Raverdeau, M.; Wilk, M.M.; Loftus, R.M.; Douglas, A.; McCormack, J.; Moran, B.; Wilkinson, M.; Mills, E.L.; et al. Suppressive effects of the obese tumor microenvironment on CD8 T cell infiltration and effector function. J. Exp. Med. 2022, 219, e20210042. [Google Scholar] [CrossRef]
- Moro-García, M.A.; Mayo, J.C.; Sainz, R.M.; Alonso-Arias, R. Influence of Inflammation in the Process of T Lymphocyte Differentiation: Proliferative, Metabolic, and Oxidative Changes. Front. Immunol. 2018, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Ding, J.; Chen, Y. Role of CD8+ T lymphocyte cells: Interplay with stromal cells in tumor microenvironment. Acta Pharm. Sin. B 2021, 11, 1365–1378. [Google Scholar] [CrossRef]
- Guo, H.; Kong, W.; Zhang, L.; Han, J.; Clark, L.H.; Yin, Y.; Fang, Z.; Sun, W.; Wang, J.; Gilliam, T.P.; et al. Reversal of obesity-driven aggressiveness of endometrial cancer by metformin. Am. J. Cancer Res. 2019, 9, 2170–2193. [Google Scholar]
- Wu, Z.; Zhang, C.; Najafi, M. Targeting of the tumor immune microenvironment by metformin. J. Cell Commun. Signal. 2021, 16, 333–348. [Google Scholar] [CrossRef]
- Kristófi, R.; Eriksson, J.W. Metformin as an anti-inflammatory agent: A short review. J. Endocrinol. 2021, 251, R11–R22. [Google Scholar] [CrossRef]
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 2021, 12, 440. [Google Scholar] [CrossRef]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; di Magliano, M.P.; Swanson, K.D.; Zheng, B. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J. Investig. Dermatol. 2017, 137, 1740–1748. [Google Scholar] [CrossRef]
- Nojima, I.; Eikawa, S.; Tomonobu, N.; Hada, Y.; Kajitani, N.; Teshigawara, S.; Miyamoto, S.; Tone, A.; Uchida, H.A.; Nakatsuka, A.; et al. Dysfunction of CD8 + PD-1 + T cells in type 2 diabetes caused by the impairment of metabolism-immune axis. Sci. Rep. 2020, 10, 14928. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef]
- Zhao, D.; Long, X.-D.; Lu, T.-F.; Wang, T.; Zhang, W.-W.; Liu, Y.-X.; Cui, X.-L.; Dai, H.-J.; Xue, F.; Xia, Q. Metformin decreases IL-22 secretion to suppress tumor growth in an orthotopic mouse model of hepatocellular carcinoma. Int. J. Cancer 2014, 136, 2556–2565. [Google Scholar] [CrossRef]
- Wang, J.-C.; Sun, X.; Ma, Q.; Fu, G.-F.; Cong, L.-L.; Zhang, H.; Fan, D.-F.; Feng, J.; Lu, S.-Y.; Liu, J.-L.; et al. Metformin’s antitumour and anti-angiogenic activities are mediated by skewing macrophage polarization. J. Cell. Mol. Med. 2018, 22, 3825–3836. [Google Scholar] [CrossRef]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol. Cell. Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef]
- Kim, J.; Kwak, H.J.; Cha, J.-Y.; Jeong, Y.-S.; Rhee, S.D.; Kim, K.R.; Cheon, H.G. Metformin Suppresses Lipopolysaccharide (LPS)-induced Inflammatory Response in Murine Macrophages via Activating Transcription Factor-3 (ATF-3) Induction. J. Biol. Chem. 2014, 289, 23246–23255. [Google Scholar] [CrossRef]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef]
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4(+)CD25(+) Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine 2017, 25, 154–164. [Google Scholar] [CrossRef]
- Pereira, F.V.; Melo, A.C.L.; Low, J.S.; de Castro, A.; Braga, T.T.; Almeida, D.C.; de Lima, A.G.U.B.; Hiyane, M.I.; Correa-Costa, M.; Andrade-Oliveira, V.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9, 25808–25825. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Cufí, S.; Vazquez-Martin, A.; Menendez, O.J.; Barrera, J.B.; Martin-Castilló, B.; Joven, J.; Menendez, J.A. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle 2012, 11, 865–870. [Google Scholar] [CrossRef]
- Cai, S.; Chen, Z.; Wang, Y.; Wang, M.; Wu, J.; Tong, Y.; Chen, L.; Lu, C.; Yang, H. Reducing PD-L1 expression with a self-assembled nanodrug: An alternative to PD-L1 antibody for enhanced chemo-immunotherapy. Theranostics 2021, 11, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Haikala, H.M.; Anttila, J.M.; Marques, E.; Raatikainen, T.; Ilander, M.; Hakanen, H.; Ala-Hongisto, H.; Savelius, M.; Balboa, D.; Von Eyss, B.; et al. Pharmacological reactivation of MYC-dependent apoptosis induces susceptibility to anti-PD-1 immunotherapy. Nat. Commun. 2019, 10, 620. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhao, Y.; Liu, G.; Zhou, H.-L.; Fan, J.; Zhang, L.; Li, Y.-L.; Wang, Y.; Liang, J.; Xu, Z.-X. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci. 2020, 256, 117923. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Liu, Y.; Chen, C.; Chi, J.; Zhong, L.; Zhao, Y.; Zhao, Y. Metformin loaded porous particles with bio-microenvironment responsiveness for promoting tumor immunotherapy. Biomater. Sci. 2020, 9, 2082–2089. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients with Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA 2022, 327, 1963–1973. [Google Scholar] [CrossRef]
- Coyle, C.; Cafferty, F.; Vale, C.; Langley, R. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442. [Google Scholar] [CrossRef]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef]
- Zhao, Y.; Gong, C.; Wang, Z.; Zhang, J.; Wang, L.; Zhang, S.; Cao, J.; Tao, Z.; Li, T.; Wang, B.; et al. A randomized phase II study of aromatase inhibitors plus metformin in pre-treated postmenopausal patients with hormone receptor positive metastatic breast cancer. Oncotarget 2017, 8, 84224–84236. [Google Scholar] [CrossRef]
- Barakat, H.E.; Hussein, R.R.S.; Elberry, A.A.; Zaki, M.A.; Ramadan, M.E. The impact of metformin use on the outcomes of locally advanced breast cancer patients receiving neoadjuvant chemotherapy: An open-labelled randomized controlled trial. Sci. Rep. 2022, 12, 7656. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Pernas, S.; Dorca, J.; Álvarez, I.; Martínez, S.; Pérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; Amillano, K.; Domínguez, S.; et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: The METTEN study. Oncotarget 2018, 9, 35687–35704. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef]
- Hadad, S.M.; Coates, P.; Jordan, L.B.; Dowling, R.J.O.; Chang, M.C.; Done, S.J.; Purdie, C.A.; Goodwin, P.J.; Stambolic, V.; Moulder-Thompson, S.; et al. Evidence for biological effects of metformin in operable breast cancer: Biomarker analysis in a pre-operative window of opportunity randomized trial. Breast Cancer Res. Treat. 2015, 150, 149–155. [Google Scholar] [CrossRef]
- Lord, S.R.; Cheng, W.-C.; Liu, D.; Gaude, E.; Haider, S.; Metcalf, T.; Patel, N.; Teoh, E.J.; Gleeson, F.; Bradley, K.; et al. Integrated Pharmacodynamic Analysis Identifies Two Metabolic Adaption Pathways to Metformin in Breast Cancer. Cell Metab. 2018, 28, 679–688.e4. [Google Scholar] [CrossRef]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Pusztai, L.; et al. Evidence for biological effects of metformin in operable breast cancer: A pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 2011, 128, 783–794. [Google Scholar] [CrossRef]
- Kim, Y.; Vagia, E.; Viveiros, P.; Kang, C.Y.; Lee, J.Y.; Gim, G.; Cho, S.; Choi, H.; Kim, L.; Park, I.; et al. Overcoming acquired resistance to PD-1 inhibitor with the addition of metformin in small cell lung cancer (SCLC). Cancer Immunol. Immunother. 2020, 70, 961–965. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Morris, P.G.; Zhou, X.K.; Gucalp, A.; Giri, D.; Harbus, M.D.; Falcone, D.J.; Krasne, M.D.; Vahdat, L.T.; Subbaramaiah, K.; et al. Menopause Is a Determinant of Breast Adipose Inflammation. Cancer Prev. Res. 2015, 8, 349–358. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Immune Changes | Metabolic Changes |
|---|---|
| Upregulation of proinflammatory signalling pathways [24] | Increased insulin and IGF levels [28] |
| Increased immune cell infiltration [29] | Insulin resistance [20,27] |
| Upregulation of WNT signalling [30] | Elevates leptin levels [31] |
| Increased synthesis of arachidonic acid and PGE2 [32] | Increases oestrogen and androgen levels [33,34] |
| Downregulation of response to antigen and mitogen stimulation [35,36] | Anti-apoptotic, promotes stemness [37] |
| Study | Sample Size | CLS− (n) | CLS+ (n) | CLS Marker | RFS, CLS+ vs. CLS− | OS, CLS+ vs. CLS− |
|---|---|---|---|---|---|---|
| Iyengar N (cohort 2) (2016) [58] | 127 | 75 | 52 | CD68 | 1.83 (1.07–3.13) a,c | not reported |
| Koru-Sengul T f (2016) [56] | 134 | NR | NR | CD40 | 5.87 (0.73–47.23) a,c | 13.59 (1.56–118.16) a,c |
| Koru-Sengul T f (2016) [56] | 134 | NR | NR | CD163 | 2.21 (0.65–7.59) a,c | 2.42 (0.54–10.89) a,c |
| Koru-Sengul T f (2016) [56] | 134 | NR | NR | CD206 | 1.17 (0.09–15.35) a,c | 0.74 (0.04–15.55) a,c |
| Cha YJ (2018) g [55] | 140 | 122 | 18 | CD163 | 105 (94–116) vs. 124 (118–131) b,d | 105 (94–116) vs. 130 (124–136) b,d |
| Cha YJ (2018) g [55] | 140 | 115 | 25 | CD68 | 106 (97–114) vs. 124 (117–131) b,d | 106 (99–114) vs. 130 (124–136) b,d |
| Cha YJ (2018) g [55] | 56 | 49 | 7 | CD68 | 76 (56–96) vs. 120 (108–132) b,d,e | 79 (63–96) vs. 125 (114–136) b,d,e |
| Maliniak M (2020) [59] | 319 | 223 | 96 | CD68 | 1.05 (0.64–1.72) a,c | 1.02 (0.55–1.87) a,c |
| Birts C (2022) [45] | 117 | 47 | 61 | CD32B | 4.2 (1.01–17.4) a,c | not reported |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savva, C.; Copson, E.; Johnson, P.W.M.; Cutress, R.I.; Beers, S.A. Obesity Is Associated with Immunometabolic Changes in Adipose Tissue That May Drive Treatment Resistance in Breast Cancer: Immune-Metabolic Reprogramming and Novel Therapeutic Strategies. Cancers 2023, 15, 2440. https://doi.org/10.3390/cancers15092440
Savva C, Copson E, Johnson PWM, Cutress RI, Beers SA. Obesity Is Associated with Immunometabolic Changes in Adipose Tissue That May Drive Treatment Resistance in Breast Cancer: Immune-Metabolic Reprogramming and Novel Therapeutic Strategies. Cancers. 2023; 15(9):2440. https://doi.org/10.3390/cancers15092440
Chicago/Turabian StyleSavva, Constantinos, Ellen Copson, Peter W. M. Johnson, Ramsey I. Cutress, and Stephen A. Beers. 2023. "Obesity Is Associated with Immunometabolic Changes in Adipose Tissue That May Drive Treatment Resistance in Breast Cancer: Immune-Metabolic Reprogramming and Novel Therapeutic Strategies" Cancers 15, no. 9: 2440. https://doi.org/10.3390/cancers15092440
APA StyleSavva, C., Copson, E., Johnson, P. W. M., Cutress, R. I., & Beers, S. A. (2023). Obesity Is Associated with Immunometabolic Changes in Adipose Tissue That May Drive Treatment Resistance in Breast Cancer: Immune-Metabolic Reprogramming and Novel Therapeutic Strategies. Cancers, 15(9), 2440. https://doi.org/10.3390/cancers15092440