
Non-Inflamed Tumor Microenvironment and Methylation/Downregulation of Antigen-Presenting Machineries in Cholangiocarcinoma

 ,
,  ,
, 
 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Data
2.2. Statistical Analysis
3. Results
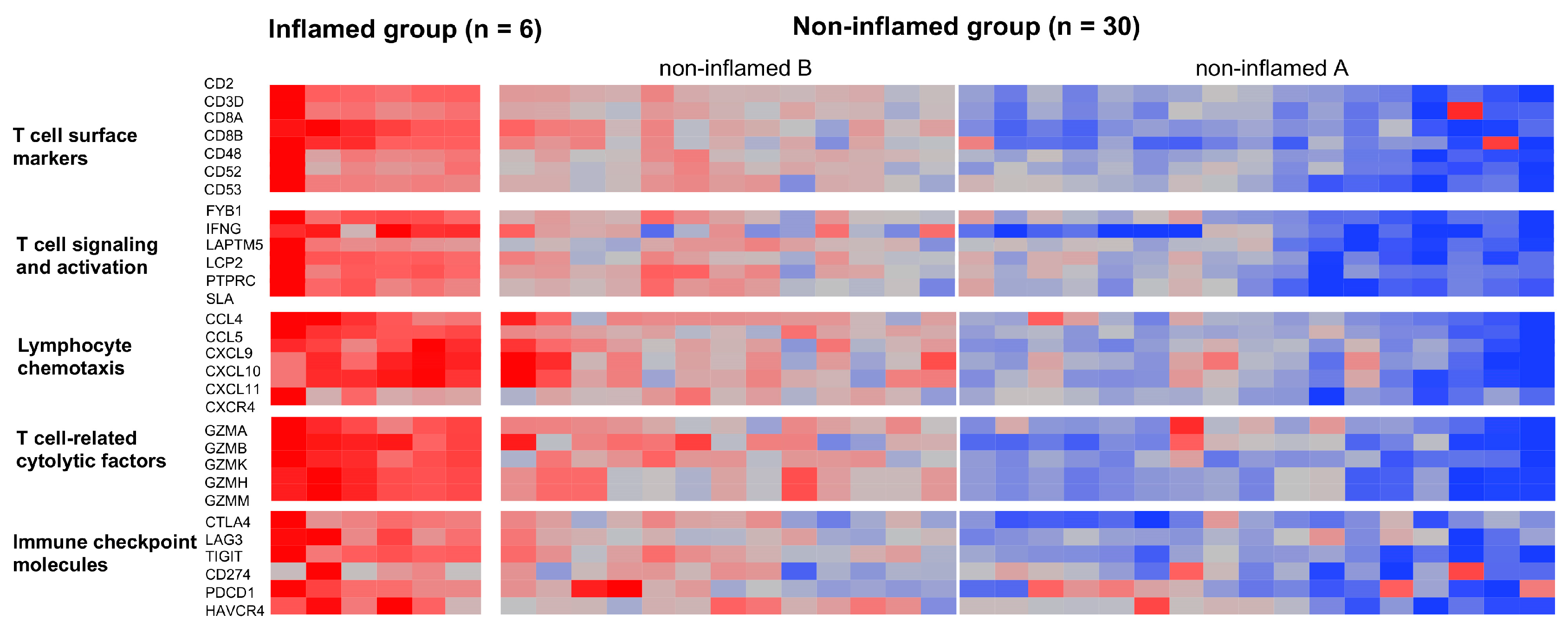
3.1. Classification of CCAs Based on Transcriptome Data
3.2. Genetic and Epigenetic Alterations Associated with the Inflammation Profile of CCAs
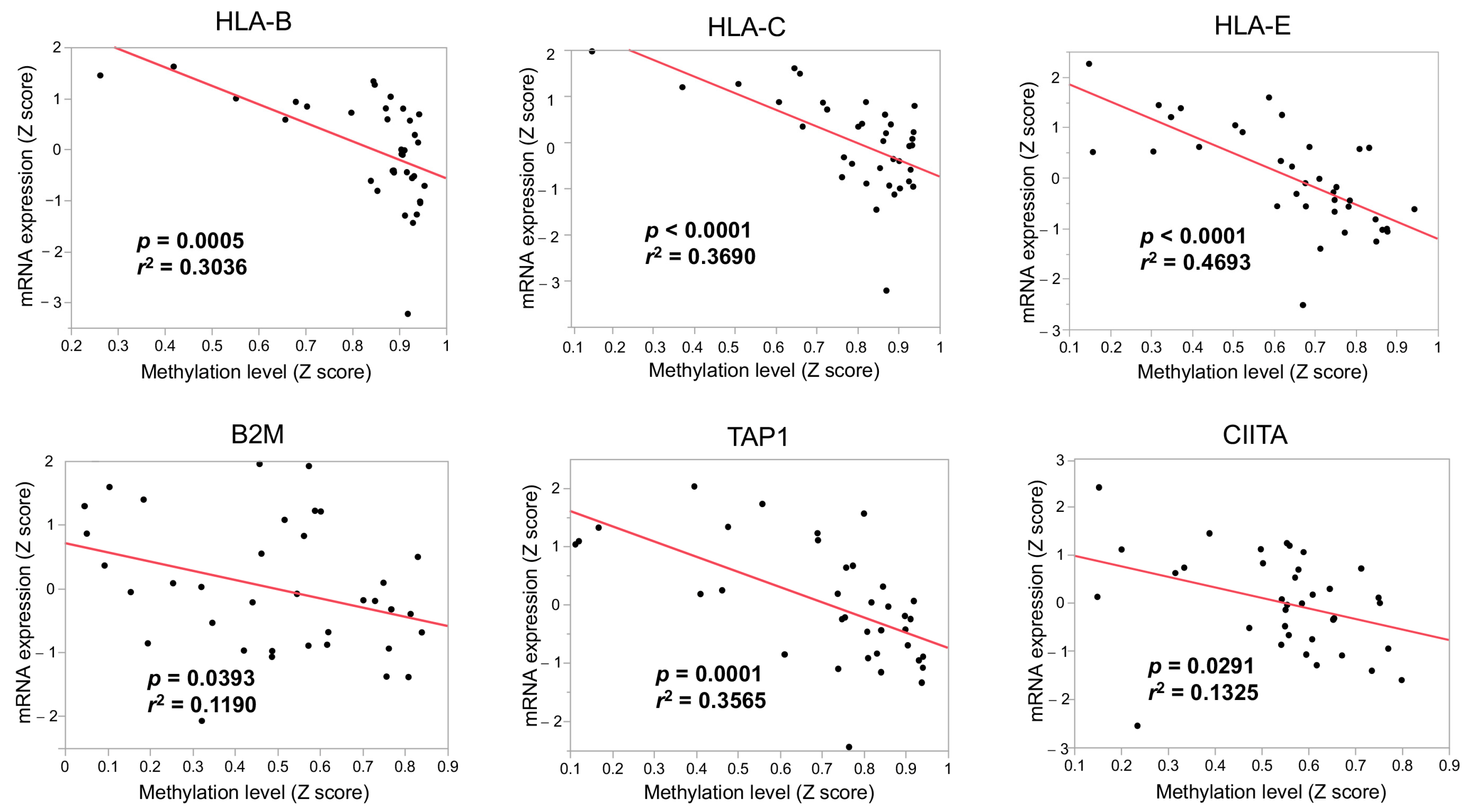
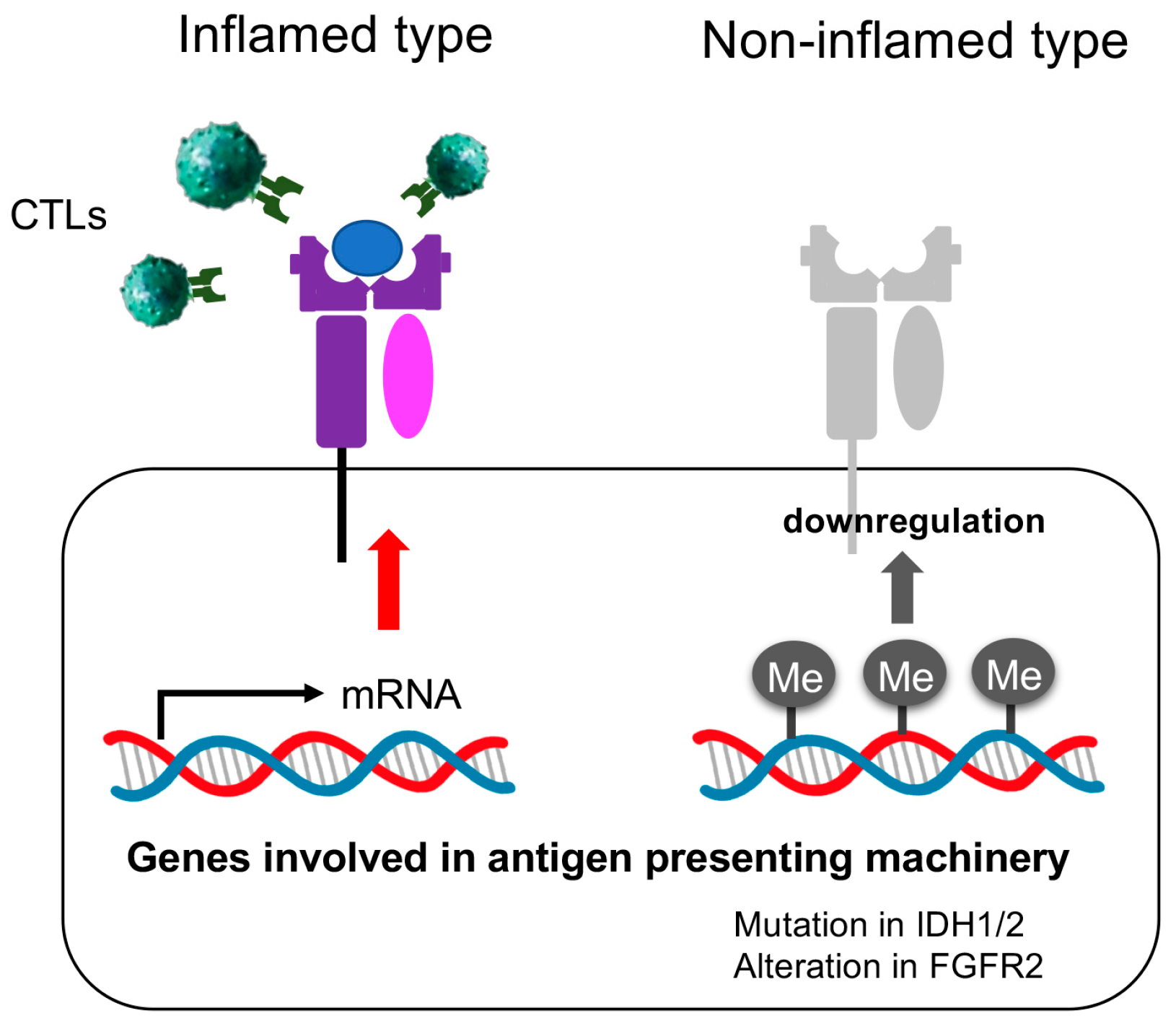
3.3. The Immunological Microenvironment of “Non-Inflamed” Is Associated with Methylation and Downregulation of Antigen-Presenting Machineries and Mutations in IDH Genes in Biliary Tract Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma-evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. New Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Ioka, T.; Kanai, M.; Kobayashi, S.; Sakai, D.; Eguchi, H.; Baba, H.; Seo, S.; Taketomi, A.; Takayama, T.; Yamaue, H.; et al. Randomized phase III study of gemcitabine, cisplatin plus S-1 versus gemcitabine, cisplatin for advanced biliary tract cancer (KHBO1401-MITSUBA). J. Hepatobiliary Pancreat. Sci. 2023, 30, 102–110. [Google Scholar] [CrossRef]
- de la Fouchardiere, C. Towards greater clarity in the treatment of cholangiocarcinoma. Lancet Oncol. 2020, 21, 738–739. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients With Advanced Cholangiocarcinoma With IDH1 Mutation: The Phase 3 Randomized Clinical ClarIDHy Trial. JAMA Oncol. 2021, 7, 1669–1677. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; Kelley, R.K.; Cassier, P.A.; et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. New Engl. J. Med. 2023, 388, 228–239. [Google Scholar] [CrossRef]
- Oh, D.-Y.; He, A.R.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Lee, M.A.M.D. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- Harding, J.J.; Khalil, D.N.; Fabris, L.; Abou-Alfa, G.K. Rational development of combination therapies for biliary tract cancers. J. Hepatol. 2023, 78, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, M.A.; Kepecs, B.; Torres-Martin, M.; Bramel, E.R.; Haber, P.K.; Merritt, E.; Rialdi, A.; Param, N.J.; Maeda, M.; Lindblad, K.E.; et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut 2022, 72, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2023, 72, 129–140. [Google Scholar] [CrossRef]
- Uson Junior, P.L.S.; Bearss, J.; Babiker, H.M.; Borad, M.J. Novel precision therapies for cholangiocarcinoma: An overview of clinical trials. Expert. Opin. Investig. Drugs 2023, 32, 69–75. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Lim, S.O.; Yamaguchi, H. Oncogenic signaling pathways associated with immune evasion and resistance to immune checkpoint inhibitors in cancer. Semin. Cancer Biol. 2020, 65, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Mody, K.; Jain, P.; El-Refai, S.M.; Azad, N.S.; Zabransky, D.J.; Baretti, M.; Shroff, R.T.; Kelley, R.K.; El-Khouiery, A.B.; Hockenberry, A.J.; et al. Clinical, Genomic, and Transcriptomic Data Profiling of Biliary Tract Cancer Reveals Subtype-Specific Immune Signatures. JCO Precis. Oncol. 2022, 6, e2100510. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.K.; Castet, F.; Torres-Martin, M.; Andreu-Oller, C.; Puigvehi, M.; Miho, M.; Radu, P.; Dufour, J.F.; Verslype, C.; Zimpel, C.; et al. Molecular Markers of Response to Anti-PD1 Therapy in Advanced Hepatocellular Carcinoma. Gastroenterology 2023, 164, 72–88.e18. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma. Cancers 2020, 12, 1274. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N. Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma. Cancers 2021, 13, 3666. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef]
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune Exclusion-Wnt/CTNNB1 Class Predicts Resistance to Immunotherapies in HCC. Clin. Cancer Res. 2019, 25, 2021–2023. [Google Scholar] [CrossRef]
- Morita, M.; Nishida, N.; Sakai, K.; Aoki, T.; Chishina, H.; Takita, M.; Ida, H.; Hagiwara, S.; Minami, Y.; Ueshima, K.; et al. Immunological Microenvironment Predicts the Survival of the Patients with Hepatocellular Carcinoma Treated with Anti-PD-1 Antibody. Liver Cancer 2021, 10, 380–393. [Google Scholar] [CrossRef]
- Jeon, Y.; Kwon, S.M.; Rhee, H.; Yoo, J.E.; Chung, T.; Woo, H.G.; Park, Y.N. Molecular and radiopathologic spectrum between HCC and intrahepatic cholangiocarcinoma. Hepatology. 2023, 77, 92–108. [Google Scholar] [CrossRef]
- Xue, R.; Chen, L.; Zhang, C.; Fujita, M.; Li, R.; Yan, S.M.; Ong, C.K.; Liao, X.; Gao, Q.; Sasagawa, S.; et al. Genomic and Transcriptomic Profiling of Combined Hepatocellular and Intrahepatic Cholangiocarcinoma Reveals Distinct Molecular Subtypes. Cancer Cell 2019, 35, 932–947.e938. [Google Scholar] [CrossRef] [PubMed]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 72, 965–981. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Peng, L.; Dong, L.; Liu, D.; Ma, J.; Lin, J.; Chen, X.; Lin, P.; Song, G.; Zhang, M.; et al. Geospatial Immune Heterogeneity Reflects the Diverse Tumor-Immune Interactions in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2022, 12, 2350–2371. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Liu, Z.; Zhang, C.; Li, Z.; Gao, J.; Zhang, C.; Cao, Q.; Cheng, J.; Liu, H.; Chen, D.; et al. IDH Mutation Subgroup Status Associates with Intratumor Heterogeneity and the Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Adv. Sci. 2021, 8, e2101230. [Google Scholar] [CrossRef]
- Wu, M.J.; Shi, L.; Dubrot, J.; Merritt, J.; Vijay, V.; Wei, T.Y.; Kessler, E.; Olander, K.E.; Adil, R.; Pankaj, A.; et al. Mutant IDH Inhibits IFNgamma-TET2 Signaling to Promote Immunoevasion and Tumor Maintenance in Cholangiocarcinoma. Cancer Discov. 2022, 12, 812–835. [Google Scholar] [CrossRef]
- Kawazu, M.; Ueno, T.; Saeki, K.; Sax, N.; Togashi, Y.; Kanaseki, T.; Chida, K.; Kishigami, F.; Sato, K.; Kojima, S.; et al. HLA Class I Analysis Provides Insight Into the Genetic and Epigenetic Background of Immune Evasion in Colorectal Cancer With High Microsatellite Instability. Gastroenterology 2022, 162, 799–812. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef]
- Gyorffy, B.; Bottai, G.; Fleischer, T.; Munkacsy, G.; Budczies, J.; Paladini, L.; Borresen-Dale, A.L.; Kristensen, V.N.; Santarpia, L. Aberrant DNA methylation impacts gene expression and prognosis in breast cancer subtypes. Int. J. Cancer 2016, 138, 87–97. [Google Scholar] [CrossRef]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef]
- Fortin, J.; Chiang, M.F.; Meydan, C.; Foox, J.; Ramachandran, P.; Leca, J.; Lemonnier, F.; Li, W.Y.; Gams, M.S.; Sakamoto, T.; et al. Distinct and opposite effects of leukemogenic Idh and Tet2 mutations in hematopoietic stem and progenitor cells. Proc. Natl. Acad. Sci. USA 2023, 120, e2208176120. [Google Scholar] [CrossRef]
- Adachi, Y.; Kamiyama, H.; Ichikawa, K.; Fukushima, S.; Ozawa, Y.; Yamaguchi, S.; Goda, S.; Kimura, T.; Kodama, K.; Matsuki, M.; et al. Inhibition of FGFR Reactivates IFNgamma Signaling in Tumor Cells to Enhance the Combined Antitumor Activity of Lenvatinib with Anti-PD-1 Antibodies. Cancer Res. 2022, 82, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Vlkova, V.; Stepanek, I.; Hruskova, V.; Senigl, F.; Mayerova, V.; Sramek, M.; Simova, J.; Bieblova, J.; Indrova, M.; Hejhal, T.; et al. Epigenetic regulations in the IFNgamma signalling pathway: IFNgamma-mediated MHC class I upregulation on tumour cells is associated with DNA demethylation of antigen-presenting machinery genes. Oncotarget 2014, 5, 6923–6935. [Google Scholar] [CrossRef] [PubMed]
- Komuta, M. Intrahepatic cholangiocarcinoma: Tumour heterogeneity and its clinical relevance. Clin. Mol. Hepatol. 2022, 28, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Han, D.H.; Cho, K.; Park, S.B.; Kim, C.; Leem, G.; Jung, D.E.; Kwon, S.S.; Kim, C.H.; Jo, J.H.; et al. Integrative analysis of multiple genomic data from intrahepatic cholangiocarcinoma organoids enables tumor subtyping. Nat. Commun. 2023, 14, 237. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflamed (n = 6) | Non-Inflamed (n = 30) | p Value by Fisher’s Exact Test | |
|---|---|---|---|
| FGF2R alterations | |||
| with (n = 7) | 0 | 7 | 0.3171 |
| without (n = 29) | 6 | 23 | |
| IDH1/2 mutations | |||
| with (n = 7) | 0 | 7 | 0.3171 |
| without (n = 29) | 6 | 23 | |
| KRAS mutations | |||
| with (n = 2) | 0 | 2 | 1.0 |
| without (n = 24) | 6 | 28 | |
| BAP1 mutations | |||
| with (n = 8) | 1 | 7 | 1.0 |
| without (n = 28) | 5 | 23 | |
| TP53 mutations | |||
| with (n = 5) | 0 | 5 | 0.5638 |
| without (n = 31) | 6 | 25 | |
| ARID1/2 mutations | |||
| with (n = 6) | 1 | 5 | 1.0 |
| without (n = 30) | 5 | 25 | |
| HLA mutation | |||
| with (n = 6) | 1 | 5 | 1.0 |
| without (n = 30) | 5 | 25 | |
| HLA copy number loss | |||
| with (n = 4) | 0 | 4 | 1.0 |
| without (n = 32) | 6 | 26 | |
| TMB score 1 | |||
| low (n = 27) | 5 | 22 | 1.0 |
| high (n = 9) | 1 | 8 | |
| Expression of antigen-presenting machineries 2 | |||
| low (n = 22) | 0 | 22 | 0.0015 |
| high (n = 14) | 6 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, N.; Aoki, T.; Morita, M.; Chishina, H.; Takita, M.; Ida, H.; Hagiwara, S.; Minami, Y.; Ueshima, K.; Kudo, M. Non-Inflamed Tumor Microenvironment and Methylation/Downregulation of Antigen-Presenting Machineries in Cholangiocarcinoma. Cancers 2023, 15, 2379. https://doi.org/10.3390/cancers15082379
Nishida N, Aoki T, Morita M, Chishina H, Takita M, Ida H, Hagiwara S, Minami Y, Ueshima K, Kudo M. Non-Inflamed Tumor Microenvironment and Methylation/Downregulation of Antigen-Presenting Machineries in Cholangiocarcinoma. Cancers. 2023; 15(8):2379. https://doi.org/10.3390/cancers15082379
Chicago/Turabian StyleNishida, Naoshi, Tomoko Aoki, Masahiro Morita, Hirokazu Chishina, Masahiro Takita, Hiroshi Ida, Satoru Hagiwara, Yasunori Minami, Kazuomi Ueshima, and Masatoshi Kudo. 2023. "Non-Inflamed Tumor Microenvironment and Methylation/Downregulation of Antigen-Presenting Machineries in Cholangiocarcinoma" Cancers 15, no. 8: 2379. https://doi.org/10.3390/cancers15082379
APA StyleNishida, N., Aoki, T., Morita, M., Chishina, H., Takita, M., Ida, H., Hagiwara, S., Minami, Y., Ueshima, K., & Kudo, M. (2023). Non-Inflamed Tumor Microenvironment and Methylation/Downregulation of Antigen-Presenting Machineries in Cholangiocarcinoma. Cancers, 15(8), 2379. https://doi.org/10.3390/cancers15082379