
Ursolic Acid Alleviates Cancer Cachexia and Prevents Muscle Wasting via Activating SIRT1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. The Conditioned Medium Collection
2.4. Cell Viability Assay
2.5. Immunofluorescence Staining and Myofiber Size Measurement
2.6. Animal Models and Treatments
2.7. Histological Examination
2.8. RNA Extraction and Quantitative Reverse Transcription PCR
2.9. Western Blot Analysis
2.10. ELISA Assay
2.11. Molecular Docking
2.12. Statistical Analysis
3. Results
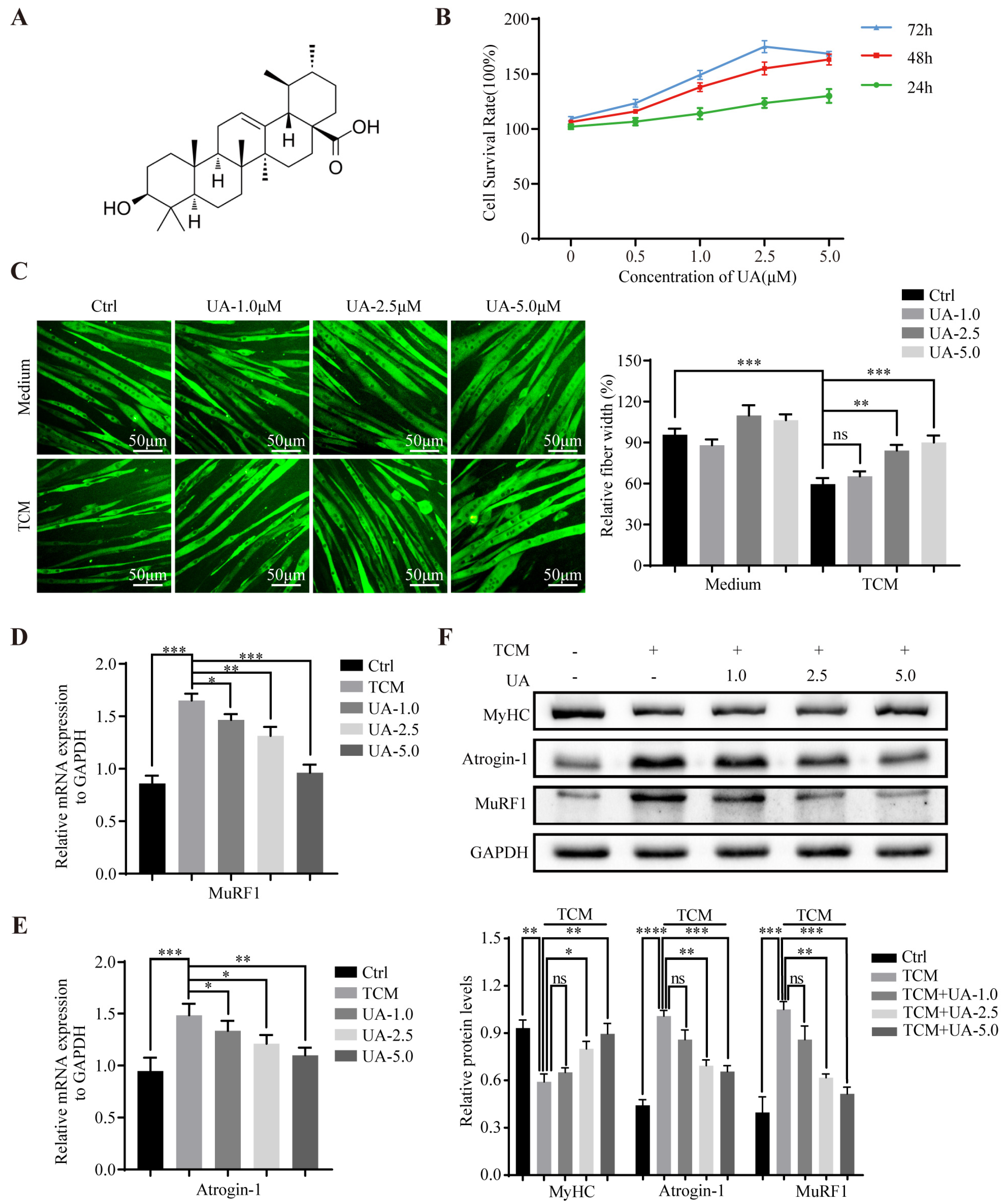
3.1. UA Alleviates TCM-Induced Myotube Atrophy In Vitro
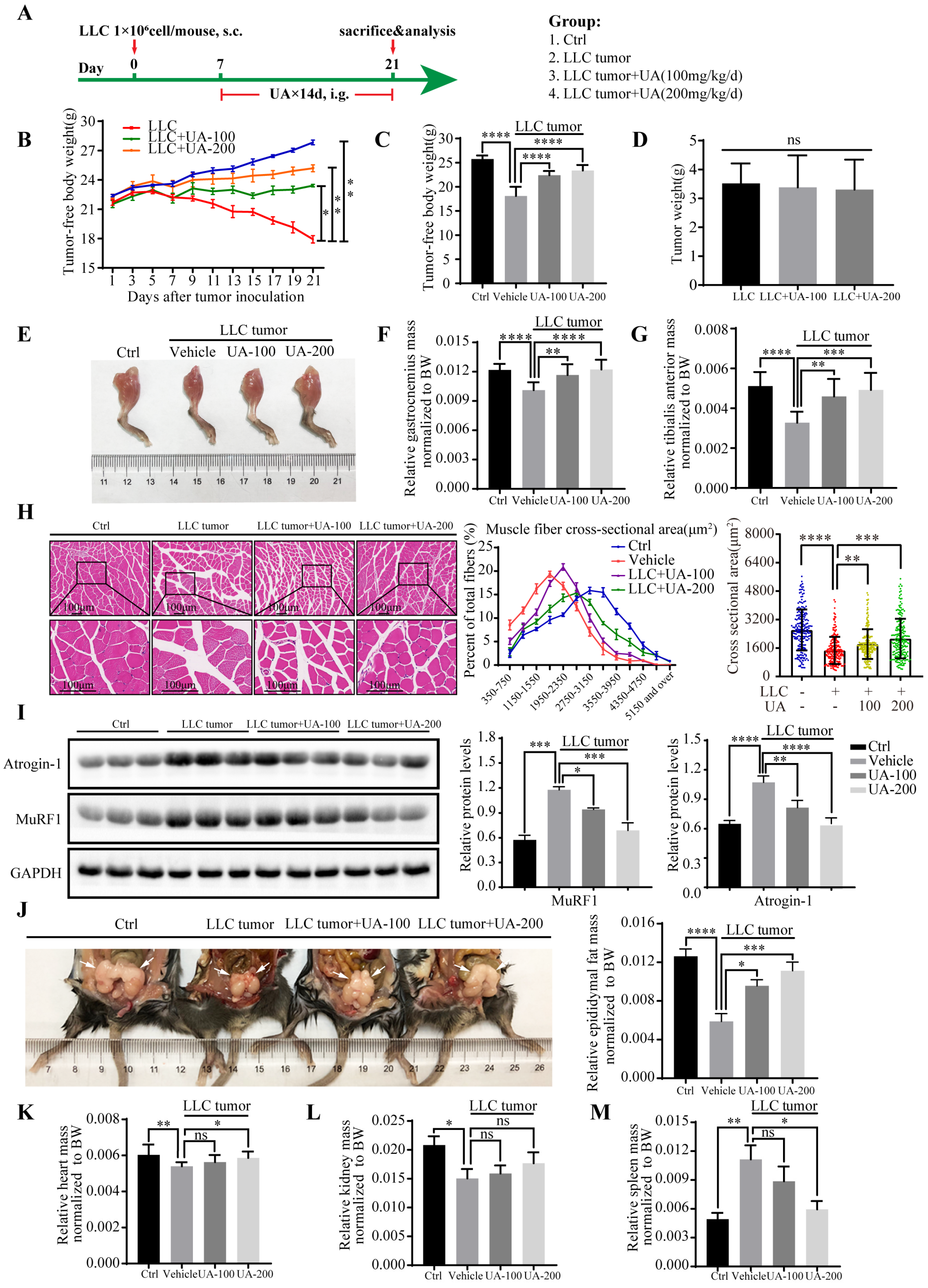
3.2. UA Alleviates Muscle Wasting and Prevents Cancer Cachexia In Vivo
3.3. UA Upregulates SIRT1 Expression and Inhibits the Expression of NOX4, FOXO1, and FOXO3a In Vitro and In Vivo
3.4. UA Suppresses Phosphorylation of STAT3 and p65 Expression In Vitro and In Vivo and Reverses Elevated Serum Cytokines in the Cancer Cachectic Model
3.5. UA Relieves TCM-Induced Myotube Wasting through SIRT1 Activation
3.6. UA Improves Muscle Wasting and Prevents Cancer Cachexia in LLC Tumor-Bearing Mice through SIRT1 Activation
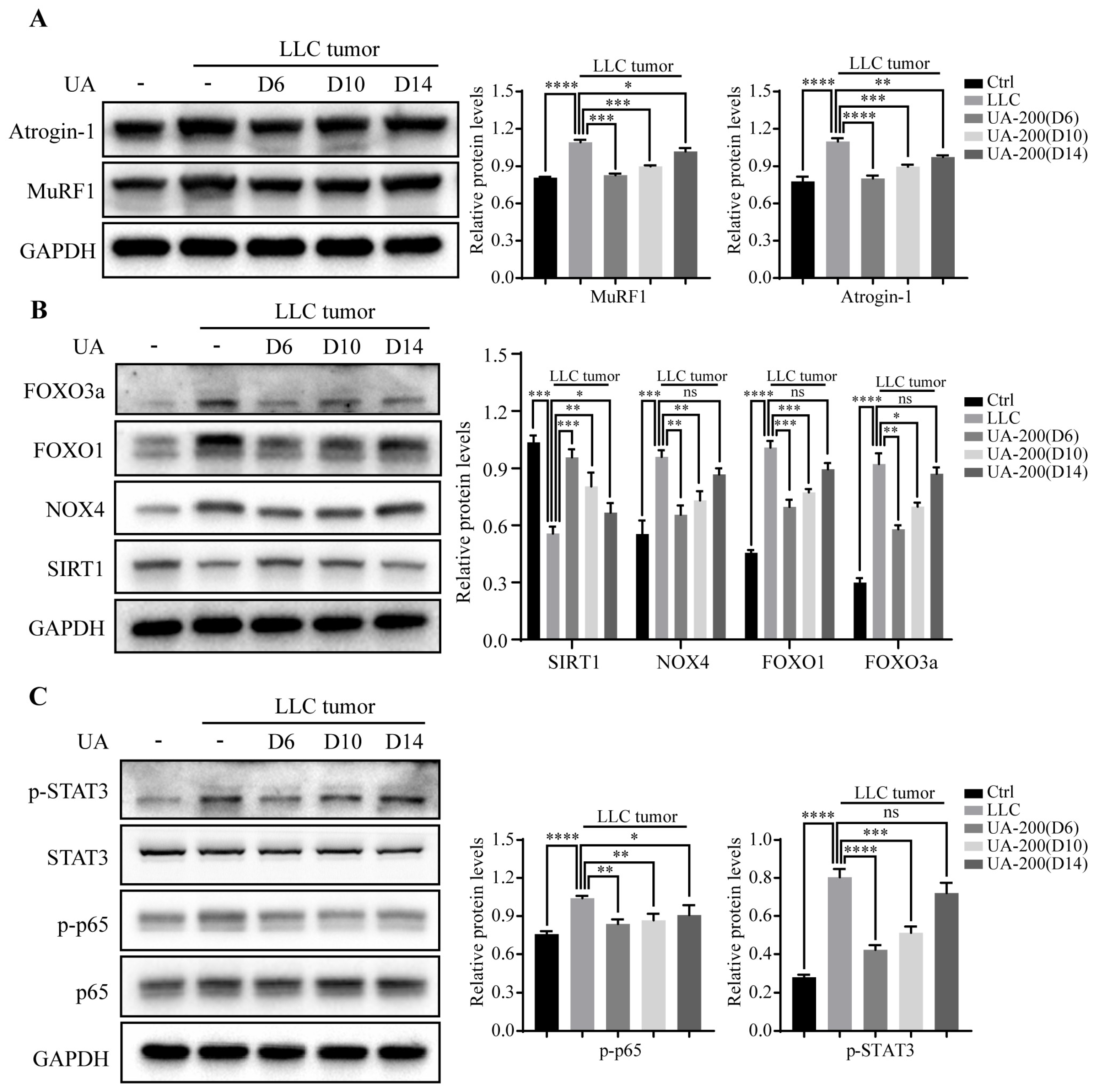
3.7. Effects of Delaying Treatment with UA until Advanced Stages of Tumor and Cachexia Progression in LLC Tumor-Bearing Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biswas, A.K.; Acharyya, S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 2020, 20, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, A.P.; Sakellariou, G.K.; Nye, G.A.; McArdle, F.; Jackson, M.J.; Griffiths, R.D.; McArdle, A. SS-31 attenuates TNF-alpha induced cytokine release from C2C12 myotubes. Redox Biol. 2015, 6, 253–259. [Google Scholar] [CrossRef]
- Webster, J.M.; Kempen, L.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Mediators of cachexia in cancer patients. Nutrition 2019, 66, 11–15. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef]
- Mourkioti, F.; Rosenthal, N. NF-kappaB signaling in skeletal muscle: Prospects for intervention in muscle diseases. J. Mol. Med. 2008, 86, 747–759. [Google Scholar] [CrossRef]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef]
- Sin, T.K.; Zhang, G.; Zhang, Z.; Zhu, J.Z.; Zuo, Y.; Frost, J.A.; Li, M.; Li, Y.P. Cancer-Induced Muscle Wasting Requires p38beta MAPK Activation of p300. Cancer Res. 2021, 81, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Rom, O.; Reznick, A.Z. The role of E3 ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass. Free Radic. Biol. Med. 2016, 98, 218–230. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef]
- Yang, Y.; Hou, H.; Haller, E.M.; Nicosia, S.V.; Bai, W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005, 24, 1021–1032. [Google Scholar] [CrossRef]
- Lee, D.; Goldberg, A.L. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in Regulating Oxidative Stress and Inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Tian, Z.; Zhou, G.; Niu, Q.; Chen, J.; Li, P.; Dong, L.; Xia, T.; Zhang, S.; Wang, A. SIRT1-dependent mitochondrial biogenesis supports therapeutic effects of resveratrol against neurodevelopment damage by fluoride. Theranostics 2020, 10, 4822–4838. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell. Signal. 2009, 21, 1356–1360. [Google Scholar] [CrossRef]
- Myers, M.J.; Shepherd, D.L.; Durr, A.J.; Stanton, D.S.; Mohamed, J.S.; Hollander, J.M.; Alway, S.E. The role of SIRT1 in skeletal muscle function and repair of older mice. J. Cachexia Sarcopenia Muscle 2019, 10, 929–949. [Google Scholar] [CrossRef]
- Sharples, A.P.; Hughes, D.C.; Deane, C.S.; Saini, A.; Selman, C.; Stewart, C.E. Longevity and skeletal muscle mass: The role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell 2015, 14, 511–523. [Google Scholar] [CrossRef]
- Pardo, P.S.; Boriek, A.M. The physiological roles of Sirt1 in skeletal muscle. Aging 2011, 3, 430–437. [Google Scholar] [CrossRef]
- Wen, W.; Chen, X.; Huang, Z.; Chen, D.; Chen, H.; Luo, Y.; He, J.; Zheng, P.; Yu, J.; Yu, B. Resveratrol regulates muscle fiber type conversion via miR-22-3p and AMPK/SIRT1/PGC-1alpha pathway. J. Nutr. Biochem. 2020, 77, 108297. [Google Scholar] [CrossRef]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef]
- Rathbone, C.R.; Booth, F.W.; Lees, S.J. Sirt1 increases skeletal muscle precursor cell proliferation. Eur. J. Cell Biol. 2009, 88, 35–44. [Google Scholar] [CrossRef]
- Dasgupta, A.; Shukla, S.K.; Vernucci, E.; King, R.J.; Abrego, J.; Mulder, S.E.; Mullen, N.J.; Graves, G.; Buettner, K.; Thakur, R.; et al. SIRT1-NOX4 signaling axis regulates cancer cachexia. J. Exp. Med. 2020, 217, e20190745. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Han, Y.M.; Lee, H.J.; Park, Y.J.; Hahm, K.B. Nicotinamide Riboside Vitamin B3 Mitigated C26 Adenocarcinoma-Induced Cancer Cachexia. Front. Pharmacol. 2021, 12, 665493. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhao, Y.; Teng, S.; Ni, Y.; Xu, S.; Wu, X.; Zhang, J.; Xu, X.; Fang, Y.; Shi, J.; et al. Atractylodin alleviates cancer anorexia-cachexia syndrome by regulating NPY through hypothalamic Sirt1/AMPK axis-induced autophagy. Biochem. Biophys. Res. Commun. 2022, 625, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ju, R.; Zhu, L.; Li, J.; Chen, W.; Zhang, D.C.; Ye, C.Y.; Guo, L. Carboxyamidotriazole alleviates muscle atrophy in tumor-bearing mice by inhibiting NF-kappaB and activating SIRT1. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 423–433. [Google Scholar] [CrossRef]
- Chu, X.; He, X.; Shi, Z.; Li, C.; Guo, F.; Li, S.; Li, Y.; Na, L.; Sun, C. Ursolic acid increases energy expenditure through enhancing free fatty acid uptake and beta-oxidation via an UCP3/AMPK-dependent pathway in skeletal muscle. Mol. Nutr. Food Res. 2015, 59, 1491–1503. [Google Scholar] [CrossRef]
- Shishodia, S.; Majumdar, S.; Banerjee, S.; Aggarwal, B.B. Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: Correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003, 63, 4375–4383. [Google Scholar]
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid: An anti- and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42. [Google Scholar] [CrossRef]
- Li, S.; Liao, X.; Meng, F.; Wang, Y.; Sun, Z.; Guo, F.; Li, X.; Meng, M.; Li, Y.; Sun, C. Therapeutic role of ursolic acid on ameliorating hepatic steatosis and improving metabolic disorders in high-fat diet-induced non-alcoholic fatty liver disease rats. PLoS ONE 2014, 9, e86724. [Google Scholar] [CrossRef]
- Lin, J.; Chen, Y.; Wei, L.; Hong, Z.; Sferra, T.J.; Peng, J. Ursolic acid inhibits colorectal cancer angiogenesis through suppression of multiple signaling pathways. Int. J. Oncol. 2013, 43, 1666–1674. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Yang, L.; Mei, Y.; Long, H.; Zhang, X.; Zhang, J.; Qimuge, S.; Su, X. Ursolic acid inhibits proliferation and induces apoptosis of cancer cells in vitro and in vivo. J. Biomed. Biotechnol. 2011, 2011, 419343. [Google Scholar] [CrossRef]
- Kunkel, S.D.; Elmore, C.J.; Bongers, K.S.; Ebert, S.M.; Fox, D.K.; Dyle, M.C.; Bullard, S.A.; Adams, C.M. Ursolic acid increases skeletal muscle and brown fat and decreases diet-induced obesity, glucose intolerance and fatty liver disease. PLoS ONE 2012, 7, e39332. [Google Scholar] [CrossRef] [PubMed]
- Bang, H.S.; Seo, D.Y.; Chung, Y.M.; Oh, K.M.; Park, J.J.; Arturo, F.; Jeong, S.H.; Kim, N.; Han, J. Ursolic Acid-induced elevation of serum irisin augments muscle strength during resistance training in men. Korean J. Physiol. Pharmacol. 2014, 18, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, I.; Afzal, M.; Rahman, S.; Iqbal, M.; Imam, F.; Anwar, F. Antiobesity potential of ursolic acid stearoyl glucoside by inhibiting pancreatic lipase. Eur. J. Pharmacol. 2013, 709, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, S.D.; Suneja, M.; Ebert, S.M.; Bongers, K.S.; Fox, D.K.; Malmberg, S.E.; Alipour, F.; Shields, R.K.; Adams, C.M. mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metab. 2011, 13, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Chen, J.A.; Xu, J.; Cao, J.; Wang, Y.; Thomas, S.S.; Hu, Z. Suppression of muscle wasting by the plant-derived compound ursolic acid in a model of chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 327–341. [Google Scholar] [CrossRef]
- Bakhtiari, N.; Hosseinkhani, S.; Soleimani, M.; Hemmati, R.; Noori-Zadeh, A.; Javan, M.; Tashakor, A. Short-term ursolic acid promotes skeletal muscle rejuvenation through enhancing of SIRT1 expression and satellite cells proliferation. Biomed. Pharmacother. 2016, 78, 185–196. [Google Scholar] [CrossRef]
- Sheng, Q.; Li, F.; Chen, G.; Li, J.; Li, J.; Wang, Y.; Lu, Y.; Li, Q.; Li, M.; Chai, K. Ursolic Acid Regulates Intestinal Microbiota and Inflammatory Cell Infiltration to Prevent Ulcerative Colitis. J. Immunol. Res. 2021, 2021, 6679316. [Google Scholar] [CrossRef]
- Chen, L.; Xu, W.; Yang, Q.; Zhang, H.; Wan, L.; Xin, B.; Zhang, J.; Guo, C. Imperatorin alleviates cancer cachexia and prevents muscle wasting via directly inhibiting STAT3. Pharmacol. Res. 2020, 158, 104871. [Google Scholar] [CrossRef]
- Oon, C.E.; Strell, C.; Yeong, K.Y.; Ostman, A.; Prakash, J. SIRT1 inhibition in pancreatic cancer models: Contrasting effects in vitro and in vivo. Eur. J. Pharmacol. 2015, 757, 59–67. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Evans, W.J.; Morley, J.E.; Argiles, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Goto, M.; Fukunishi, S.; Asai, A.; Nishiguchi, S.; Higuchi, K. Cancer Cachexia: Its Mechanism and Clinical Significance. Int. J. Mol. Sci. 2021, 22, 8491. [Google Scholar] [CrossRef] [PubMed]
- Baazim, H.; Antonio-Herrera, L.; Bergthaler, A. The interplay of immunology and cachexia in infection and cancer. Nat. Rev. Immunol. 2022, 22, 309–321. [Google Scholar] [CrossRef]
- Lee, J.Y.; Choi, J.K.; Jeong, N.H.; Yoo, J.; Ha, Y.S.; Lee, B.; Choi, H.; Park, P.H.; Shin, T.Y.; Kwon, T.K.; et al. Anti-inflammatory effects of ursolic acid-3-acetate on human synovial fibroblasts and a murine model of rheumatoid arthritis. Int. Immunopharmacol. 2017, 49, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Prasad, N.R. Effect of ursolic acid, a triterpenoid antioxidant, on ultraviolet-B radiation-induced cytotoxicity, lipid peroxidation and DNA damage in human lymphocytes. Chem. Biol. Interact. 2008, 176, 99–107. [Google Scholar] [CrossRef]
- Yeh, C.T.; Wu, C.H.; Yen, G.C. Ursolic acid, a naturally occurring triterpenoid, suppresses migration and invasion of human breast cancer cells by modulating c-Jun N-terminal kinase, Akt and mammalian target of rapamycin signaling. Mol. Nutr. Food Res. 2010, 54, 1285–1295. [Google Scholar] [CrossRef]
- Li, Y.; Xing, D.; Chen, Q.; Chen, W.R. Enhancement of chemotherapeutic agent-induced apoptosis by inhibition of NF-kappaB using ursolic acid. Int. J. Cancer 2010, 127, 462–473. [Google Scholar] [CrossRef]
- Leng, S.; Hao, Y.; Du, D.; Xie, S.; Hong, L.; Gu, H.; Zhu, X.; Zhang, J.; Fan, D.; Kung, H.F. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int. J. Cancer 2013, 133, 2781–2790. [Google Scholar] [CrossRef]
- Zhao, C.; Yin, S.; Dong, Y.; Guo, X.; Fan, L.; Ye, M.; Hu, H. Autophagy-dependent EIF2AK3 activation compromises ursolic acid-induced apoptosis through upregulation of MCL1 in MCF-7 human breast cancer cells. Autophagy 2013, 9, 196–207. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Chen, W.Y.; Wang, D.H.; Yen, R.C.; Luo, J.; Gu, W.; Baylin, S.B. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 2005, 123, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Carson, J.A. Lewis lung carcinoma regulation of mechanical stretch-induced protein synthesis in cultured myotubes. Am. J. Physiol. Cell Physiol. 2016, 310, C66–C79. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.; Iwanowycz, S.; Saaoud, F.; Wang, J.; Wang, Y.; Sergin, I.; Razani, B.; Fan, D. Ursolic acid enhances macrophage autophagy and attenuates atherogenesis. J. Lipid Res. 2016, 57, 1006–1016. [Google Scholar] [CrossRef]
- Checker, R.; Sandur, S.K.; Sharma, D.; Patwardhan, R.S.; Jayakumar, S.; Kohli, V.; Sethi, G.; Aggarwal, B.B.; Sainis, K.B. Potent anti-inflammatory activity of ursolic acid, a triterpenoid antioxidant, is mediated through suppression of NF-kappaB, AP-1 and NF-AT. PLoS ONE 2012, 7, e31318. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, W.; Ouyang, Z.; Liao, Z.; Li, L.; Zhang, Y.; Gao, J.; Ma, L.; Yu, S. Ursolic Acid Alleviates Cancer Cachexia and Prevents Muscle Wasting via Activating SIRT1. Cancers 2023, 15, 2378. https://doi.org/10.3390/cancers15082378
Tao W, Ouyang Z, Liao Z, Li L, Zhang Y, Gao J, Ma L, Yu S. Ursolic Acid Alleviates Cancer Cachexia and Prevents Muscle Wasting via Activating SIRT1. Cancers. 2023; 15(8):2378. https://doi.org/10.3390/cancers15082378
Chicago/Turabian StyleTao, Weili, Ze Ouyang, Zhiqi Liao, Lu Li, Yujie Zhang, Jiali Gao, Li Ma, and Shiying Yu. 2023. "Ursolic Acid Alleviates Cancer Cachexia and Prevents Muscle Wasting via Activating SIRT1" Cancers 15, no. 8: 2378. https://doi.org/10.3390/cancers15082378
APA StyleTao, W., Ouyang, Z., Liao, Z., Li, L., Zhang, Y., Gao, J., Ma, L., & Yu, S. (2023). Ursolic Acid Alleviates Cancer Cachexia and Prevents Muscle Wasting via Activating SIRT1. Cancers, 15(8), 2378. https://doi.org/10.3390/cancers15082378