

Sequence-Based Platforms for Discovering Biomarkers in Liquid Biopsy of Non-Small-Cell Lung Cancer

 , ,
, ,  ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
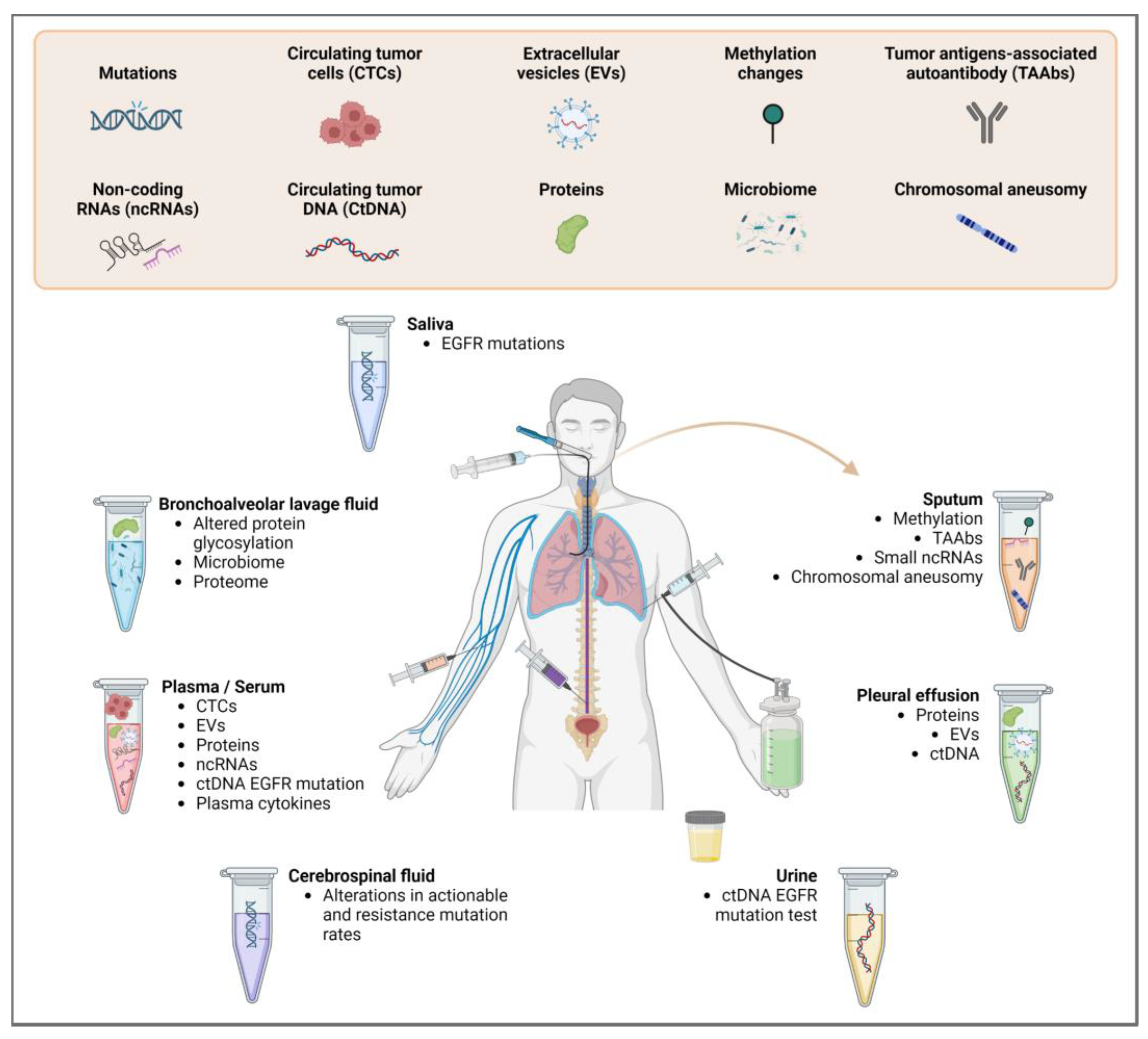
2. Sample Types and Materials for Biomarker Analysis
2.1. Circulating Tumor Cells (CTCs)
2.2. Circulating Tumor DNA (ctDNA)
2.3. Circulating Tumor RNA (ctRNA)
2.4. Extracellular Vesicles (EVs)
3. Commonly Used Techniques for Biomarker Discovery in Lung Liquid Biopsy
3.1. PCR-Based Approaches
3.2. NGS-Based Approaches
3.3. Clinically Validated Platforms for Biomarker Detection

{kind=link}
| Technology | Brief Description | References |
|---|---|---|
| Non Targeted | ||
| WES | Whole Exome Sequencing sequences all exons in ctDNA for mutation detection. Less expensive than WGS (lower coverage). Sample requirement not always feasible in liquid biopsy. | [107,108,109] |
| Digital Karyotyping | Uses WGS to sequence short DNA tags and then aligns these tags to the reference genome to identify genomic alterations, e.g., CNVs, SNVs and SVs. The short DNA tags are typically generated by restriction enzyme digestion. Requires high-quality genomic DNA. | [43,110,111,112] |
| FAST-SeqS | Fast Aneuploidy Screening Test-sequencing System uses individual primer pairs to amplify the repeat regions of interest. The WGS version, called mFAST-SeqS, identifies any somatic mutations in the tumor and then uses those mutations as unique markers for monitoring the disease. | [113,114,115] |
| PARE | Personalized Analysis of Rearranged Ends uses WGS data to identify rearranged ends in ctDNA. Detects structural variations, e.g., translocations and inversions. | [111,116,117] |
| Targeted panel | ||
| Tam-seq | Tagged-Amplicon deep sequencing uses primers targeting regions of interest for a pre-amplification step. Templates undergo individual amplification, aiding purification. | [44] |
| Safe-SeqS | Safe-Sequencing System is a method for profiling low-frequency mutations. The method combines PCR amplification of targeted genomic regions with UMIs and deep sequencing to achieve high accuracy and sensitivity. The use of UMIs reduces errors introduced by PCR amplification and sequencing. | [118] |
| CAPP-Seq | Cancer Personalized Profiling by Deep Sequencing uses a library that generates many hybrid affinities captures of recurrently mutated genomic regions to create the selector, which is used to identify individual-specific mutations in the tumor DNA. The selector is then applied to ctDNA for quantification. | [119] |
| Ion AmpliSeq™ | Customized multiplex PCR amplifies target regions for analysis with the Ion Torrent sequencing platform. | [120] |
| Guardant360® | Analyzes 73 genes commonly mutated in cancer. Digital sequencing technology for mutation detection with 99.5% sensitivity and 99.999% specificity. FDA approval for use in patients with advanced cancer without treatment options. | [100,101] |
| Foundation One®CDx | Analyzes 324 genes and selects genomic signatures, including MSI and TMB. Detects single nucleotide variants, small in/dels, copy number alterations and gene fusions. FDA-approved for use in patients with solid tumors, including NSCLC, to sort patients for specific targeted therapies. | [102,103] |
| iDES | In Integrated Digital Error Suppression, DNA is tagged with UIDs and tracked through library preparation and sequencing for error correction. By incorporating UIDs into NGS, iDES can improve the accuracy and sensitivity of NGS assays, particularly in low-frequency variant detection. | [121] |
| TEC-Seq | Targeted Error Correction Sequencing is a method for profiling low-frequency mutations in cfDNA. Utilizes molecular barcoding to distinguish true mutations from false positive variants. Before any amplification, DNA fragments are tagged with different “exogenous” DNA barcodes. Additionally, the start and end genome mapping positions of paired-end sequenced fragments are used as “endogenous barcodes” to differentiate between individual molecules. This combination of barcodes enables tracking each fragment, allowing for the detection of rare mutations with high accuracy and sensitivity. | [98] |
4. Emerging Methods for Liquid Biopsy Biomarker Discovery
4.1. Long-Read Sequencing
4.2. DNA Methylation Markers
4.3. Single-Cell Sequencing
4.4. Fragmentomics
5. Bioinformatics Pipelines for Analyzing Liquid Biopsy NGS Data
5.1. Sequence Data Processing
5.2. Sequence Data Interpretation
5.2.1. Biological Interpretation of the Sequencing Data
5.2.2. Bioinformatic Platforms for Analyzing Long-Read Sequence Data
5.2.3. Analyzing DNA Whole-Genome Methylation Data
5.2.4. Analyzing Single-Cell Sequence Data
5.2.5. Other Software for Analyzing Liquid Biopsy Samples
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, J.-B.; Hou, L.-K.; Yu, F.; Zhang, J.; Wu, W.; Tang, X.-M.; Sun, F.; Lu, H.-M.; Deng, J.; et al. Liquid Biopsy in Lung Cancer: Significance in Diagnostics, Prediction, and Treatment Monitoring. Mol. Cancer 2022, 21, 25. [Google Scholar] [CrossRef]
- Shen, H.; Jin, Y.; Zhao, H.; Wu, M.; Zhang, K.; Wei, Z.; Wang, X.; Wang, Z.; Li, Y.; Yang, F.; et al. Potential Clinical Utility of Liquid Biopsy in Early-Stage Non-Small Cell Lung Cancer. BMC Med. 2022, 20, 480. [Google Scholar] [CrossRef]
- Gristina, V.; Barraco, N.; La Mantia, M.; Castellana, L.; Insalaco, L.; Bono, M.; Perez, A.; Sardo, D.; Inguglia, S.; Iacono, F.; et al. Clinical Potential of Circulating Cell-Free DNA (CfDNA) for Longitudinally Monitoring Clinical Outcomes in the First-Line Setting of Non-Small-Cell Lung Cancer (NSCLC): A Real-World Prospective Study. Cancers 2022, 14, 6013. [Google Scholar] [CrossRef] [PubMed]
- Guibert, N.; Pradines, A.; Favre, G.; Mazieres, J. Current and Future Applications of Liquid Biopsy in Nonsmall Cell Lung Cancer from Early to Advanced Stages. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2020, 29, 190052. [Google Scholar] [CrossRef]
- Bracht, J.W.P.; Mayo-de-Las-Casas, C.; Berenguer, J.; Karachaliou, N.; Rosell, R. The Present and Future of Liquid Biopsies in Non-Small Cell Lung Cancer: Combining Four Biosources for Diagnosis, Prognosis, Prediction, and Disease Monitoring. Curr. Oncol. Rep. 2018, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M.; et al. Liquid Biopsy: A Step Closer to Transform Diagnosis, Prognosis and Future of Cancer Treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef]
- Hasenleithner, S.O.; Speicher, M.R. A Clinician’s Handbook for Using CtDNA throughout the Patient Journey. Mol. Cancer 2022, 21, 81. [Google Scholar] [CrossRef]
- Saarenheimo, J.; Eigeliene, N.; Andersen, H.; Tiirola, M.; Jekunen, A. The Value of Liquid Biopsies for Guiding Therapy Decisions in Non-Small Cell Lung Cancer. Front. Oncol. 2019, 9, 129. [Google Scholar] [CrossRef]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of Applications of High-Throughput Sequencing in Personalized Medicine: Barriers and Facilitators of Future Progress in Research and Clinical Application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, H. Next-Generation Sequencing in Liquid Biopsy: Cancer Screening and Early Detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid Biopsy and Minimal Residual Disease—Latest Advances and Implications for Cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Lin, C.; Liu, X.; Zheng, B.; Ke, R.; Tzeng, C.-M. Liquid Biopsy, CtDNA Diagnosis through NGS. Life 2021, 11, 890. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, N.; Kohsaka, S.; Kurokawa, K.; Shinno, Y.; Takeda Nakamura, I.; Ueno, T.; Kojima, S.; Kawazu, M.; Suehara, Y.; Ishijima, M.; et al. Highly Sensitive Fusion Detection Using Plasma Cell-Free RNA in Non-Small-Cell Lung Cancers. Cancer Sci. 2021, 112, 4393–4403. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating Liquid Biopsies into the Management of Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Frullanti, E. Different Liquid Biopsies for the Management of Non-Small Cell Lung Cancer in the Mutational Oncology Era. Med. Sci. 2023, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Lianidou, E.; Pantel, K. Liquid Biopsies. Genes. Chromosomes Cancer 2019, 58, 219–232. [Google Scholar] [CrossRef]
- Soyano, A.E.; Dholaria, B.; Marin-Acevedo, J.A.; Diehl, N.; Hodge, D.; Luo, Y.; Manochakian, R.; Chumsri, S.; Adjei, A.; Knutson, K.L.; et al. Peripheral Blood Biomarkers Correlate with Outcomes in Advanced Non-Small Cell Lung Cancer Patients Treated with Anti-PD-1 Antibodies. J. Immunother. Cancer 2018, 6, 129. [Google Scholar] [CrossRef]
- Underwood, J.J.; Quadri, R.S.; Kalva, S.P.; Shah, H.; Sanjeevaiah, A.R.; Beg, M.S.; Sutphin, P.D. Liquid Biopsy for Cancer: Review and Implications for the Radiologist. Radiology 2020, 294, 5–17. [Google Scholar] [CrossRef]
- Michela, B. Liquid Biopsy: A Family of Possible Diagnostic Tools. Diagnostics 2021, 11, 1391. [Google Scholar] [CrossRef]
- Fernández-Lázaro, D.; García Hernández, J.L.; García, A.C.; Córdova Martínez, A.; Mielgo-Ayuso, J.; Cruz-Hernández, J.J. Liquid Biopsy as Novel Tool in Precision Medicine: Origins, Properties, Identification and Clinical Perspective of Cancer’s Biomarkers. Diagnostics 2020, 10, 215. [Google Scholar] [CrossRef]
- Young, R.; Pailler, E.; Billiot, F.; Drusch, F.; Barthelemy, A.; Oulhen, M.; Besse, B.; Soria, J.-C.; Farace, F.; Vielh, P. Circulating Tumor Cells in Lung Cancer. Acta Cytol. 2012, 56, 655–660. [Google Scholar] [CrossRef]
- Lindsay, C.R.; Faugeroux, V.; Michiels, S.; Pailler, E.; Facchinetti, F.; Ou, D.; Bluthgen, M.V.; Pannet, C.; Ngo-Camus, M.; Bescher, G.; et al. A Prospective Examination of Circulating Tumor Cell Profiles in Non-Small-Cell Lung Cancer Molecular Subgroups. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1523–1531. [Google Scholar] [CrossRef]
- Tamminga, M.; de Wit, S.; Schuuring, E.; Timens, W.; Terstappen, L.W.M.M.; Hiltermann, T.J.N.; Groen, H.J.M. Circulating Tumor Cells in Lung Cancer Are Prognostic and Predictive for Worse Tumor Response in Both Targeted- and Chemotherapy. Transl. Lung Cancer Res. 2019, 8, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Shao, W.; Li, Z.; Zhao, R.; Ye, Z. Strategies for Enrichment of Circulating Tumor Cells. Transl. Cancer Res. 2020, 9, 2012–2025. [Google Scholar] [CrossRef]
- Sharma, S.; Zhuang, R.; Long, M.; Pavlovic, M.; Kang, Y.; Ilyas, A.; Asghar, W. Circulating Tumor Cell Isolation, Culture, and Downstream Molecular Analysis. Biotechnol. Adv. 2018, 36, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.; Chen, C.; Zhang, J.; Xu, L.; Zhang, X.; Li, Z.; Chen, Y.; Zhou, J.; Ji, F.; Wang, L. Detection of Circulating Tumor Cells: Opportunities and Challenges. Biomark. Res. 2022, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Wang, M. Circulating Tumor Cells in Patients with Lung Cancer: Developments and Applications for Precision Medicine. Future Oncol. Lond. Engl. 2019, 15, 2531–2542. [Google Scholar] [CrossRef]
- Bhagwat, N.; Dulmage, K.; Pletcher, C.H.; Wang, L.; DeMuth, W.; Sen, M.; Balli, D.; Yee, S.S.; Sa, S.; Tong, F.; et al. An Integrated Flow Cytometry-Based Platform for Isolation and Molecular Characterization of Circulating Tumor Single Cells and Clusters. Sci. Rep. 2018, 8, 5035. [Google Scholar] [CrossRef]
- Basu, S.; Campbell, H.M.; Dittel, B.N.; Ray, A. Purification of Specific Cell Population by Fluorescence Activated Cell Sorting (FACS). J. Vis. Exp. 2010, 41, e1546. [Google Scholar] [CrossRef]
- Volovetskiy, A.B.; Malinina, P.A.; Kapitannikova, A.Y.; Smetanina, S.V.; Kruglova, I.A.; Maslennikova, A.V. Isolation of Circulating Tumor Cells from Peripheral Blood Samples of Cancer Patients Using Microfluidic Technology. Sovrem. Tekhnol. Med. 2020, 12, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Zamarchi, R. Single-Cell Analysis of Circulating Tumor Cells: How Far Have We Come in the -Omics Era? Front. Genet. 2019, 10, 958. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.-M.; Fehm, T.; Orsini, M.; Cayrefourcq, L.; Maudelonde, T.; Pantel, K.; Alix-Panabières, C. Prognostic Relevance of Viable Circulating Tumor Cells Detected by EPISPOT in Metastatic Breast Cancer Patients. Clin. Chem. 2014, 60, 214–221. [Google Scholar] [CrossRef]
- Eslami-S, Z.; Cortés-Hernández, L.E.; Thomas, F.; Pantel, K.; Alix-Panabières, C. Functional Analysis of Circulating Tumour Cells: The KEY to Understand the Biology of the Metastatic Cascade. Br. J. Cancer 2022, 127, 800–810. [Google Scholar] [CrossRef]
- Park, E.S.; Yan, J.P.; Ang, R.A.; Lee, J.H.; Deng, X.; Duffy, S.P.; Beja, K.; Annala, M.; Black, P.C.; Chi, K.N.; et al. Isolation and Genome Sequencing of Individual Circulating Tumor Cells Using Hydrogel Encapsulation and Laser Capture Microdissection. Lab. Chip 2018, 18, 1736–1749. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Herrero, E.; Serna-Blasco, R.; Robado de Lope, L.; González-Rumayor, V.; Romero, A.; Provencio, M. Circulating Tumor DNA as a Cancer Biomarker: An Overview of Biological Features and Factors That May Impact on CtDNA Analysis. Front. Oncol. 2022, 12, 943253. [Google Scholar] [CrossRef]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002198. [Google Scholar] [CrossRef]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating Genomic Features for Non-Invasive Early Lung Cancer Detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic CtDNA Analysis Depicts Early-Stage Lung Cancer Evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Hai, L.; Li, L.; Liu, Z.; Tong, Z.; Sun, Y. Whole-Genome Circulating Tumor DNA Methylation Landscape Reveals Sensitive Biomarkers of Breast Cancer. MedComm 2022, 3, e134. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and Quantification of Mutations in the Plasma of Patients with Colorectal Tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed]
- de Kock, R.; van den Borne, B.; Youssef-El Soud, M.; Belderbos, H.; Brunsveld, L.; Scharnhorst, V.; Deiman, B. Therapy Monitoring of EGFR-Positive Non-Small-Cell Lung Cancer Patients Using DdPCR Multiplex Assays. J. Mol. Diagn. 2021, 23, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.A.; Jiang, P.; Zheng, Y.W.L.; Liao, G.J.W.; Sun, H.; Wong, J.; Siu, S.S.N.; Chan, W.C.; Chan, S.L.; Chan, A.T.C.; et al. Cancer Genome Scanning in Plasma: Detection of Tumor-Associated Copy Number Aberrations, Single-Nucleotide Variants, and Tumoral Heterogeneity by Massively Parallel Sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, Z.; Dai, Y.; Zhu, Q.; Chen, L.-A. Update on Liquid Biopsy in Clinical Management of Non-Small Cell Lung Cancer. OncoTargets Ther. 2019, 12, 5097–5109. [Google Scholar] [CrossRef]
- Tzimagiorgis, G.; Michailidou, E.Z.; Kritis, A.; Markopoulos, A.K.; Kouidou, S. Recovering Circulating Extracellular or Cell-Free RNA from Bodily Fluids. Cancer Epidemiol. 2011, 35, 580–589. [Google Scholar] [CrossRef]
- Ishiba, T.; Hoffmann, A.-C.; Usher, J.; Elshimali, Y.; Sturdevant, T.; Dang, M.; Jaimes, Y.; Tyagi, R.; Gonzales, R.; Grino, M.; et al. Frequencies and Expression Levels of Programmed Death Ligand 1 (PD-L1) in Circulating Tumor RNA (CtRNA) in Various Cancer Types. Biochem. Biophys. Res. Commun. 2018, 500, 621–625. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, T.; Yang, X.; Zhou, Q.; Zhu, S.; Zeng, J.; Chen, H.; Sun, J.; Li, L.; Xu, J.; et al. Polyadenylation Ligation-Mediated Sequencing (PALM-Seq) Characterizes Cell-Free Coding and Non-Coding RNAs in Human Biofluids. Clin. Transl. Med. 2022, 12, e987. [Google Scholar] [CrossRef]
- Kopreski, M.S.; Benko, F.A.; Kwak, L.W.; Gocke, C.D. Detection of Tumor Messenger RNA in the Serum of Patients with Malignant Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 1961–1965. [Google Scholar]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-Free Nucleic Acids as Biomarkers in Cancer Patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Cabús, L.; Lagarde, J.; Curado, J.; Lizano, E.; Pérez-Boza, J. Current Challenges and Best Practices for Cell-Free Long RNA Biomarker Discovery. Biomark. Res. 2022, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.; Fontanini, G. Next Generation Sequencing for Gene Fusion Analysis in Lung Cancer: A Literature Review. Diagnostics 2020, 10, 521. [Google Scholar] [CrossRef] [PubMed]
- Mensah, M.; Borzi, C.; Verri, C.; Suatoni, P.; Conte, D.; Pastorino, U.; Orazio, F.; Sozzi, G.; Boeri, M. MicroRNA Based Liquid Biopsy: The Experience of the Plasma MiRNA Signature Classifier (MSC) for Lung Cancer Screening. J. Vis. Exp. 2017, 128, 56326. [Google Scholar] [CrossRef]
- Costa, C.; Giménez-Capitán, A.; Karachaliou, N.; Rosell, R. Comprehensive Molecular Screening: From the RT-PCR to the RNA-Seq. Transl. Lung Cancer Res. 2013, 2, 87–91. [Google Scholar] [CrossRef]
- Forder, A.; Hsing, C.-Y.; Trejo Vazquez, J.; Garnis, C. Emerging Role of Extracellular Vesicles and Cellular Communication in Metastasis. Cells 2021, 10, 3429. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Li, Y.; Wang, M.; Gu, J.; Xu, W.; Cai, H.; Fang, X.; Zhang, X. Exosomes as a New Frontier of Cancer Liquid Biopsy. Mol. Cancer 2022, 21, 56. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 Identifies Cancer Exosomes and Detects Early Pancreatic Cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Vassalli, G. Exosomes: Therapy Delivery Tools and Biomarkers of Diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-Derived Exosomal MiR-1247-3p Induces Cancer-Associated Fibroblast Activation to Foster Lung Metastasis of Liver Cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle Formation during Reticulocyte Maturation. Association of Plasma Membrane Activities with Released Vesicles (Exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Kanwar, S.S.; Dunlay, C.J.; Simeone, D.M.; Nagrath, S. Microfluidic Device (ExoChip) for on-Chip Isolation, Quantification and Characterization of Circulating Exosomes. Lab Chip 2014, 14, 1891–1900. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Mannavola, F.; Pellè, E.; Quaresmini, D.; Tucci, M.; Silvestris, F. Liquid Biopsy of Cancer: A Multimodal Diagnostic Tool in Clinical Oncology. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhou, M.; Xu, Y.; Gu, X.; Zou, M.; Abudushalamu, G.; Yao, Y.; Fan, X.; Wu, G. Clinical Application and Detection Techniques of Liquid Biopsy in Gastric Cancer. Mol. Cancer 2023, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid Biopsy: Current Technology and Clinical Applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Paweletz, C.P.; Sacher, A.G.; Raymond, C.K.; Alden, R.S.; O’Connell, A.; Mach, S.L.; Kuang, Y.; Gandhi, L.; Kirschmeier, P.; English, J.M.; et al. Bias-Corrected Targeted Next-Generation Sequencing for Rapid, Multiplexed Detection of Actionable Alterations in Cell-Free DNA from Advanced Lung Cancer Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 915–922. [Google Scholar] [CrossRef]
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating Tumor DNA and Liquid Biopsy in Oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P150044 (accessed on 1 March 2023).
- EMA Iressa. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/iressa (accessed on 5 March 2023).
- EMA Tagrisso. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/tagrisso (accessed on 5 March 2023).
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 1647–1662. [Google Scholar] [CrossRef]
- Fleischhacker, M.; Schmidt, B. Circulating Nucleic Acids (CNAs) and Cancer—A Survey. Biochim. Biophys. Acta 2007, 1775, 181–232. [Google Scholar] [CrossRef]
- Ma, M.; Zhu, H.; Zhang, C.; Sun, X.; Gao, X.; Chen, G. “Liquid Biopsy”-CtDNA Detection with Great Potential and Challenges. Ann. Transl. Med. 2015, 3, 235. [Google Scholar] [CrossRef] [PubMed]
- Köhn, L.; Johansson, M.; Grankvist, K.; Nilsson, J. Liquid Biopsies in Lung Cancer-Time to Implement Research Technologies in Routine Care? Ann. Transl. Med. 2017, 5, 278. [Google Scholar] [CrossRef]
- Mosko, M.J.; Nakorchevsky, A.A.; Flores, E.; Metzler, H.; Ehrich, M.; van den Boom, D.J.; Sherwood, J.L.; Nygren, A.O.H. Ultrasensitive Detection of Multiplexed Somatic Mutations Using MALDI-TOF Mass Spectrometry. J. Mol. Diagn. 2016, 18, 23–31. [Google Scholar] [CrossRef]
- Milbury, C.A.; Chen, C.C.; Mamon, H.; Liu, P.; Santagata, S.; Makrigiorgos, G.M. Multiplex Amplification Coupled with COLD-PCR and High Resolution Melting Enables Identification of Low-Abundance Mutations in Cancer Samples with Low DNA Content. J. Mol. Diagn. 2011, 13, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.-L.G.; Pallisgaard, N.; Vogelius, I.; Jakobsen, A. Quantitative Cell-Free DNA, KRAS, and BRAF Mutations in Plasma from Patients with Metastatic Colorectal Cancer during Treatment with Cetuximab and Irinotecan. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1177–1185. [Google Scholar] [CrossRef]
- Watanabe, K.; Fukuhara, T.; Tsukita, Y.; Morita, M.; Suzuki, A.; Tanaka, N.; Terasaki, H.; Nukiwa, T.; Maemondo, M. EGFR Mutation Analysis of Circulating Tumor DNA Using an Improved PNA-LNA PCR Clamp Method. Can. Respir. J. 2016, 2016, 5297329. [Google Scholar] [CrossRef]
- Oh, J.E.; Lim, H.S.; An, C.H.; Jeong, E.G.; Han, J.Y.; Lee, S.H.; Yoo, N.J. Detection of Low-Level KRAS Mutations Using PNA-Mediated Asymmetric PCR Clamping and Melting Curve Analysis with Unlabeled Probes. J. Mol. Diagn. 2010, 12, 418–424. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-Molecule PCR on Microparticles in Water-in-Oil Emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; He, Q.; Liang, H.; Cheng, B.; Li, J.; Xiong, S.; Zhao, Y.; Guo, M.; Liu, Z.; He, J.; et al. Diagnostic Accuracy of Droplet Digital PCR and Amplification Refractory Mutation System PCR for Detecting EGFR Mutation in Cell-Free DNA of Lung Cancer: A Meta-Analysis. Front. Oncol. 2020, 10, 290. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, J.; Zhao, J.; Ren, P.; Wang, Z.; Wei, B.; Dong, B.; Sun, R.; Wang, X.; Groen, H.J.M.; et al. Developing Ultrasensitive Library-Aliquot-Based Droplet Digital PCR for Detecting T790M in Plasma-Circulating Tumor DNA of Non-Small-Cell-Lung-Cancer Patients. Anal. Chem. 2018, 90, 11203–11209. [Google Scholar] [CrossRef]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming Single DNA Molecules into Fluorescent Magnetic Particles for Detection and Enumeration of Genetic Variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex Picodroplet Digital PCR to Detect KRAS Mutations in Circulating DNA from the Plasma of Colorectal Cancer Patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef]
- Mouliere, F.; El Messaoudi, S.; Gongora, C.; Guedj, A.-S.; Robert, B.; Del Rio, M.; Molina, F.; Lamy, P.-J.; Lopez-Crapez, E.; Mathonnet, M.; et al. Circulating Cell-Free DNA from Colorectal Cancer Patients May Reveal High KRAS or BRAF Mutation Load. Transl. Oncol. 2013, 6, 319–328. [Google Scholar] [CrossRef]
- Mouliere, F.; El Messaoudi, S.; Pang, D.; Dritschilo, A.; Thierry, A.R. Multi-Marker Analysis of Circulating Cell-Free DNA toward Personalized Medicine for Colorectal Cancer. Mol. Oncol. 2014, 8, 927–941. [Google Scholar] [CrossRef]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET Receptor Drives Resistance to Anti-EGFR Therapies in Colorectal Cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Pu, H.; Liu, Q.; Guo, Z.; Luo, D. Circulating Tumor DNA-A Novel Biomarker of Tumor Progression and Its Favorable Detection Techniques. Cancers 2022, 14, 6025. [Google Scholar] [CrossRef]
- Wang, W.; Song, Z.; Zhang, Y. A Comparison of DdPCR and ARMS for Detecting EGFR T790M Status in CtDNA from Advanced NSCLC Patients with Acquired EGFR-TKI Resistance. Cancer Med. 2017, 6, 154–162. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Blood Tests That Can Help Guide Cancer Treatment—NCI. Available online: https://www.cancer.gov/news-events/cancer-currents-blog/2020/fda-guardant-360-foundation-one-cancer-liquid-biopsy (accessed on 1 March 2023).
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Belic, J.; Koch, M.; Ulz, P.; Auer, M.; Gerhalter, T.; Mohan, S.; Fischereder, K.; Petru, E.; Bauernhofer, T.; Geigl, J.B.; et al. Rapid Identification of Plasma DNA Samples with Increased CtDNA Levels by a Modified FAST-SeqS Approach. Clin. Chem. 2015, 61, 838–849. [Google Scholar] [CrossRef]
- Castro-Giner, F.; Gkountela, S.; Donato, C.; Alborelli, I.; Quagliata, L.; Ng, C.K.Y.; Piscuoglio, S.; Aceto, N. Cancer Diagnosis Using a Liquid Biopsy: Challenges and Expectations. Diagnostics 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- McCoach, C.E.; Blakely, C.M.; Banks, K.C.; Levy, B.; Chue, B.M.; Raymond, V.M.; Le, A.T.; Lee, C.E.; Diaz, J.; Waqar, S.N.; et al. Clinical Utility of Cell-Free DNA for the Detection of ALK Fusions and Genomic Mechanisms of ALK Inhibitor Resistance in Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2758–2770. [Google Scholar] [CrossRef]
- Baer, C.; Kern, W.; Koch, S.; Nadarajah, N.; Schindela, S.; Meggendorfer, M.; Haferlach, C.; Haferlach, T. Ultra-Deep Sequencing Leads to Earlier and More Sensitive Detection of the Tyrosine Kinase Inhibitor Resistance Mutation T315I in Chronic Myeloid Leukemia. Haematologica 2016, 101, 830–838. [Google Scholar] [CrossRef]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct Detection of Early-Stage Cancers Using Circulating Tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Imperial, R.; Nazer, M.; Ahmed, Z.; Kam, A.E.; Pluard, T.J.; Bahaj, W.; Levy, M.; Kuzel, T.M.; Hayden, D.M.; Pappas, S.G.; et al. Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (CtDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers 2019, 11, 1399. [Google Scholar] [CrossRef] [PubMed]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.B.; Kopetz, E.S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef] [PubMed]
- Solutions—Guardant360. Available online: https://www.guardanthealthamea.com/solutions/ (accessed on 1 March 2023).
- Milbury, C.A.; Creeden, J.; Yip, W.-K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J.; et al. Clinical and Analytical Validation of FoundationOne®CDx, a Comprehensive Genomic Profiling Assay for Solid Tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef] [PubMed]
- FoundationOne CDx|Foundation Medicine. Available online: https://www.foundationmedicine.com/test/foundationone-cdx (accessed on 1 March 2023).
- Torres, S.; González, Á.; Cunquero Tomas, A.J.; Calabuig Fariñas, S.; Ferrero, M.; Mirda, D.; Sirera, R.; Jantus-Lewintre, E.; Camps, C. A Profile on Cobas® EGFR Mutation Test v2 as Companion Diagnostic for First-Line Treatment of Patients with Non-Small Cell Lung Cancer. Expert Rev. Mol. Diagn. 2020, 20, 575–582. [Google Scholar] [CrossRef]
- Remon, J.; Hendriks, L.E.L.; Cardona, A.F.; Besse, B. EGFR Exon 20 Insertions in Advanced Non-Small Cell Lung Cancer: A New History Begins. Cancer Treat. Rev. 2020, 90, 102105. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Amivantamab-Vmjw for Metastatic Non-Small Cell Lung Cancer|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-amivantamab-vmjw-metastatic-non-small-cell-lung-cancer (accessed on 6 April 2023).
- Koeppel, F.; Blanchard, S.; Jovelet, C.; Genin, B.; Marcaillou, C.; Martin, E.; Rouleau, E.; Solary, E.; Soria, J.-C.; André, F.; et al. Whole Exome Sequencing for Determination of Tumor Mutation Load in Liquid Biopsy from Advanced Cancer Patients. PLoS ONE 2017, 12, e0188174. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-Invasive Analysis of Acquired Resistance to Cancer Therapy by Sequencing of Plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.K.; Angus, L.; Nasserinejad, K.; Jager, A.; Jansen, M.P.H.M.; Martens, J.W.M.; Sleijfer, S. Whole Exome Sequencing of Cell-Free DNA—A Systematic Review and Bayesian Individual Patient Data Meta-Analysis. Cancer Treat. Rev. 2020, 83, 101951. [Google Scholar] [CrossRef]
- Wang, T.-L.; Maierhofer, C.; Speicher, M.R.; Lengauer, C.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. Digital Karyotyping. Proc. Natl. Acad. Sci. USA 2002, 99, 16156–16161. [Google Scholar] [CrossRef] [PubMed]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of Chromosomal Alterations in the Circulation of Cancer Patients with Whole-Genome Sequencing. Sci. Transl. Med. 2012, 4, 162ra154. [Google Scholar] [CrossRef]
- Diaz, L.A.; Sausen, M.; Fisher, G.A.; Velculescu, V.E. Insights into Therapeutic Resistance from Whole-Genome Analyses of Circulating Tumor DNA. Oncotarget 2013, 4, 1856–1857. [Google Scholar] [CrossRef]
- Kinde, I.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. FAST-SeqS: A Simple and Efficient Method for the Detection of Aneuploidy by Massively Parallel Sequencing. PLoS ONE 2012, 7, e41162. [Google Scholar] [CrossRef] [PubMed]
- Mendelaar, P.A.J.; Robbrecht, D.G.J.; Rijnders, M.; de Wit, R.; de Weerd, V.; Deger, T.; Westgeest, H.M.; Aarts, M.J.B.; Voortman, J.; Martens, J.W.M.; et al. Genome-Wide Aneuploidy Detected by MFast-SeqS in Circulating Cell-Free DNA Is Associated with Poor Response to Pembrolizumab in Patients with Advanced Urothelial Cancer. Mol. Oncol. 2022, 16, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- Belic, J.; Koch, M.; Ulz, P.; Auer, M.; Gerhalter, T.; Mohan, S.; Fischereder, K.; Petru, E.; Bauernhofer, T.; Geigl, J.B.; et al. MFast-SeqS as a Monitoring and Pre-Screening Tool for Tumor-Specific Aneuploidy in Plasma DNA. Adv. Exp. Med. Biol. 2016, 924, 147–155. [Google Scholar] [CrossRef]
- Leary, R.J.; Kinde, I.; Diehl, F.; Schmidt, K.; Clouser, C.; Duncan, C.; Antipova, A.; Lee, C.; McKernan, K.; De La Vega, F.M.; et al. Development of Personalized Tumor Biomarkers Using Massively Parallel Sequencing. Sci. Transl. Med. 2010, 2, 20ra14. [Google Scholar] [CrossRef]
- McBride, D.J.; Orpana, A.K.; Sotiriou, C.; Joensuu, H.; Stephens, P.J.; Mudie, L.J.; Hämäläinen, E.; Stebbings, L.A.; Andersson, L.C.; Flanagan, A.M.; et al. Use of Cancer-Specific Genomic Rearrangements to Quantify Disease Burden in Plasma from Patients with Solid Tumors. Genes. Chromosomes Cancer 2010, 49, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and Quantification of Rare Mutations with Massively Parallel Sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Rothé, F.; Laes, J.-F.; Lambrechts, D.; Smeets, D.; Vincent, D.; Maetens, M.; Fumagalli, D.; Michiels, S.; Drisis, S.; Moerman, C.; et al. Plasma Circulating Tumor DNA as an Alternative to Metastatic Biopsies for Mutational Analysis in Breast Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated Digital Error Suppression for Improved Detection of Circulating Tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Pollard, M.O.; Gurdasani, D.; Mentzer, A.J.; Porter, T.; Sandhu, M.S. Long Reads: Their Purpose and Place. Hum. Mol. Genet. 2018, 27, R234–R241. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Lo, Y.M.D.; Han, D.S.C.; Jiang, P.; Chiu, R.W.K. Epigenetics, Fragmentomics, and Topology of Cell-Free DNA in Liquid Biopsies. Science 2021, 372, eaaw3616. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.C.Y.; Jiang, P.; Peng, W.; Cheng, S.H.; Cheung, Y.T.T.; Tse, O.Y.O.; Shang, H.; Poon, L.C.; Leung, T.Y.; Chan, K.C.A.; et al. Single-Molecule Sequencing Reveals a Large Population of Long Cell-Free DNA Molecules in Maternal Plasma. Proc. Natl. Acad. Sci. USA 2021, 118, e2114937118. [Google Scholar] [CrossRef]
- Choy, L.Y.L.; Peng, W.; Jiang, P.; Cheng, S.H.; Yu, S.C.Y.; Shang, H.; Olivia Tse, O.Y.; Wong, J.; Wong, V.W.S.; Wong, G.L.H.; et al. Single-Molecule Sequencing Enables Long Cell-Free DNA Detection and Direct Methylation Analysis for Cancer Patients. Clin. Chem. 2022, 68, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.C.Y.; Deng, J.; Qiao, R.; Cheng, S.H.; Peng, W.; Lau, S.L.; Choy, L.Y.L.; Leung, T.Y.; Wong, J.; Wong, V.W.-S.; et al. Comparison of Single Molecule, Real-Time Sequencing and Nanopore Sequencing for Analysis of the Size, End-Motif, and Tissue-of-Origin of Long Cell-Free DNA in Plasma. Clin. Chem. 2023, 69, 168–179. [Google Scholar] [CrossRef]
- Peters, B.A.; Kermani, B.G.; Sparks, A.B.; Alferov, O.; Hong, P.; Alexeev, A.; Jiang, Y.; Dahl, F.; Tang, Y.T.; Haas, J.; et al. Accurate Whole-Genome Sequencing and Haplotyping from 10 to 20 Human Cells. Nature 2012, 487, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping Germline and Cancer Genomes with High-Throughput Linked-Read Sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, X.; Zheng, J.; Dong, D. DNA Methylome Profiling of Circulating Tumor Cells in Lung Cancer at Single Base-Pair Resolution. Oncogene 2021, 40, 1884–1895. [Google Scholar] [CrossRef]
- Gonçalves, E.; Gonçalves-Reis, M.; Pereira-Leal, J.B.; Cardoso, J. DNA Methylation Fingerprint of Hepatocellular Carcinoma from Tissue and Liquid Biopsies. Sci. Rep. 2022, 12, 11512. [Google Scholar] [CrossRef]
- Luo, H.; Wei, W.; Ye, Z.; Zheng, J.; Xu, R.-H. Liquid Biopsy of Methylation Biomarkers in Cell-Free DNA. Trends Mol. Med. 2021, 27, 482–500. [Google Scholar] [CrossRef]
- Zhang, Y.; Bewerunge-Hudler, M.; Schick, M.; Burwinkel, B.; Herpel, E.; Hoffmeister, M.; Brenner, H. Blood-Derived DNA Methylation Predictors of Mortality Discriminate Tumor and Healthy Tissue in Multiple Organs. Mol. Oncol. 2020, 14, 2111–2123. [Google Scholar] [CrossRef]
- Sproul, D.; Meehan, R.R. Genomic Insights into Cancer-Associated Aberrant CpG Island Hypermethylation. Brief. Funct. Genom. 2013, 12, 174–190. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. TIG 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Balgkouranidou, I.; Chimonidou, M.; Milaki, G.; Tsaroucha, E.; Kakolyris, S.; Georgoulias, V.; Lianidou, E. SOX17 Promoter Methylation in Plasma Circulating Tumor DNA of Patients with Non-Small Cell Lung Cancer. Clin. Chem. Lab. Med. 2016, 54, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ricarte Filho, J.; Mallisetty, A.; Villani, C.; Kottorou, A.; Rodgers, K.; Chen, C.; Ito, T.; Holmes, K.; Gastala, N.; et al. Detection of Promoter DNA Methylation in Urine and Plasma Aids the Detection of Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, A.; Liu, M.C.; Klein, E.A.; Venn, O.; Hubbell, E.; Beausang, J.F.; Gross, S.; Melton, C.; Fields, A.P.; Liu, Q.; et al. Evaluation of Cell-Free DNA Approaches for Multi-Cancer Early Detection. Cancer Cell 2022, 40, 1537–1549.e12. [Google Scholar] [CrossRef]
- Jamieson, E.; Korologou-Linden, R.; Wootton, R.E.; Guyatt, A.L.; Battram, T.; Burrows, K.; Gaunt, T.R.; Tobin, M.D.; Munafò, M.; Davey Smith, G.; et al. Smoking, DNA Methylation, and Lung Function: A Mendelian Randomization Analysis to Investigate Causal Pathways. Am. J. Hum. Genet. 2020, 106, 315–326. [Google Scholar] [CrossRef]
- Lissa, D.; Robles, A.I. Sputum-Based DNA Methylation Biomarkers to Guide Lung Cancer Screening Decisions. J. Thorac. Dis. 2017, 9, 4308–4310. [Google Scholar] [CrossRef]
- Li, P.; Liu, S.; Du, L.; Mohseni, G.; Zhang, Y.; Wang, C. Liquid Biopsies Based on DNA Methylation as Biomarkers for the Detection and Prognosis of Lung Cancer. Clin. Epigenet. 2022, 14, 118. [Google Scholar] [CrossRef]
- Loyfer, N.; Magenheim, J.; Peretz, A.; Cann, G.; Bredno, J.; Klochendler, A.; Fox-Fisher, I.; Shabi-Porat, S.; Hecht, M.; Pelet, T.; et al. A DNA Methylation Atlas of Normal Human Cell Types. Nature 2023, 613, 355–364. [Google Scholar] [CrossRef]
- Chen, Q.-F.; Gao, H.; Pan, Q.-Y.; Wang, Y.-J.; Zhong, X.-N. Analysis at the Single-Cell Level Indicates an Important Role of Heterogeneous Global DNA Methylation Status on the Progression of Lung Adenocarcinoma. Sci. Rep. 2021, 11, 23337. [Google Scholar] [CrossRef]
- Yong, W.-S.; Hsu, F.-M.; Chen, P.-Y. Profiling Genome-Wide DNA Methylation. Epigenet. Chromatin 2016, 9, 26. [Google Scholar] [CrossRef]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome Analysis Using MeDIP-Seq with Low DNA Concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liao, K.; Yang, X.; Wu, C.; Wu, W. Using Single-Cell Sequencing Technology to Detect Circulating Tumor Cells in Solid Tumors. Mol. Cancer 2021, 20, 104. [Google Scholar] [CrossRef]
- Lim, S.B.; Di Lee, W.; Vasudevan, J.; Lim, W.-T.; Lim, C.T. Liquid Biopsy: One Cell at a Time. NPJ Precis. Oncol. 2019, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, Z.; Shi, Q. Liquid Biopsy Based Single-Cell Transcriptome Profiling Characterizes Heterogeneity of Disseminated Tumor Cells from Lung Adenocarcinoma. Proteomics 2020, 20, e1900224. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global Characterization of T Cells in Non-Small-Cell Lung Cancer by Single-Cell Sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, I.; Cerezo-Wallis, D.; Hidalgo, A. Understanding NSCLC, One Cell at a Time. Cancer Cell 2022, 40, 1459–1461. [Google Scholar] [CrossRef]
- Keller, L.; Pantel, K. Unravelling Tumour Heterogeneity by Single-Cell Profiling of Circulating Tumour Cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef]
- Pei, H.; Li, L.; Han, Z.; Wang, Y.; Tang, B. Recent Advances in Microfluidic Technologies for Circulating Tumor Cells: Enrichment, Single-Cell Analysis, and Liquid Biopsy for Clinical Applications. Lab Chip 2020, 20, 3854–3875. [Google Scholar] [CrossRef]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible Copy Number Variation Patterns among Single Circulating Tumor Cells of Lung Cancer Patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088. [Google Scholar] [CrossRef]
- Kim, O.; Lee, D.; Chungwon Lee, A.; Lee, Y.; Bae, H.J.; Lee, H.-B.; Kim, R.N.; Han, W.; Kwon, S. Whole Genome Sequencing of Single Circulating Tumor Cells Isolated by Applying a Pulsed Laser to Cell-Capturing Microstructures. Small Weinh. Bergstr. Ger. 2019, 15, e1902607. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, C.; Lu, S.; Zhou, W.; Tang, F.; Xie, X.S.; Huang, Y. Uniform and Accurate Single-Cell Sequencing Based on Emulsion Whole-Genome Amplification. Proc. Natl. Acad. Sci. USA 2015, 112, 11923–11928. [Google Scholar] [CrossRef]
- Kojima, M.; Harada, T.; Fukazawa, T.; Kurihara, S.; Saeki, I.; Takahashi, S.; Hiyama, E. Single-Cell DNA and RNA Sequencing of Circulating Tumor Cells. Sci. Rep. 2021, 11, 22864. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current Best Practices in Single-Cell RNA-Seq Analysis: A Tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Park, S.-M.; Wong, D.J.; Ooi, C.C.; Kurtz, D.M.; Vermesh, O.; Aalipour, A.; Suh, S.; Pian, K.L.; Chabon, J.J.; Lee, S.H.; et al. Molecular Profiling of Single Circulating Tumor Cells from Lung Cancer Patients. Proc. Natl. Acad. Sci. USA 2016, 113, E8379–E8386. [Google Scholar] [CrossRef]
- Lim, S.B.; Yeo, T.; Lee, W.D.; Bhagat, A.A.S.; Tan, S.J.; Tan, D.S.W.; Lim, W.-T.; Lim, C.T. Addressing Cellular Heterogeneity in Tumor and Circulation for Refined Prognostication. Proc. Natl. Acad. Sci. USA 2019, 116, 17957–17962. [Google Scholar] [CrossRef]
- Zhitnyuk, Y.V.; Koval, A.P.; Alferov, A.A.; Shtykova, Y.A.; Mamedov, I.Z.; Kushlinskii, N.E.; Chudakov, D.M.; Shcherbo, D.S. Deep CfDNA Fragment End Profiling Enables Cancer Detection. Mol. Cancer 2022, 21, 26. [Google Scholar] [CrossRef]
- Gianni, C.; Palleschi, M.; Merloni, F.; Di Menna, G.; Sirico, M.; Sarti, S.; Virga, A.; Ulivi, P.; Cecconetto, L.; Mariotti, M.; et al. Cell-Free DNA Fragmentomics: A Promising Biomarker for Diagnosis, Prognosis and Prediction of Response in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 14197. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R. Circulating DNA Fragmentomics and Cancer Screening. Cell Genom. 2023, 3, 100242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Z.; Tang, W.; Wang, X.; Liu, R.; Bao, H.; Chen, X.; Wei, Y.; Wu, S.; Bao, H.; et al. Ultrasensitive and Affordable Assay for Early Detection of Primary Liver Cancer Using Plasma Cell-Free DNA Fragmentomics. Hepatol. Baltim. Md 2022, 76, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.C.; Lo, Y.M.D. Cell-Free DNA Fragmentomics in Liquid Biopsy. Diagnostics 2022, 12, 978. [Google Scholar] [CrossRef]
- Ivanov, M.; Baranova, A.; Butler, T.; Spellman, P.; Mileyko, V. Non-Random Fragmentation Patterns in Circulating Cell-Free DNA Reflect Epigenetic Regulation. BMC Genom. 2015, 16 (Suppl. S13), S1. [Google Scholar] [CrossRef]
- Liu, Y. At the Dawn: Cell-Free DNA Fragmentomics and Gene Regulation. Br. J. Cancer 2022, 126, 379–390. [Google Scholar] [CrossRef]
- Stroun, M.; Anker, P.; Lyautey, J.; Lederrey, C.; Maurice, P.A. Isolation and Characterization of DNA from the Plasma of Cancer Patients. Eur. J. Cancer Clin. Oncol. 1987, 23, 707–712. [Google Scholar] [CrossRef]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-Wide Cell-Free DNA Fragmentation in Patients with Cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced Detection of Circulating Tumor DNA by Fragment Size Analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Mei, W.; Ma, K.; Zeng, C. Circulating Tumor DNA and Minimal Residual Disease (MRD) in Solid Tumors: Current Horizons and Future Perspectives. Front. Oncol. 2021, 11, 763790. [Google Scholar] [CrossRef]
- Vessies, D.C.L.; Schuurbiers, M.M.F.; van der Noort, V.; Schouten, I.; Linders, T.C.; Lanfermeijer, M.; Ramkisoensing, K.L.; Hartemink, K.J.; Monkhorst, K.; van den Heuvel, M.M.; et al. Combining Variant Detection and Fragment Length Analysis Improves Detection of Minimal Residual Disease in Postsurgery Circulating Tumour DNA of Stage II-IIIA NSCLC Patients. Mol. Oncol. 2022, 16, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Wang, Z.; Ma, X.; Guo, W.; Zhang, X.; Tang, W.; Chen, X.; Wang, X.; Chen, Y.; Mo, S.; et al. Letter to the Editor: An Ultra-Sensitive Assay Using Cell-Free DNA Fragmentomics for Multi-Cancer Early Detection. Mol. Cancer 2022, 21, 129. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, F.; Li, M.; Bao, H.; Chen, X.; Zhu, M.; Liu, R.; Xu, X.; Yang, S.; Wu, X.; et al. Multi-Dimensional Cell-Free DNA Fragmentomic Assay for Detection of Early-Stage Lung Cancer. Am. J. Respir. Crit. Care Med. 2022. [Google Scholar] [CrossRef]
- Guo, W.; Chen, X.; Liu, R.; Liang, N.; Ma, Q.; Bao, H.; Xu, X.; Wu, X.; Yang, S.; Shao, Y.; et al. Sensitive Detection of Stage I Lung Adenocarcinoma Using Plasma Cell-Free DNA Breakpoint Motif Profiling. EBioMedicine 2022, 81, 104131. [Google Scholar] [CrossRef]
- Moorthie, S.; Hall, A.; Wright, C.F. Informatics and Clinical Genome Sequencing: Opening the Black Box. Genet. Med. Off. J. Am. Coll. Med. Genet. 2013, 15, 165–171. [Google Scholar] [CrossRef]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef]
- BCL Convert Support. Available online: https://support.illumina.com/sequencing/sequencing_software/bcl-convert.html (accessed on 1 March 2023).
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ File Format for Sequences with Quality Scores, and the Solexa/Illumina FASTQ Variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- de Sena Brandine, G.; Smith, A.D. Falco: High-Speed FastQC Emulation for Quality Control of Sequencing Data. F1000Research 2019, 8, 1874. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A Toolkit for Quality Control of next Generation Sequencing Data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 March 2023).
- Reinert, K.; Langmead, B.; Weese, D.; Evers, D.J. Alignment of Next-Generation Sequencing Reads. Annu. Rev. Genom. Hum. Genet. 2015, 16, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Salzberg, S.L. How to Map Billions of Short Reads onto Genomes. Nat. Biotechnol. 2009, 27, 455–457. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genom. Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Park, C.; Bennett, C.; Thornton, M.; Kim, D. Rapid and Accurate Alignment of Nucleotide Conversion Sequencing Reads with HISAT-3N. Genom. Res. 2021, 31, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genom. Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant Detection in Massively Parallel Sequencing of Individual and Pooled Samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to Accessing Data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genom. Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genom. Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genom. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A Novel Biological Module-Centric Algorithm to Functionally Analyze Large Gene Lists. Genom. Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA Update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A Novel and Versatile Variant Caller for next-Generation Sequencing in Cancer Research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and Accurate Calling of Germline and Somatic Variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genom. Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Alser, M.; Rotman, J.; Deshpande, D.; Taraszka, K.; Shi, H.; Baykal, P.I.; Yang, H.T.; Xue, V.; Knyazev, S.; Singer, B.D.; et al. Technology Dictates Algorithms: Recent Developments in Read Alignment. Genom. Biol. 2021, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- CCS Home|CCS Docs. Available online: https://ccs.how/ (accessed on 5 March 2023).
- Oxford Nanopore Technologies. Available online: https://github.com/nanoporetech (accessed on 1 March 2023).
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-Value and M-Value Methods for Quantifying Methylation Levels by Microarray Analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating High-Throughput Genomic Analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Bioconductor—BiocViews. Available online: http://www.bioconductor.org/packages/release/BiocViews.html#___DNAMethylation (accessed on 1 March 2023).
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of Sequencing-Based Methods to Profile DNA Methylation and Identification of Monoallelic Epigenetic Modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Beck, D.; Ben Maamar, M.; Skinner, M.K. Genome-Wide CpG Density and DNA Methylation Analysis Method (MeDIP, RRBS, and WGBS) Comparisons. Epigenetics 2022, 17, 518–530. [Google Scholar] [CrossRef]
- Konwar, C.; Del Gobbo, G.; Yuan, V.; Robinson, W.P. Considerations When Processing and Interpreting Genomics Data of the Placenta. Placenta 2019, 84, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genom. Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Campbell, K.R.; Lun, A.T.L.; Wills, Q.F. Scater: Pre-Processing, Quality Control, Normalization and Visualization of Single-Cell RNA-Seq Data in R. Bioinformatics 2017, 33, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- GitHub–Lmcinnes/Umap: Uniform Manifold Approximation and Projection. Available online: https://github.com/lmcinnes/umap (accessed on 1 March 2023).
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The Human Cell Atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A Manually Curated Resource of Cell Markers in Human and Mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. Scmap: Projection of Single-Cell RNA-Seq Data across Data Sets. Nat. Methods 2018, 15, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The Dynamics and Regulators of Cell Fate Decisions Are Revealed by Pseudotemporal Ordering of Single Cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Bendall, S.C.; Davis, K.L.; Amir, E.-A.D.; Tadmor, M.D.; Simonds, E.F.; Chen, T.J.; Shenfeld, D.K.; Nolan, G.P.; Pe’er, D. Single-Cell Trajectory Detection Uncovers Progression and Regulatory Coordination in Human B Cell Development. Cell 2014, 157, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell Lineage and Pseudotime Inference for Single-Cell Transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genom. Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinform. Oxf. Engl. 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed Graph Embedding Resolves Complex Single-Cell Trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.-A.; Trapnell, C. Single-Cell MRNA Quantification and Differential Analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Schep, A.N.; Wu, B.; Buenrostro, J.D.; Greenleaf, W.J. ChromVAR: Inferring Transcription-Factor-Associated Accessibility from Single-Cell Epigenomic Data. Nat. Methods 2017, 14, 975–978. [Google Scholar] [CrossRef]
- Hie, B.; Bryson, B.; Berger, B. Efficient Integration of Heterogeneous Single-Cell Transcriptomes Using Scanorama. Nat. Biotechnol. 2019, 37, 685–691. [Google Scholar] [CrossRef]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Katsman, E.; Orlanski, S.; Martignano, F.; Fox-Fisher, I.; Shemer, R.; Dor, Y.; Zick, A.; Eden, A.; Petrini, I.; Conticello, S.G.; et al. Detecting Cell-of-Origin and Cancer-Specific Methylation Features of Cell-Free DNA from Nanopore Sequencing. Genom. Biol. 2022, 23, 158. [Google Scholar] [CrossRef]
- GitHub—Methylgrammarlab/Cfdna-Ont. Available online: https://github.com/methylgrammarlab/cfdna-ont (accessed on 5 March 2023).
- GitHub—Mouliere-Lab/FrEIA: Fragment End Integrated Analysis Tool. Available online: https://github.com/mouliere-lab/FrEIA (accessed on 5 March 2023).
- Moldovan, N.; van der Pol, Y.; van den Ende, T.; Boers, D.; Verkuijlen, S.; Creemers, A.; Ramaker, J.; Vu, T.; Fransen, M.F.; Pegtel, M.; et al. Genome-Wide Cell-Free DNA Termini in Patients with Cancer. medRxiv 2021. [Google Scholar] [CrossRef]
- GitHub—Friend1ws/Nanomonsv: SV Detection Tool for Nanopore Sequence Reads. Available online: https://github.com/friend1ws/nanomonsv (accessed on 5 March 2023).
- Shiraishi, Y.; Koya, J.; Chiba, K.; Okada, A.; Arai, Y.; Saito, Y.; Shibata, T.; Kataoka, K. Precise Characterization of Somatic Complex Structural Variations from Paired Long-Read Sequencing Data with Nanomonsv. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.H.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate Characterization of Patients’ Genomic Structural Variants Using Low-Depth Nanopore Sequencing. Genom. Biol. 2020, 21, 56. [Google Scholar] [CrossRef]
- Zafar, H.; Wang, Y.; Nakhleh, L.; Navin, N.; Chen, K. Monovar: Single-Nucleotide Variant Detection in Single Cells. Nat. Methods 2016, 13, 505–507. [Google Scholar] [CrossRef]
- Ross, E.M.; Markowetz, F. OncoNEM: Inferring Tumor Evolution from Single-Cell Sequencing Data. Genom. Biol. 2016, 17, 69. [Google Scholar] [CrossRef]
- Jahn, K.; Kuipers, J.; Beerenwinkel, N. Tree Inference for Single-Cell Data. Genom. Biol. 2016, 17, 86. [Google Scholar] [CrossRef] [PubMed]
- Grün, D.; Muraro, M.J.; Boisset, J.-C.; Wiebrands, K.; Lyubimova, A.; Dharmadhikari, G.; van den Born, M.; van Es, J.; Jansen, E.; Clevers, H.; et al. De Novo Prediction of Stem Cell Identity Using Single-Cell Transcriptome Data. Cell Stem Cell 2016, 19, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Tsoucas, D.; Yuan, G.-C. Recent Progress in Single-Cell Cancer Genomics. Curr. Opin. Genet. Dev. 2017, 42, 22–32. [Google Scholar] [CrossRef]
- Garvin, T.; Aboukhalil, R.; Kendall, J.; Baslan, T.; Atwal, G.S.; Hicks, J.; Wigler, M.; Schatz, M.C. Interactive Analysis and Assessment of Single-Cell Copy-Number Variations. Nat. Methods 2015, 12, 1058–1060. [Google Scholar] [CrossRef]
- Deger, T.; Mendelaar, P.A.J.; Kraan, J.; Prager-van der Smissen, W.J.C.; van der Vlugt-Daane, M.; Bindels, E.M.J.; Sieuwerts, A.M.; Sleijfer, S.; Wilting, S.M.; Hollestelle, A.; et al. A Pipeline for Copy Number Profiling of Single Circulating Tumour Cells to Assess Intrapatient Tumour Heterogeneity. Mol. Oncol. 2022, 16, 2981–3000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wei, L.; Huang, J.; Zhong, B.; Li, J.; Xu, H.; He, S.; Liu, Y.; Liu, J.; Lv, H.; et al. CfDNApipe: A Comprehensive Quality Control and Analysis Pipeline for Cell-Free DNA High-Throughput Sequencing Data. Bioinformtics 2021, 37, 4251–4252. [Google Scholar] [CrossRef]
- CfDNApipe. Available online: https://xwanglabthu.github.io/cfDNApipe/ (accessed on 1 March 2023).

| Tool | Description | Resource | References |
|---|---|---|---|
| Variant calling | |||
| GATK | Genome Analysis Toolkit has multiple applications, e.g., variant discovery, genotyping and mutation detection, quality control, coverage analysis and error correction. | https://gatk.broadinstitute.org/hc/en-us (accessed on 1 March 2023) | [198] |
| VarScan | Detects and characterizes variants, e.g., SNPs, indels and somatic mutations in tumor-normal pairs. Identifies low-frequency variants using Bayesian algorithms and statistical models for sensitivity and specificity. | https://varscan.sourceforge.net/ (accessed on 1 March 2023) | [199] |
| Vardict | Detects SNVs, indels and CNVs from tumors and tumor-normal pairs. Uses a combination of local realignment, base quality score recalibration and variant calling algorithms to identify variants. Handles data with high variability, e.g., low-coverage or high tumor heterogeneity. | https://github.com/AstraZeneca-NGS/VarDict (accessed on 1 March 2023) | [216] |
| Samtools | Analyzes alignment files in multiple formats, e.g., BAM, SAM and CRAM. Performs file conversion, sorting, indexing, filtering and merging. Quality control, coverage analysis and variant calling for reference genomes and alignment algorithms. | https://www.htslib.org/ (accessed on 1 March 2023) | [200] |
| Strelka2 | Heuristic approach to detect SNVs, indels and structural variants. Employs a combination of probabilistic and machine learning methods to detect somatic mutations while minimizing false positives. Uses local assembly-based variant calling to improve variant detection sensitivity in regions with low read coverage or high levels of noise. | https://github.com/Illumina/strelka (accessed on 1 March 2023) | [217] |
| ANNOVAR | Enables genetic variants annotation in various genome builds, e.g., RefSeq, dbNSFP and gnomAD. Allows filtering and prioritization of variants based on the functional impact, population frequency, etc. | https://annovar.openbioinformatics.org/en/latest/ (accessed on 1 March 2023) | [207] |
| Variant annotation | |||
| SnpEff | Annotation and functional analysis of genetic variants. Predict the effects of genetic variants on genes, transcripts and regulatory regions and classify variants based on their impact. | http://pcingola.github.io/SnpEff/ (accessed on 1 March 2023) | [208] |
| VEP | Variant Effect Predictor performs analysis, annotation and prioritization of genomic variants in coding and non-coding regions | https://useast.ensembl.org/info/docs/tools/vep/index.html (accessed on 1 March 2023) | [209] |
| Functional interpretation | |||
| GSEA | Gene Set Enrichment Analysis identifies enriched biological pathways, functions and processes based on the expression profiles of genes in a sample or dataset. | https://www.gsea-msigdb.org/gsea/index.jsp (accessed on 1 March 2023) | [210] |
| KEGG | Kyoto Encyclopedia of Genes and Genomes is a data and knowledge base of biological systems, e.g., metabolic pathways, regulatory networks and genetic information. A comprehensive set of reference genomes, gene annotations and pathway maps. | https://www.genome.jp/kegg/ (accessed on 1 March 2023) | [211] |
| Cytoscape | Open-source software for the visualization, analysis and interpretation of complex biological networks. Utilizes various data types, e.g., genetic, genomic, proteomic and metabolomic. | https://cytoscape.org/ (accessed on 1 March 2023) | [212] |
| DAVID | Resource for functional annotation and analysis of biological data. A comprehensive set of functional annotation tools, including gene ontology/pathway analysis and functional annotation clustering. | https://david.ncifcrf.gov/ (accessed on 1 March 2023) | [213] |
| Enrichr | Web-based analysis tool. Provides visualization summaries of collective functions of gene lists. Integrates public databases and annotations for identification and annotation of biological pathways, functions and processes associated with a set of genes or proteins. | https://maayanlab.cloud/Enrichr/ (accessed on 1 March 2023) | [214] |
| GeneMania | Identifies and analyzes functional gene networks. Uses combinations of functional genomics data sources, including protein-protein interactions, co-expression, genetic interactions and pathways to construct gene networks related to the biological function or disease. | http://genemania.org/ (accessed on 1 March 2023) | [215,218] |
| IPA | Ingenuity Pathway Analysis identifies key biological pathways, networks and functions associated with gene or protein sets. A range of visualization and reporting features. Supports various input and output file formats. | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ (accessed on 1 March 2023) | Not applicable |
| Tool | Brief Description | Resource | References |
|---|---|---|---|
| Seurat | R-based platform for raw data processing, paired sample analysis and visualizations. Uses machine learning and clustering algorithms to identify biological features. Assesses cellular heterogeneity via normalization, dimensionality reduction and integration tools. | http://satijalab.org/ (accessed on 1 March 2023) | [230,242] |
| Monocle | R-based scRNA-Seq analysis software. It uses algorithms and machine learning to determine cell developmental trajectories, identify molecular pathways and track changes in gene expression. | http://cole-trapnell-lab.github.io/monocle-release/ (accessed on 1 March 2023) | [237,243,244] |
| ChromVAR | R package for analyzing variations in chromatin accessibility in scATAC-Seq data to identify associated motifs or genomic annotations. It uses visualization techniques to detect and highlight changes in gene expression and provides users with powerful statistical methods. It is also capable of detecting and correlating molecular pathways. | https://greenleaflab.github.io/chromVAR/ (accessed on 1 March 2023) | [245] |
| DRAGEN Single-Cell RNA Pipeline | Cloud-based platform to analyze scRNA-Seq data: aligning and mapping reads, detecting features and biomarkers and generating visualizations. It processes multiplexed scRNA-Seq datasets from reads to a cell-by-gene UMI count gene expression matrix. Features splice-aware RNAseq alignment and matching to annotated genes for transcript reads, cell-barcode and UMI error correction and QC metrics. | http://illumina.com/ (accessed on 1 March 2023) | Not applicable |
| Tapestri | Pipeline to analyze scRNA-Seq data generated by the Tapestri platform. It Includes sequence import, data analysis and visualization capabilities. The software enables variant identification, including SNVs and CNVs, at clonal and subclonal levels. | https://support.missionbio.com/hc/en-us/categories/360002505454-Tapestri-Insights (accessed on 1 March 2023) | Not applicable |
| Scanorama | Integrates data from heterogenous scRNA-seq experiments via detecting common cell types among datasets. Identifies datasets, e.g., cells with similar transcriptional profiles, and leverages the matches for batch correction and integration. Can handle different dataset sizes and sources and does not require all datasets to share a cell population. | https://cb.csail.mit.edu/cb/scanorama/ (accessed on 1 March 2023) | [246] |
| scmap v1.1.5 | An R package that projects cells from a scRNA-Seq data set onto cell types or individual cells from various experiments. It is a widely applicable projection method, detecting the best-matching cell type or individual cell in the reference. It allows fast feature selection, centroid calculation and index creation. | https://scmap.sanger.ac.uk/scmap/ (accessed on 1 March 2023) | [236] |
| Scrublet v0.1 | Single-Cell Remover of Doublets, acronym Scrublet, is a framework for predicting the effect of multiplets in analysis and also identifies problematic multiplets. It can identify neotypic multiplets for an analyzed dataset. The Scrublet classifier can implement arbitrary functions for preprocessing and embedding of single-cell data. | https://github.com/AllonKleinLab/scrublet (accessed on 1 March 2023) | [247] |
| CellRanger v2.2.0 | A set of analysis pipelines that can process Chromium single-cell data to align reads, generate feature-barcode matrices, perform clustering, amongst other tasks. It contains five pipelines for the 3′ Single Cell Gene Expression Solutions and similar products. | https://support.10xgenomics.com/single-cell-gene-expression/software (accessed on 1 March 2023) | Not applicable |
| CITE-seq-count v1.2 | Python package that aids counting antibody tags from CITE-Seq or cell hashing experiments. | https://github.com/Hoohm/CITE-seq-Count (accessed on 1 March 2023) | [248] |
| Drop-seq tools v2.0.0 | Java tool to analyze profiling of individual cells (UMI cells encapsulated in droplets for oligodT sequencing). Libraries produce paired-end reads (read 1: cell barcode and UMI; read 2: cDNA sequence) for identity and abundance of the transcripts in each cell. | https://github.com/broadinstitute/Drop-seq (accessed on 1 March 2023) | Not applicable |
| deepTools v3.1.2 | Galaxy-based web server for processing and visualizing deeply sequenced data. Allows completion of bioinformatic workflows to integrative analyses. Supports four tasks: quality control, data processing and normalization, data integration and visualization. | Not applicable | [249] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brockley, L.J.; Souza, V.G.P.; Forder, A.; Pewarchuk, M.E.; Erkan, M.; Telkar, N.; Benard, K.; Trejo, J.; Stewart, M.D.; Stewart, G.L.; et al. Sequence-Based Platforms for Discovering Biomarkers in Liquid Biopsy of Non-Small-Cell Lung Cancer. Cancers 2023, 15, 2275. https://doi.org/10.3390/cancers15082275
Brockley LJ, Souza VGP, Forder A, Pewarchuk ME, Erkan M, Telkar N, Benard K, Trejo J, Stewart MD, Stewart GL, et al. Sequence-Based Platforms for Discovering Biomarkers in Liquid Biopsy of Non-Small-Cell Lung Cancer. Cancers. 2023; 15(8):2275. https://doi.org/10.3390/cancers15082275
Chicago/Turabian StyleBrockley, Liam J., Vanessa G. P. Souza, Aisling Forder, Michelle E. Pewarchuk, Melis Erkan, Nikita Telkar, Katya Benard, Jessica Trejo, Matt D. Stewart, Greg L. Stewart, and et al. 2023. "Sequence-Based Platforms for Discovering Biomarkers in Liquid Biopsy of Non-Small-Cell Lung Cancer" Cancers 15, no. 8: 2275. https://doi.org/10.3390/cancers15082275
APA StyleBrockley, L. J., Souza, V. G. P., Forder, A., Pewarchuk, M. E., Erkan, M., Telkar, N., Benard, K., Trejo, J., Stewart, M. D., Stewart, G. L., Reis, P. P., Lam, W. L., & Martinez, V. D. (2023). Sequence-Based Platforms for Discovering Biomarkers in Liquid Biopsy of Non-Small-Cell Lung Cancer. Cancers, 15(8), 2275. https://doi.org/10.3390/cancers15082275