
Development of a Novel Circulating Autoantibody Biomarker Panel for the Identification of Patients with ‘Actionable’ Pulmonary Nodules

 , , ,
, , , 
Abstract
Simple Summary
Abstract
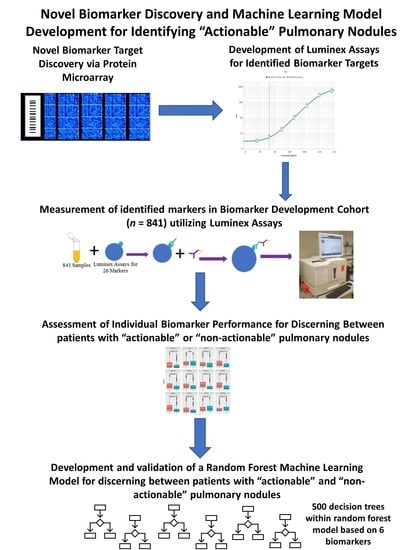
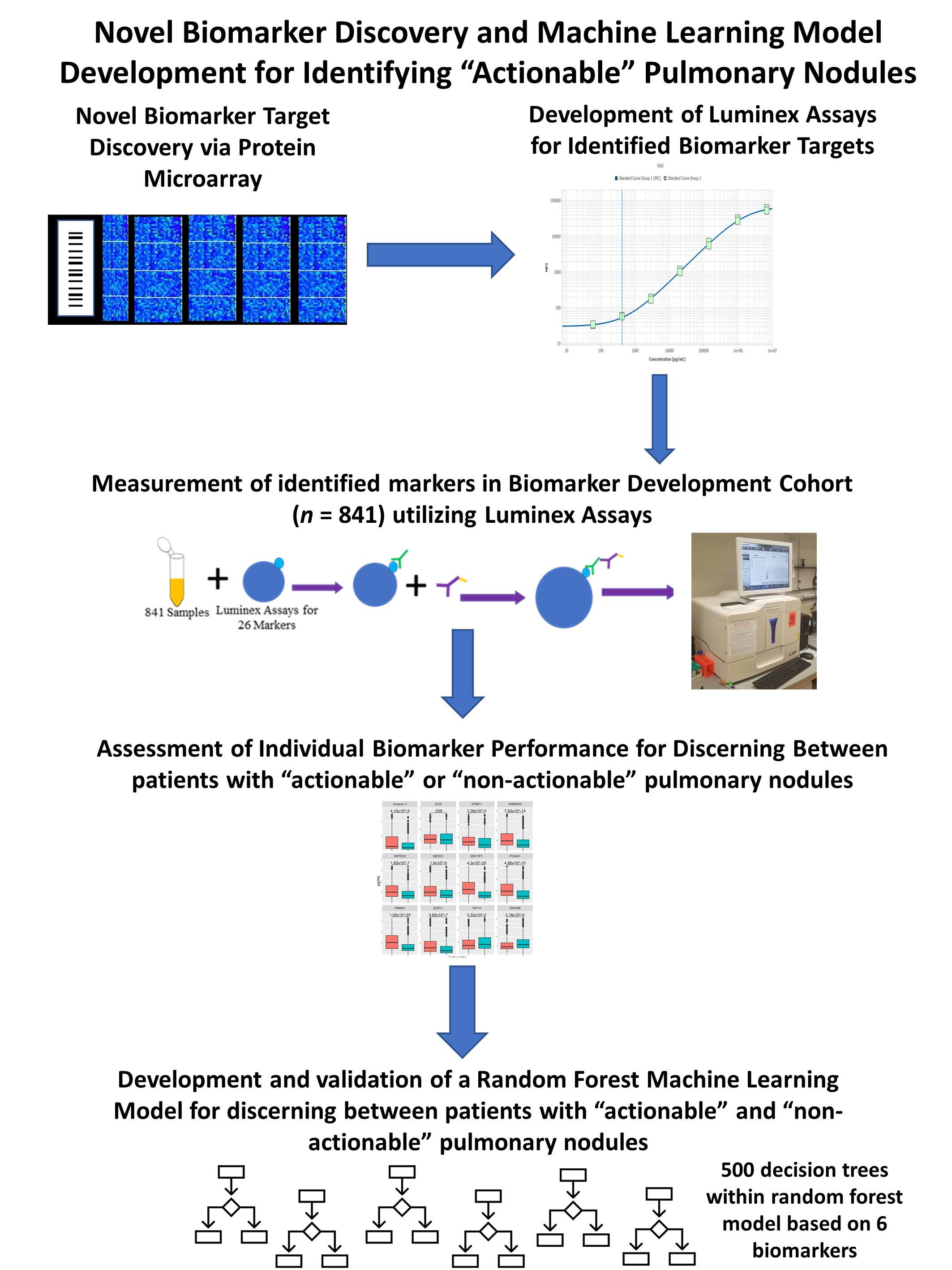
1. Introduction
2. Materials and Methods
2.1. Patient Cohorts
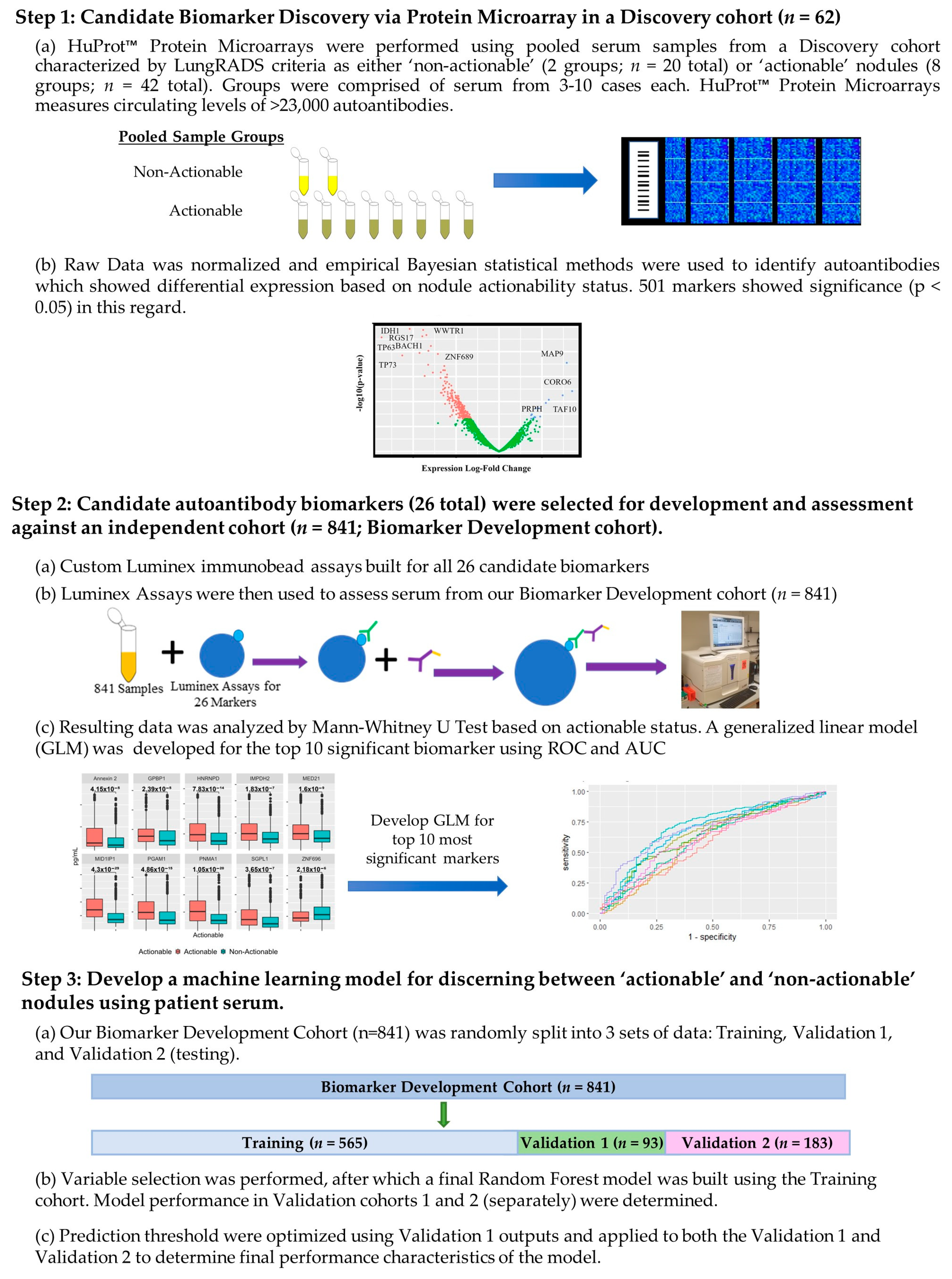
2.2. High-Density Protein Microarrays (Illustrated in Step 1 of Figure 1)
2.3. Custom Luminex Immunobead Assay Development (Development of Assays Used in Figure 1 Step 2 (a))
2.4. Cohort Testing (Figure 1 Step 2 (a) Testing of a Large Cohort)
2.5. Luminex Data Pre-Processing and Analysis (Figure 1 Step 2b) Assessment of Targets Individual Performance for Discerning Actionable from Non-Actionable Samples)
2.6. Development of a Multianalyte Panel for Patient Risk Stratification (Figure 1 Step 3)
3. Results
3.1. Patient Population for the HuProtTM Microarrays for the Discovery of Novel Lung Cancer Early Detection Targets
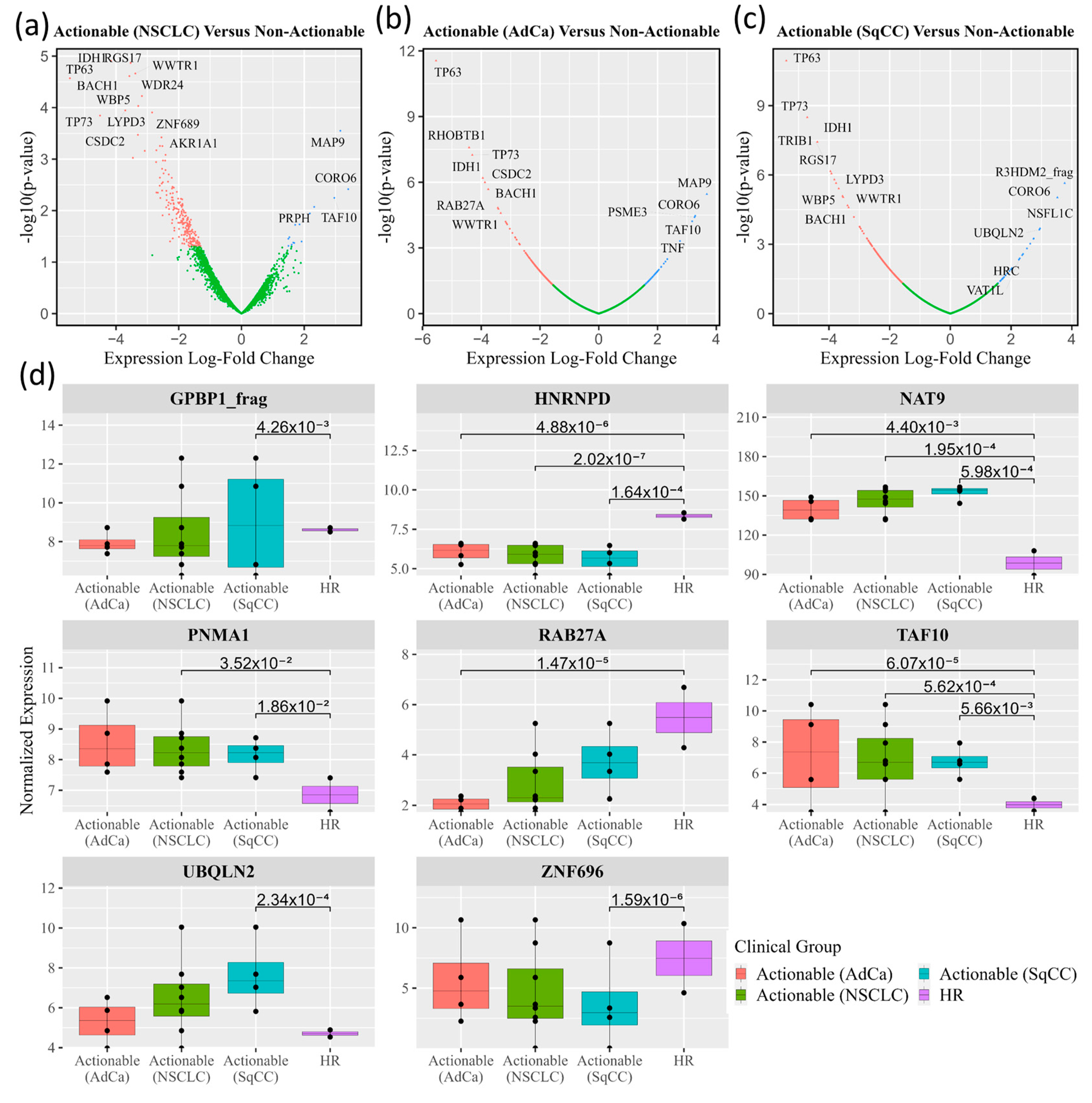
3.2. Autoantibodies with Differential Signal in Patients with ‘Actionable’ vs. ‘Non-Actionable’ Nodules via HuProt™ Protein Microarrays
3.3. Charactersitics of the Biomarker Development Cohort with Subgroups for the Classification Model Development and Assessment Provided
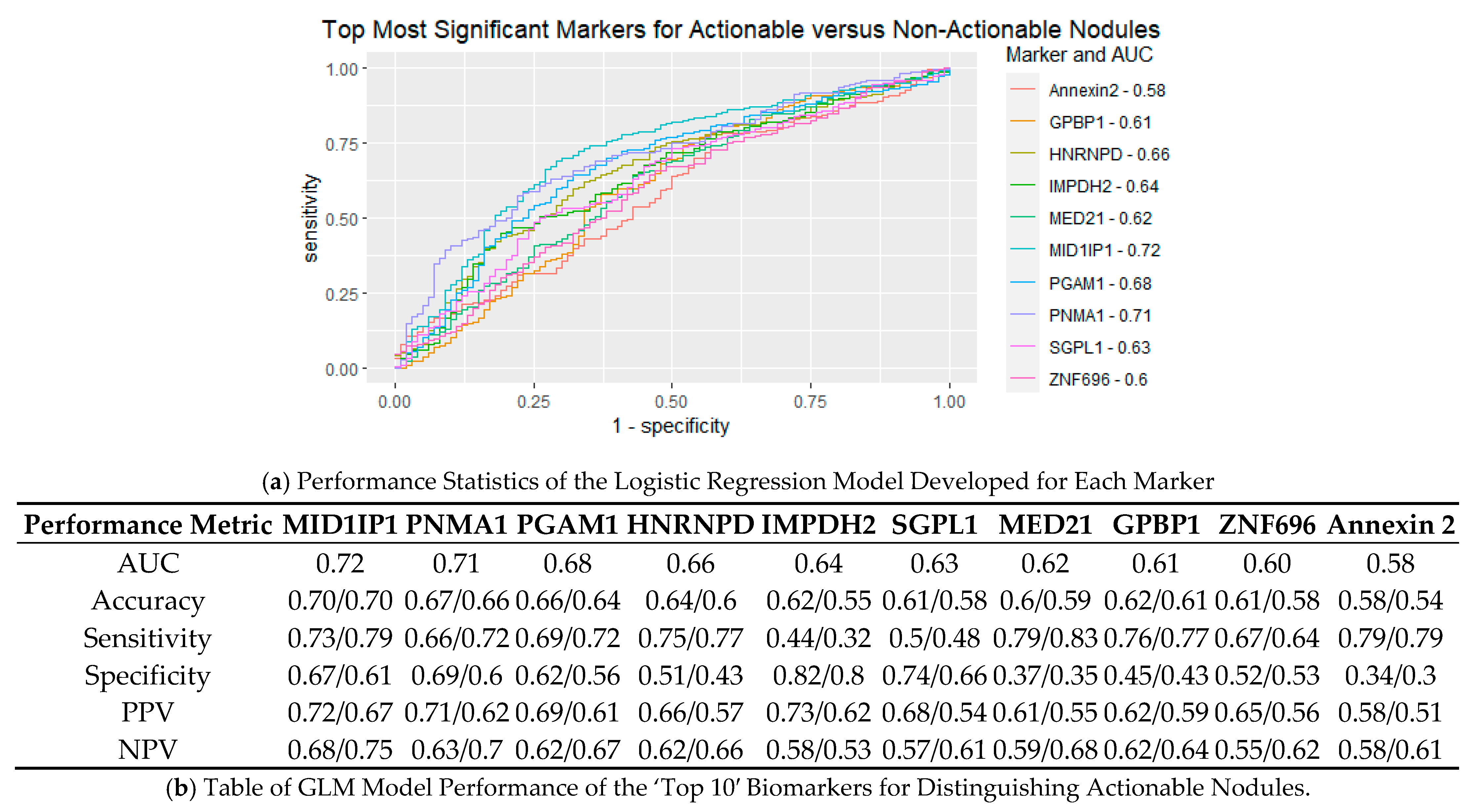
3.4. Performance of Logistic Regression Produced from Top Biomarkers
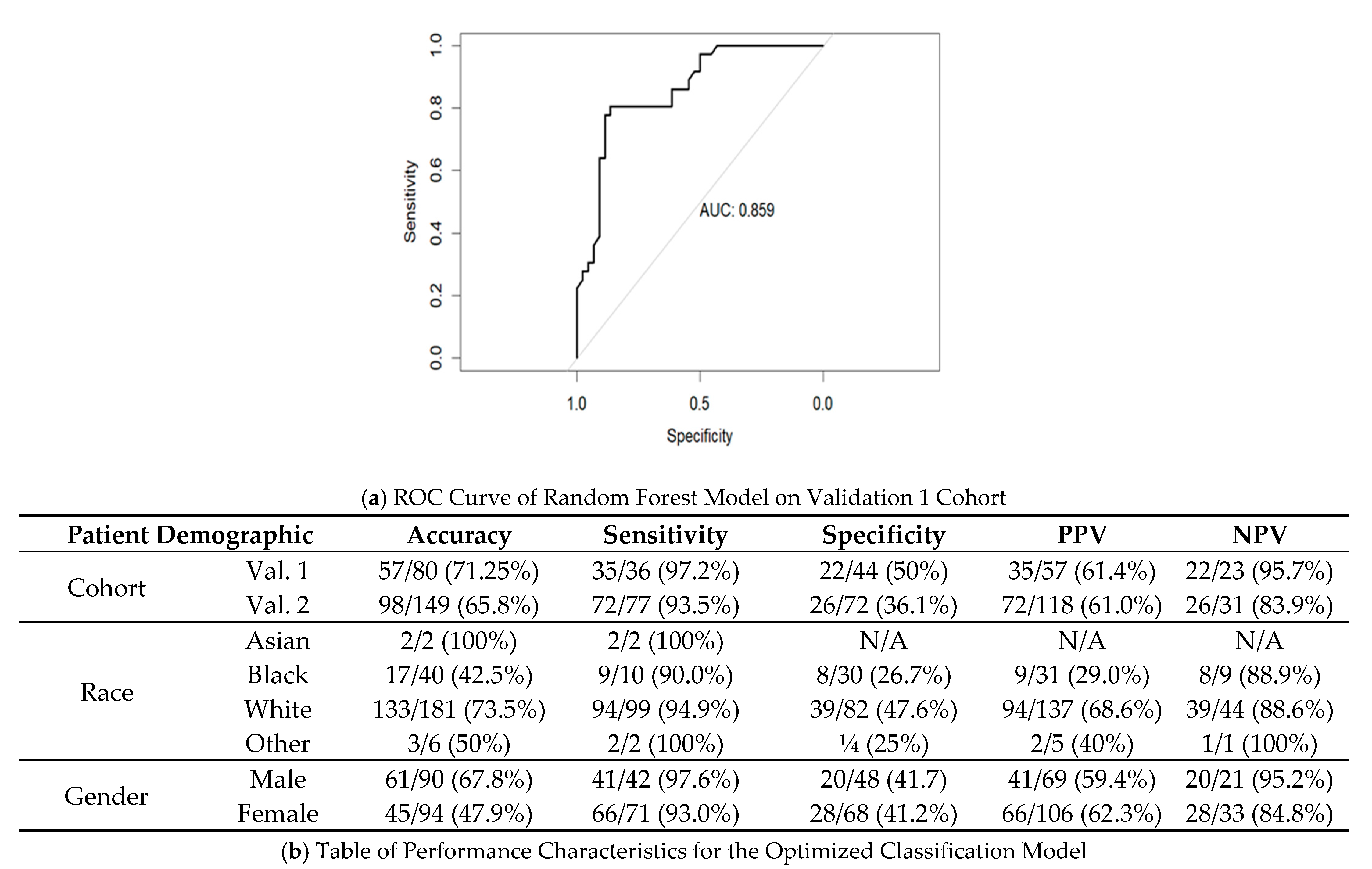
3.5. Creation of Random Forest Model for Determining Actionable versus Non-Actionable Nodules
3.6. Development of an Preliminary Biomarker Panel via Machine Learning
3.7. Performance of Final Optimized Panel for Patient Risk Startification
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Uniprot | Single-Plex/Multiplex | Antibody Catalog | Source |
|---|---|---|---|---|
| Annexin 1 | P04083 | 1 | 21990-1-AP | Proteintech |
| NIP30 | Q9GZU8 | 1 | LS-C667168 | Lifespan Biosciences |
| Annexin 2 | P07355 | 2 | 11256-1-AP | Proteintech |
| CFAP36 | Q96G28 | 3 | LS-C664461 | Lifespan Biosciences |
| MID1IP1 | Q9NPA3 | 3 | LS-C80861 | Lifespan Biosciences |
| DCD | P81605 | 4 | LS-C754340 | Lifespan Biosciences |
| MED21 | Q13503 | 4 | CSB-PA070304 | Cusabio |
| TAF10 | Q12962 | 4 | H00006881-D01P | Novus Biological |
| ZNF696 | Q9H7X3 | 4 | LS-C101596 | Lifespan Biosciences |
| Dr1 | Q01658 | 5 | LS-C755318 | Lifespan Biosciences |
| HSP70 | P0DMV9 | 5 | 14887-1-AP | Proteintech |
| KEAP1 | Q14145 | 6 | TA590238 | OriGene |
| GPBP1 | Q86WP2 | 7 | LS-C753825 | Lifespan Biosciences |
| MYBPH | Q13203 | 7 | LS-C500819 | Lifespan Biosciences |
| PGAM1 | P18669 | 7 | 16126-1-AP | Proteintech |
| HNRNPD | Q14103-1 | 8 | LS-C211799 | Lifespan Biosciences |
| IKZF5 | P04083 | 9 | HPA051574 | Atlas Antibodies |
| NAT9 | Q9BTE0 | 9 | ABIN631510 | Antibodies-online |
| PNMA1 | Q86WP2 | 9 | H00009240-D01P | Novus Biological |
| IMPDH2 | P04083 | 10 | LS-C666439 | Lifespan Biosciences |
| NAP1L5 | P04083 | 11 | LS-C680924 | Lifespan Biosciences |
| RAB27A | Q86WP2 | 12 | LS-C662585 | Lifespan Biosciences |
| SGPL1 | O95470 | 12 | H00008879-D01P | Novus Biological |
| TP53 | Q9NPA3 | 12 | PAB12719 | Abnova |
| Ubiquillin 1 | Q9NPA3 | 13 | 23516-1-AP | Proteintech |
| Ubiquillin 2 | Q9NPA3 | 14 | LS-C661407 | Lifespan Biosciences |
| IgG Goat anti-Human, R-PE, Polyclonal | RRID: AB_2795648 | N/A | OB204009 | Fisher Scientific |
| IgG Goat anti-Rabbit, R-PE, Polyclonal | P01870 | N/A | OB403009 | Fisher Scientific |
| Stage | AdCa | AdCa/SqCC | SqCC | NSCLC | Carcinoid | Large Cell |
|---|---|---|---|---|---|---|
| T1a | 24 | 1 | 5 | 1 | 2 | 4 |
| Not Available | 0 | 0 | 1 | 0 | 1 | 0 |
| N0 | 24 | 1 | 4 | 0 | 1 | 4 |
| N1 | 0 | 0 | 0 | 1 | 0 | 0 |
| T1b | 65 | 4 | 16 | 0 | 2 | 1 |
| Not Available | 2 | 0 | 1 | 0 | 0 | 0 |
| N0 | 63 | 4 | 15 | 0 | 2 | 1 |
| N1 | 0 | 0 | 0 | 0 | 0 | 0 |
| T1c | 16 | 1 | 14 | 0 | 2 | 4 |
| Not Available | 1 | 0 | 0 | 0 | 0 | 0 |
| N0 | 15 | 1 | 14 | 0 | 2 | 4 |
| N1 | 0 | 0 | 0 | 0 | 0 | 0 |
| T2a | 25 | 1 | 9 | 0 | 1 | 2 |
| Not Available | 0 | 0 | 0 | 0 | 0 | 0 |
| N0 | 24 | 1 | 9 | 0 | 1 | 2 |
| N1 | 0 | 0 | 0 | 0 | 0 | 0 |
| N1, M1b | 1 | 0 | 0 | 0 | 0 | 0 |
| T2b | 4 | 0 | 9 | 1 | 0 | 0 |
| Not Available | 0 | 0 | 0 | 0 | 0 | 0 |
| N0 | 4 | 0 | 9 | 1 | 0 | 0 |
| N1 | 0 | 0 | 0 | 0 | 0 | 0 |
| T3 | 11 | 2 | 14 | 0 | 1 | 1 |
| Not Available | 1 | 0 | 1 | 0 | 0 | 0 |
| N0 | 10 | 2 | 13 | 0 | 1 | 1 |
| N1 | 0 | 0 | 0 | 0 | 0 | 0 |
| T4 | 6 | 0 | 5 | 0 | 0 | 0 |
| Not Available | 1 | 0 | 2 | 0 | 0 | 0 |
| N0 | 5 | 0 | 3 | 0 | 0 | 0 |
| N1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Not Available | 10 | 0 | 1 | 0 | 0 | 0 |
| SUBGROUP | DIAGNOSIS | IMPDH2 | HNRNPD | PGAM1 | PNMA1 | MIDIP1 |
|---|---|---|---|---|---|---|
| MALIGNANCY | Lung Metastasis | 0.67 (4/6) | 1 (8/8) | 0.88 (7/8) | 0.86 (6/7) | 0.75 (6/8) |
| AdCa | 0.48 (47/97) | 0.81 (114/140) | 0.74 (105/142) | 0.68 (92/135) | 0.8 (118/148) | |
| Mixed AdCa/SqCC | 0.6 (3/5) | 0.75 (6/8) | 0.71 (5/7) | 0.63 (5/8) | 0.67 (6/9) | |
| SqCC | 0.57 (24/42) | 0.94 (61/65) | 0.88 (58/66) | 0.82 (53/65) | 0.85 (57/67) | |
| NSCLC (Unspecified) | 0 (0/1) | 0 (0/1) | 0.5 (1/2) | 0.5 (1/2) | 0.5 (1/2) | |
| Carcinoid | 0 (0/4) | 0.5 (4/8) | 0.43 (3/7) | 0.38 (3/8) | 0.5 (4/8) | |
| Small-Cell Lung Cancer | 0.33 (2/6) | 0.44 (4/9) | 0.44 (4/9) | 0.44 (4/9) | 0.56 (5/9) | |
| Total Performance | 0.5 (80/161) | 0.82 (197/239) | 0.76 (183/241) | 0.7 (164/234) | 0.78 (197/251) | |
| BENIGN | Granuloma | 0.46 (6/13) | 0.96 (24/25) | 0.77 (20/26) | 0.77 (20/26) | 0.88 (23/26) |
| Hamartoma | 0.5 (3/6) | 0.86 (12/14) | 0.75 (9/12) | 0.92 (12/13) | 0.92 (12/13) | |
| Fibrosis/Scarring/Inflammation | 0.5 (12/24) | 0.72 (33/46) | 0.74 (34/46) | 0.76 (31/41) | 0.76 (34/45) | |
| Infection/Pneumonia | 0.33 (1/3) | 1 (3/3) | 1 (3/3) | 1 (3/3) | 1 (4/4) | |
| Other Non-Malignant Nodule | 0.17 (1/6) | 0.86 (6/7) | 0.83 (5/6) | 0.83 (5/6) | 0.71 (5/7) | |
| Total Performance | 0.44 (23/52) | 0.82 (78/95) | 0.76 (71/93) | 0.8 (71/89) | 0.82 (78/95) | |
| CONTROL | Stable or Resolving | 0.73 (72/99) | 0.52 (71/137) | 0.5 (72/143) | 0.59 (86/145) | 0.59 (86/147) |
| Interval Increase in Size | 0.25 (1/4) | 0.33 (2/6) | 0.83 (5/6) | 0.5 (3/6) | 0.67 (4/6) | |
| New Nodule/Unknown Growth | 0.66 (103/156) | 0.5 (113/228) | 0.6 (144/239) | 0.65 (156/241) | 0.66 (159/242) | |
| Mix of New and Stable Nodules | 1 (1/1) | 1 (1/1) | 1 (1/1) | 0 (0/1) | 1 (1/1) | |
| No Finding | 0.7 (21/30) | 0.27 (13/48) | 0.56 (24/43) | 0.59 (29/49) | 0.57 (26/46) | |
| Total Performance | 0.68 (198/290) | 0.48 (200/420) | 0.57 (246/432) | 0.62 (274/442) | 0.62 (276/442) |
| Training Cohort | Validation 1 Cohort | Validation 2 (Testing) Cohort | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Non-Actionable | Actionable | Total | Non-Actionable | Actionable | Total | Non-Actionable | Actionable | |
| n = 565 | n = 260 | n = 305 | n = 93 | n = 48 | n = 45 | n = 183 | n = 84 | n = 99 | |
| Gender | |||||||||
| Male (%) | 242 (42.8%) | 105 (40.4%) | 137 (44.9%) | 38 (40.9%) | 22 (45.8%) | 16 (35.6%) | 73 (40.0%) | 34 (40.5%) | 39 (39.3%) |
| Age, years Median | 67 | 64 | 69 | 67 | 63 | 71 | 67 | 65 | 68 |
| Minimum | 41 | 48 | 41 | 47 | 47 | 51 | 44 | 53 | 44 |
| Maximum | 87 | 82 | 87 | 83 | 77 | 83 | 88 | 82 | 88 |
| Diagnosis | |||||||||
| NSCLC | 159 | 1 | 158 | 31 | 0 | 31 | 55 | 0 | 55 |
| AdCa | 1 | 103 | 0 | 21 | 0 | 36 | |||
| SqCC | 0 | 47 | 0 | 9 | 0 | 17 | |||
| AdCa/SqCC Mixed | 0 | 7 | 0 | 1 | 0 | 1 | |||
| NSCLC (Not Specified) | 0 | 1 | 0 | 0 | 0 | 1 | |||
| Malignancy, non-NSCLC | 22 | 1 | 21 | 2 | 0 | 2 | 5 | 0 | 5 |
| Carcinoid (G1/G2) | 0 | 6 | 0 | 1 | 0 | 1 | |||
| Large-Cell/SCLC (G3) | 0 | 9 | 0 | 1 | 0 | 2 | |||
| Metastasis (Not Lung Cancer) | 1 | 6 | 0 | 0 | 0 | 2 | |||
| Benign | 73 | 6 | 67 | 6 | 2 | 4 | 22 | 3 | 19 |
| Granuloma | 1 | 21 | 0 | 1 | 1 | 5 | |||
| Hamartoma | 0 | 12 | 0 | 0 | 0 | 2 | |||
| Fibrosis/Scar/Inflammation | 5 | 25 | 2 | 2 | 2 | 10 | |||
| Infection/Org. Pneumonia | 0 | 4 | 0 | 0 | 0 | 1 | |||
| Other | 0 | 5 | 0 | 1 | 0 | 1 | |||
| Not Assessed * | 311 | 252 | 59 | 54 | 46 | 8 | 101 | 81 | 20 |
| Stable or Resolving | 74 | 18 | 17 | 5 | 32 | 8 | |||
| Interval Increase in Size | 2 | 3 | 0 | 0 | 0 | 1 | |||
| New Nodule/Unknown Growth | 143 | 36 | 20 | 3 | 39 | 11 | |||
| Mix of New/Stable Nodules | 1 | 0 | 0 | 0 | 0 | 0 | |||
| No Nodule/Non-Specified | 32 | 2 | 9 | 0 | 10 | 0 | |||
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- United States Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. National Program of Cancer Registries and Surveillance, Epidemiology, and End Results Program SEER*Stat Database: NPCR and SEER Incidence–U.S. Cancer Statistics 2001–2019 Public Use Research Database, 2021 Submission (2001–2019). 2022. Available online: https://seer.cancer.gov/statfacts/html/lungb.html (accessed on 4 April 2023).
- Pinsky, P.F. Assessing the Benefits and Harms of Low-Dose Computed Tomography Screening for Lung Cancer. Lung Cancer Manag. 2014, 3, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; Sicks, J.D. Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef] [PubMed]
- US Preventive Services Task Force; Krist, A.H.; Davidson, K.W.; Mangione, C.M.; Barry, M.J.; Cabana, M.; Caughey, A.B.; Davis, E.M.; Donahue, K.E.; Doubeni, C.A.; et al. Screening for Lung Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2021, 325, 962. [Google Scholar] [CrossRef] [PubMed]
- Tailor, T.D.; Tong, B.C.; Gao, J.; Choudhury, K.R.; Rubin, G.D. A Geospatial Analysis of Factors Affecting Access to CT Facilities: Implications for Lung Cancer Screening. J. Am. Coll. Radiol. 2019, 16, 1663–1668. [Google Scholar] [CrossRef]
- Yong, P.C.; Sigel, K.; Rehmani, S.; Wisnivesky, J.; Kale, M.S. Lung Cancer Screening Uptake in the United States. Chest 2020, 157, 236–238. [Google Scholar] [CrossRef]
- American College of Radiology Committee on Lung-RADS®. Lung-RADS® V2022; American College of Radiology Committee on Lung-RADS®: Reston, VA, USA, 2022. [Google Scholar]
- Silva, M.; Milanese, G.; Sestini, S.; Sabia, F.; Jacobs, C.; van Ginneken, B.; Prokop, M.; Schaefer-Prokop, C.M.; Marchianò, A.; Sverzellati, N.; et al. Lung Cancer Screening by Nodule Volume in Lung-RADS v1.1: Negative Baseline CT Yields Potential for Increased Screening Interval. Eur. Radiol. 2021, 31, 1956–1968. [Google Scholar] [CrossRef]
- Borgia, J.A.; Basu, S.; Faber, L.P.; Kim, A.W.; Coon, J.S.; Kaiser-Walters, K.A.; Fhied, C.; Thomas, S.; Rouhi, O.; Warren, W.H.; et al. Establishment of a Multi-Analyte Serum Biomarker Panel to Identify Lymph Node Metastases in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2009, 4, 338–347. [Google Scholar] [CrossRef]
- Gowen, M.F.; Giles, K.M.; Simpson, D.; Tchack, J.; Zhou, H.; Moran, U.; Dawood, Z.; Pavlick, A.C.; Hu, S.; Wilson, M.A.; et al. Baseline Antibody Profiles Predict Toxicity in Melanoma Patients Treated with Immune Checkpoint Inhibitors. J. Transl. Med. 2018, 16, 82. [Google Scholar] [CrossRef]
- Johannet, P.; Liu, W.; Fenyo, D.; Wind-Rotolo, M.; Krogsgaard, M.; Mehnert, J.M.; Weber, J.S.; Zhong, J.; Osman, I. Baseline Serum Autoantibody Signatures Predict Recurrence and Toxicity in Melanoma Patients Receiving Adjuvant Immune Checkpoint Blockade. Clin. Cancer Res. 2022, 28, 4121–4130. [Google Scholar] [CrossRef]
- Fishman, D.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. PAWER: Protein Array Web ExploreR. BMC Bioinform. 2020, 21, 411. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Farlow, E.C.; Patel, K.; Basu, S.; Lee, B.-S.; Kim, A.W.; Coon, J.S.; Faber, L.P.; Bonomi, P.; Liptay, M.J.; Borgia, J.A. Development of a Multiplexed Tumor-Associated Autoantibody-Based Blood Test for the Detection of Non–Small Cell Lung Cancer. Clin. Cancer Res. 2010, 16, 3452. [Google Scholar] [CrossRef] [PubMed]
- Fhied, C.; Kanangat, S.; Borgia, J.A. Development of a Bead-Based Immunoassay to Routinely Measure Vimentin Autoantibodies in the Clinical Setting. J. Immunol. Methods 2014, 407, 9–14. [Google Scholar] [CrossRef]
- Fhied, C.L.; Tarhoni, I.; Gerard, D.; Lewin, G.M.; Moudgalya, H.; Schneider, J.R.; Borgia, J.A. Dynamic Monitoring of Seroconversion Using a Multianalyte Immunobead Assay for COVID-19. JoVE 2022, 180, 63352. [Google Scholar] [CrossRef]
- Tarhoni, I.; Fhied, C.L.; Pool, M.; Liptay, M.J.; Bonomi, P.; Seder, C.W.; Borgia, J.A. Development of Bead Based Multiplexed Immunoassay for Evaluation of Midkine, Syndecan-1, and ANGPTL4 in Patient Serum. J. Immunoass. Immunochem. 2018, 39, 84–98. [Google Scholar] [CrossRef]
- Suprun, M.; Getts, R.; Raghunathan, R.; Grishina, G.; Witmer, M.; Gimenez, G.; Sampson, H.A.; Suárez-Fariñas, M. Novel Bead-Based Epitope Assay Is a Sensitive and Reliable Tool for Profiling Epitope-Specific Antibody Repertoire in Food Allergy. Sci. Rep. 2019, 9, 18425. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Use R! Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. JOSS 2019, 4, 1686. [Google Scholar] [CrossRef]
- Kassambara, A. Rstatix: Pipe-Friendly Framework for Basic Statistical Tests. R Package Version 0.7.2. Available online: https://rpkgs.datanovia.com/rstatix/ (accessed on 4 April 2023).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kuhn, M. Caret Package. J. Stat. Softw. 2008, 28, 1–16. [Google Scholar]
- Jemal, A.; Fedewa, S.A. Lung Cancer Screening with Low-Dose Computed Tomography in the United States—2010 to 2015. JAMA Oncol. 2017, 3, 1278. [Google Scholar] [CrossRef] [PubMed]
- Kastner, J.; Hossain, R.; Jeudy, J.; Dako, F.; Mehta, V.; Dalal, S.; Dharaiya, E.; White, C. Lung-RADS Version 1.0 versus Lung-RADS Version 1.1: Comparison of Categories Using Nodules from the National Lung Screening Trial. Radiology 2021, 300, 199–206. [Google Scholar] [CrossRef]
- Platt, J.L.; Garcia de Mattos Barbosa, M.; Cascalho, M. The Five Dimensions of B Cell Tolerance. Immunol. Rev. 2019, 292, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Kashaninejad, N.; Masud, M.K.; Yamauchi, Y.; Nguyen, N.-T.; Shiddiky, M.J.A. Autoantibodies as Diagnostic and Prognostic Cancer Biomarker: Detection Techniques and Approaches. Biosens. Bioelectron. 2019, 139, 111315. [Google Scholar] [CrossRef]
- Yang, B.; Li, X.; Ren, T.; Yin, Y. Autoantibodies as Diagnostic Biomarkers for Lung Cancer: A Systematic Review. Cell. Death Discov. 2019, 5, 126. [Google Scholar] [CrossRef]
- Huang, H.; Luo, W.; Ni, Y.; Sun, S.; Wang, C.; Zhang, L. The Diagnostic Efficiency of Seven Autoantibodies in Lung Cancer. Eur. J. Cancer Prev. 2020, 29, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Wang, W.; Meng, W.; Ding, M.; Ma, S.; Wang, X. Development of a Multiplex Autoantibody Test for Detection of Lung Cancer. PLoS ONE 2014, 9, e95444. [Google Scholar] [CrossRef] [PubMed]
- Borg, M.; Wen, S.W.C.; Nederby, L.; Hansen, T.F.; Jakobsen, A.; Andersen, R.F.; Weinreich, U.M.; Hilberg, O. Performance of the EarlyCDT® Lung Test in Detection of Lung Cancer and Pulmonary Metastases in a High-Risk Cohort. Lung Cancer 2021, 158, 85–90. [Google Scholar] [CrossRef]
- Lam, S.; Boyle, P.; Healey, G.F.; Maddison, P.; Peek, L.; Murray, A.; Chapman, C.J.; Allen, J.; Wood, W.C.; Sewell, H.F.; et al. Early CDT-Lung: An Immunobiomarker Test as an Aid to Early Detection of Lung Cancer. Cancer Prev. Res. 2011, 4, 1126–1134. [Google Scholar] [CrossRef]
- Rissanen, E.; Heikkinen, S.; Seppä, K.; Ryynänen, H.; Eriksson, J.G.; Härkänen, T.; Jousilahti, P.; Knekt, P.; Koskinen, S.; Männistö, S.; et al. Incidence Trends and Risk Factors of Lung Cancer in Never Smokers: Pooled Analyses of Seven Cohorts. Int. J. Cancer 2021, 149, 2010–2019. [Google Scholar] [CrossRef] [PubMed]





| Group | Actionable | Non-Actionable | ||
|---|---|---|---|---|
| AdCa | SqCC | |||
| (n = 25) | (n = 17) | (n = 20) | ||
| Age (years) | Median (Range) | 70 (56–83) | 75 (60–79) | 65 (58–71) |
| Gender | Male (%) | 13 (50%) | 8 (47.1%) | 10 (52.6%) |
| Lesion Size (mm) | Median (range) | 24.8 (11–38) | 28 (11–35) | 3 (2–10) |
| AJCC Stage | IA1 | 1 | N/A | |
| IA2 | 8 | 6 | ||
| IA3 | 4 | 7 | ||
| IB | 12 | 3 | ||
| IIB | N/A | 1 | ||
| Single-Plex Assay | Multiplex Assay |
|---|---|
| Annexin 2 | Annexin 1 and NIP30 |
| KEAP1 | CFAP36, MID1IP1 |
| HNRNPD | DCD, MED21, TAF10, ZNF696 |
| IMPDH2 | Dr1, HSP70 |
| NAP1L5 | GPBP1, MYBPH, PGAM1 |
| Ubiquillin 1 | IKZF5, NAT9, PNMA1 |
| Ubiquilllin 2 | RAB27A, SGPL1, TP53 |
| Patient Demographic | Total | Non-Actionable | Actionable |
|---|---|---|---|
| n = 841 | n = 392 | n = 449 | |
| Gender | |||
| Male (%) | 353 (41.97%) | 161 (41.07%) | 192 (42.76%) |
| Age, years Median | 67 | 65 | 69 |
| Minimum | 41 | 47 | 41 |
| Maximum | 88 | 82 | 88 |
| Diagnosis | |||
| NSCLC | 245 | 1 | 244 |
| AdCa | 1 | 160 | |
| SqCC | 0 | 73 | |
| AdCa/SqCC Mixed | 0 | 9 | |
| NSCLC (Not Specified) | 0 | 2 | |
| Malignancy, non-NSCLC | 29 | 1 | 28 |
| Carcinoid (G1/G2) | 0 | 8 | |
| Large-Cell/SCLC (G3) | 0 | 12 | |
| Metastasis (Not Lung Cancer) | 1 | 8 | |
| Benign | 101 | 11 | 90 |
| Granuloma | 2 | 27 | |
| Hamartoma | 0 | 14 | |
| Fibrosis/Scar/Inflammation | 9 | 37 | |
| Infection/Org. Pneumonia | 0 | 5 | |
| Other | 0 | 7 | |
| Not Assessed * | 466 | 379 | 87 |
| Stable or Resolving | 123 | 31 | |
| Interval Increase in Size | 2 | 4 | |
| New Nodule/Unknown Growth | 202 | 50 | |
| Mix of New/Stable Nodules | 1 | 0 | |
| No Nodule/Non-Specified | 51 | 2 |
| Protein Name | Uniprot ID | Non-Actionable Median (Range), ng/mL | Actionable Median (Range), ng/mL | p-Value |
|---|---|---|---|---|
| MID1IP1 | Q9NPA3 | 71.25 (17.85–388.06) | 142.49 (0.14–397.30) | 4.30 × 10−29 |
| PNMA1 | Q8ND90 | 15.59 (0.19–94.31) | 30.99 (0.25–93.93) | 1.05 × 10−20 |
| PGAM1 | P18669 | 5.25 (0.11–30.96) | 9.74 (0.02–31.27) | 4.86 × 10−15 |
| HNRNPD | Q14103-1 | 13.23 (0.19–85.85) | 23.71 (0.09–90.12) | 7.83 × 10−14 |
| MED21 | Q13503 | 45.12 (1.59–229.99) | 65.03 (0.1–231.13) | 1.60 × 10−9 |
| IMPDH2 | P12268 | 316.41 (30.23–1685.81) | 481.49 (8.89–1686.36) | 1.83 × 10−7 |
| SGPL1 | O95470 | 7866.32 (6.02–77,865.4) | 14,005.25 (0.15–78,349.22) | 3.65 × 10−7 |
| ZNF696 | Q9H7X3 | 112.12 (4.38–404.82) | 87.71 (21.5–393.02) | 2.18 × 10−6 |
| GPBP1 | Q86WP2 | 42.09 (0.18–275.87) | 65.18 (0.05–278.53) | 2.39 × 10−5 |
| Annexin 2 | P07355 | 17.27 (0.01–228.71) | 26.81 (0.28–245.76) | 4.15 × 10−5 |
| NAT9 | Q9BTE0 | 1155.48 (45.72–4082.57) | 1551.26 (7.44–4304.02) | 8.67 × 10−5 |
| TP53 | P04637 | 43.78 (0.75–234.93) | 59.04 (0–236.63) | 0.0003 |
| Annexin 1 | P04083 | 0.96 (0.01–8.44) | 1.34 (0–8.51) | 0.014 |
| NIP30 | Q9GZU8 | 4.91 (0.03–29.18) | 5.96 (0.01–29.71) | 0.014 |
| TAF10 | Q12962 | 164.68 (5.48–584.71) | 147.65 (26.31–567.49) | 0.023 |
| RAB27A | P51159 | 0.54 (0.01–2.70) | 0.67 (0.01–2.73) | 0.027 |
| KEAP1 | Q14145 | 6.12 (0.06–29.3) | 8.24 (0.01–30.98) | 0.041 |
| Ubiquillin 1 | Q9UMX0 | 4.73 (0.2–42.17) | 484.48 (9.02–5403.48) | 0.077 |
| HSP70 | P0DMV9 | 0.7 (0–2.73) | 0.8 (0.09–2.74) | 0.163 |
| MYBPH | Q13203 | 436.92 (3.36–1721.33) | 385.88 (0.66–1760.32) | 0.211 |
| NAP1L5 | Q96NT1 | 587.41 (2.02–2535.53) | 632.27 (0.51–2399.45) | 0.234 |
| IKZF5 | Q9H5V7 | 46.96 (0.06–331.96) | 49.68 (0.07–338.83) | 0.258 |
| Ubiquillin2 | Q9UHD9 | 99.87 (2.39–499.05) | 84.37 (0.65–506.10) | 0.454 |
| Dr1 | Q01658 | 18.05 (0.01–97.89) | 20.58 (0.02–94.76) | 0.507 |
| DCD | P81605 | 2022.34 (71.6–6775.07) | 2120.54 (345.25–6768.67) | 0.539 |
| CFAP36 | Q96G28 | 27.08 (0.04–109.11) | 27.74 (0.03–112.46) | 0.631 |
| Subgroups | Overall Accuracy | Accuracy (Actionable) | Accuracy (Non-Actionable) | ||
|---|---|---|---|---|---|
| Combined | Validation 1 | Validation 2 | Validation 1 | Validation 2 | |
| Metastasis | 1/1 (100%) | NA | 1/1 (100%) | NA | NA |
| AdCa | 42/44 (95.5%) | 15/16 (93.75%) | 27/28 (96.43%) | NA | NA |
| AdCa/SqCC Mixed | 1/1 (100%) | 1/1 (100%) | NA | NA | NA |
| SqCC | 18/20 (90.0%) | 6/6 (100%) | 12/14 (85.7%) | NA | NA |
| NSCLC (General) | 1/1 (100%) | NA | NA|1/1 (100%) | NA | NA |
| Carcinoid | 2/2 (100%) | 1/1 (100%) | 1/1 (100%) | NA | NA |
| Small-Cell | 2/2 (100%) | 1/1 (100%) | 1/1 (100%) | NA | NA |
| Malignancy Totals | 67/71 (94.4%) | 24/25 (96.0%) | 43/46 (93.5%) | NA | NA |
| Granuloma | 3/3 (100%) | 1/1 (100%) | 2/2 (100%) | NA | NA |
| Hamartoma | 2/2 (100%) | NA | 2/2 (100%) | NA | NA |
| Fibrosis/Scarring/Inflammation | 12/16 (75%) | 2/2 (100%) | 10/10 (100%) | 0/2 (0%) | |0/2 (0%) |
| Infection/Pneumonia | 1/1 (100%) | NA | 1/1 (100%) | NA | NA |
| Other Non-Malig. Nodule | 2/2 (100%) | 1/1 (100%) | 1/1 (100%) | NA | NA |
| Benign Totals | 20/24 (83.3%) | 4/4 (100%) | 16/16 (100%) | 0/2 (0%) | 0/2 (0%) |
| Stable or Resolving Nodule | 24/57 (42.1%) | 5/5 (100%) | 6/7 (85.7%) | 6/16 (37.5%) | 7/29 (24.1%) |
| Interval Increase in Size | 0/1 (0%) | NA|0/1 (0%) | 0/1 (0%) | NA | NA |
| New Nodule/Unknown Growth | 37/61 (60.7%) | 3/3 (100%) | 7/7 (100%) | 11/19 (57.9%) | 16/32 (50%) |
| No Noted Nodule | 8/16 (50%) | NA | NA | 5/7 (71.4%) | 3/9 (33.3%) |
| Control Totals | 69/135 (51.1%) | 8/8 (100%) | 13/15 (86.7%) | 22/42 (52.4%) | 26/70 (37.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auger, C.; Moudgalya, H.; Neely, M.R.; Stephan, J.T.; Tarhoni, I.; Gerard, D.; Basu, S.; Fhied, C.L.; Abdelkader, A.; Vargas, M.; et al. Development of a Novel Circulating Autoantibody Biomarker Panel for the Identification of Patients with ‘Actionable’ Pulmonary Nodules. Cancers 2023, 15, 2259. https://doi.org/10.3390/cancers15082259
Auger C, Moudgalya H, Neely MR, Stephan JT, Tarhoni I, Gerard D, Basu S, Fhied CL, Abdelkader A, Vargas M, et al. Development of a Novel Circulating Autoantibody Biomarker Panel for the Identification of Patients with ‘Actionable’ Pulmonary Nodules. Cancers. 2023; 15(8):2259. https://doi.org/10.3390/cancers15082259
Chicago/Turabian StyleAuger, Claire, Hita Moudgalya, Matthew R. Neely, Jeremy T. Stephan, Imad Tarhoni, David Gerard, Sanjib Basu, Cristina L. Fhied, Ahmed Abdelkader, Moises Vargas, and et al. 2023. "Development of a Novel Circulating Autoantibody Biomarker Panel for the Identification of Patients with ‘Actionable’ Pulmonary Nodules" Cancers 15, no. 8: 2259. https://doi.org/10.3390/cancers15082259
APA StyleAuger, C., Moudgalya, H., Neely, M. R., Stephan, J. T., Tarhoni, I., Gerard, D., Basu, S., Fhied, C. L., Abdelkader, A., Vargas, M., Hu, S., Hulett, T., Liptay, M. J., Shah, P., Seder, C. W., & Borgia, J. A. (2023). Development of a Novel Circulating Autoantibody Biomarker Panel for the Identification of Patients with ‘Actionable’ Pulmonary Nodules. Cancers, 15(8), 2259. https://doi.org/10.3390/cancers15082259