
R-Loops at Chromosome Ends: From Formation, Regulation, and Cellular Consequence


{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
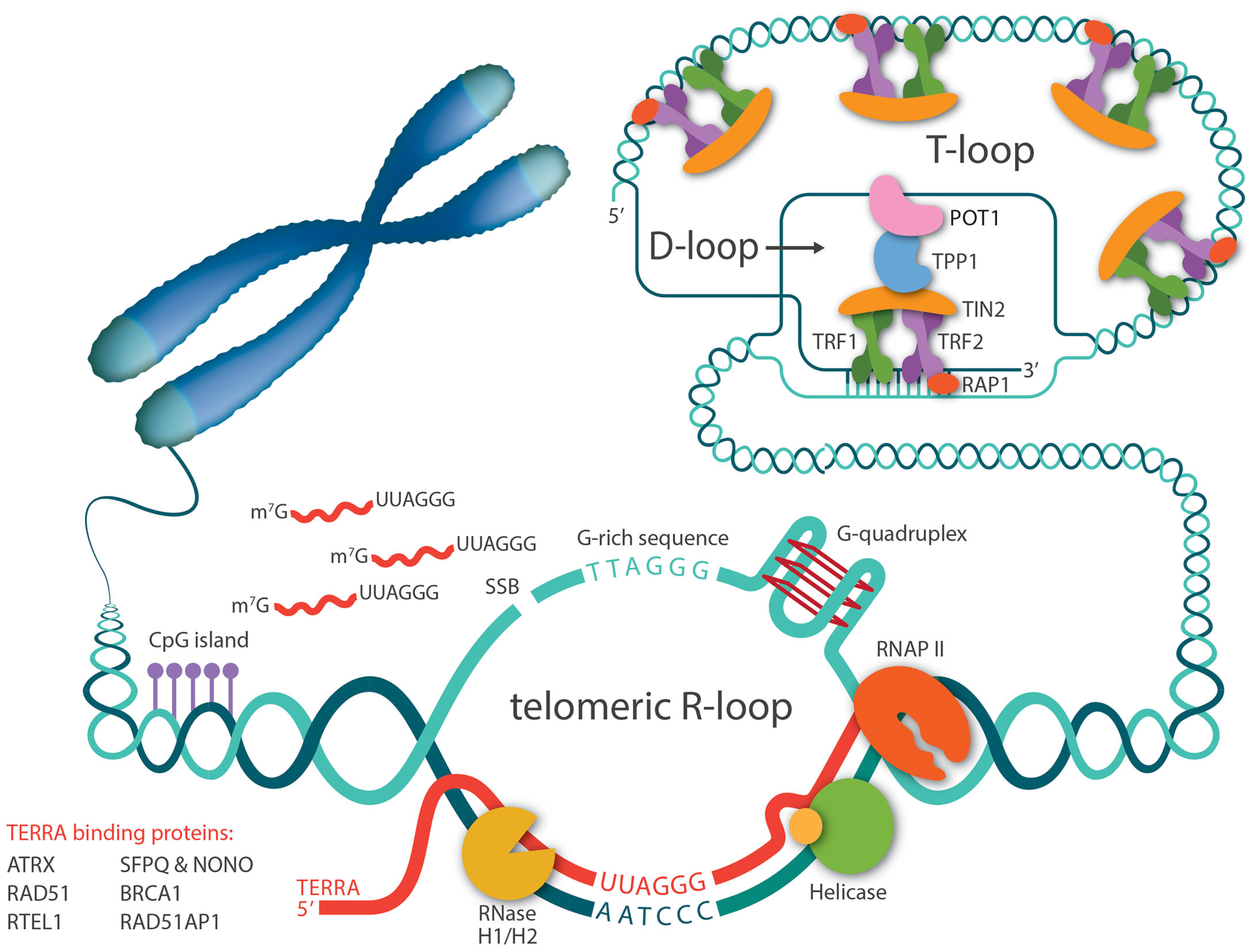
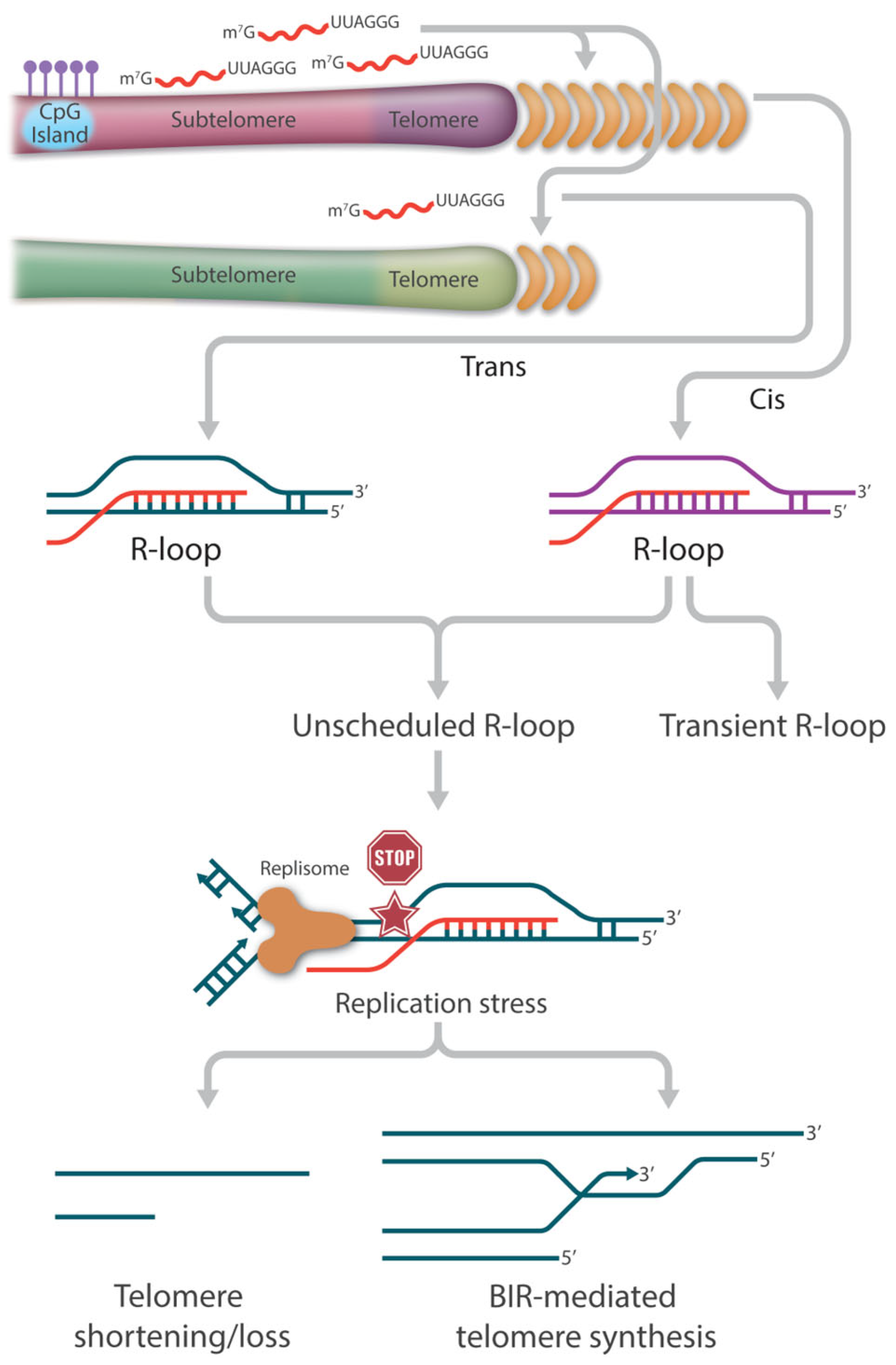
2. TERRA Is the Central Player in Telomeric R-Loop Formation
3. Proteins That Regulate R-Loop Levels at Telomeres
3.1. General R-Loop Regulating Factors
3.2. TERRA Binding Proteins
3.2.1. ATRX
3.2.2. SFPQ and NONO
3.2.3. RAD51
3.2.4. BRCA1
3.2.5. RTEL1
3.2.6. RAD51AP1
3.3. TERRA RNA Modification Proteins
3.4. Shelterins
3.5. Fanconi Anemia (FA) Pathway Proteins
4. Consequence of R-Loops at Telomeres
4.1. Survival Pathways in Yeast and Trypanosomes (Non-Mammalian Cells)
4.2. Survival Pathways in ALT Synthesis in Neoplasia
4.3. Disease Connection of TERRA R-Loops in ICF Syndrome
4.4. Connection of Oxidative DNA Damage and R-Loop Accumulation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- White, R.L.; Hogness, D.S. R loop mapping of the 18S and 28S sequences in the long and short repeating units of Drosophila melanogaster rDNA. Cell 1977, 10, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Drolet, M.; Phoenix, P.; Menzel, R.; Massé, E.; Liu, L.F.; Crouch, R.J. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. USA 1995, 92, 3526–3530. [Google Scholar] [CrossRef] [PubMed]
- Boguslawski, S.J.; Smith, D.E.; Michalak, M.A.; Mickelson, K.E.; Yehle, C.O.; Patterson, W.L.; Carrico, R.J. Characterization of monoclonal antibody to DNA· RNA and its application to immunodetection of hybrids. J. Immunol. Methods 1986, 89, 123–130. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chédin, F. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef]
- Sanz, L.A.; Chédin, F. High-resolution, strand-specific R-loop mapping via S9. 6-based DNA–RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2019, 14, 1734–1755. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Jenjaroenpun, P.; Kwoh, C.K.; Kuznetsov, V. Quantitative model of R-loop forming structures reveals a novel level of RNA–DNA interactome complexity. Nucleic Acids Res. 2012, 40, e16. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.J.; Hamperl, S.; Swigut, T.; Cimprich, K.A. qDRIP: A method to quantitatively assess RNA–DNA hybrid formation genome-wide. Nucleic Acids Res. 2020, 48, e84. [Google Scholar] [CrossRef]
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair 2018, 71, 69–81. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-loops as cellular regulators and genomic threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. R loops: From physiological to pathological roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Palancade, B.; Rothstein, R. The ultimate (mis) match: When DNA meets RNA. Cells 2021, 10, 1433. [Google Scholar] [CrossRef]
- Petermann, E.; Lan, L.; Zou, L. Sources, resolution and physiological relevance of R-loops and RNA–DNA hybrids. Nat. Rev. Mol. Cell Biol. 2022, 23, 521–540. [Google Scholar] [CrossRef]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA: RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef]
- Gómez-González, B.; García-Rubio, M.; Bermejo, R.; Gaillard, H.; Shirahige, K.; Marín, A.; Foiani, M.; Aguilera, A. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 2011, 30, 3106–3119. [Google Scholar] [CrossRef]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Cohen, S.; Puget, N.; Lin, Y.-L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA: DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef] [PubMed]
- Mischo, H.E.; Gomez-Gonzalez, B.; Grzechnik, P.; Rondon, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Huang, J.T.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef] [PubMed]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef]
- Tedeschi, F.A.; Cloutier, S.C.; Tran, E.J.; Jankowsky, E. The DEAD-box protein Dbp2p is linked to noncoding RNAs, the helicase Sen1p, and R-loops. RNA 2018, 24, 1693–1705. [Google Scholar] [CrossRef]
- Yu, Z.; Mersaoui, S.Y.; Guitton-Sert, L.; Coulombe, Y.; Song, J.; Masson, J.-Y.; Richard, S. DDX5 resolves R-loops at DNA double-strand breaks to promote DNA repair and avoid chromosomal deletions. NAR Cancer 2020, 2, zcaa028. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. Transcription–replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef]
- Bayona-Feliu, A.; Barroso, S.; Muñoz, S.; Aguilera, A. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription–replication conflicts. Nat. Genet. 2021, 53, 1050–1063. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.Y.; Gong, F.; Battenhouse, A.M.; Boutz, D.R.; Bashyal, A.; Refvik, S.T.; Chiang, C.-M.; Xhemalce, B.; Paull, T.T. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev. 2019, 33, 1751–1774. [Google Scholar] [CrossRef]
- Lam, F.C.; Kong, Y.W.; Huang, Q.; Vu Han, T.-L.; Maffa, A.D.; Kasper, E.M.; Yaffe, M.B. BRD4 prevents the accumulation of R-loops and protects against transcription–replication collision events and DNA damage. Nat. Commun. 2020, 11, 4083. [Google Scholar] [CrossRef]
- Roy, D.; Yu, K.; Lieber, M.R. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol. Cell. Biol. 2008, 28, 50–60. [Google Scholar] [CrossRef]
- Roy, D.; Lieber, M.R. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol. Cell. Biol. 2009, 29, 3124–3133. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef]
- Wahba, L.; Costantino, L.; Tan, F.J.; Zimmer, A.; Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016, 30, 1327–1338. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chédin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Chedin, F.; Hsieh, C.-L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J. The mitochondrial R-loop. Nucleic Acids Res. 2019, 47, 5480–5489. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Groh, M.; Gromak, N. Out of balance: R-loops in human disease. PLoS Genet. 2014, 10, e1004630. [Google Scholar] [CrossRef]
- Khan, E.S.; Danckwardt, S. Pathophysiological Role and Diagnostic Potential of R-Loops in Cancer and Beyond. Genes 2022, 13, 2181. [Google Scholar] [CrossRef]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef]
- O’sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- D’Adda di Fagagna, F.; Teo, S.H.; Jackson, S.P. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004, 18, 1781–1799. [Google Scholar] [CrossRef]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat–containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef]
- Schoeftner, S.; Blasco, M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008, 10, 228–236. [Google Scholar] [CrossRef]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef]
- Pfeiffer, V.; Crittin, J.; Grolimund, L.; Lingner, J. The THO complex component Thp2 counteracts telomeric R-loops and telomere shortening. EMBO J. 2013, 32, 2861–2871. [Google Scholar] [CrossRef]
- Balk, B.; Maicher, A.; Dees, M.; Klermund, J.; Luke-Glaser, S.; Bender, K.; Luke, B. Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat. Struct. Mol. Biol. 2013, 20, 1199–1205. [Google Scholar] [CrossRef]
- Nanavaty, V.; Sandhu, R.; Jehi, S.E.; Pandya, U.M.; Li, B. Trypanosoma brucei RAP1 maintains telomere and subtelomere integrity by suppressing TERRA and telomeric RNA: DNA hybrids. Nucleic Acids Res. 2017, 45, 5785–5796. [Google Scholar] [CrossRef]
- Graf, M.; Bonetti, D.; Lockhart, A.; Serhal, K.; Kellner, V.; Maicher, A.; Jolivet, P.; Teixeira, M.T.; Luke, B. Telomere length determines TERRA and R-loop regulation through the cell cycle. Cell 2017, 170, 72–85.e14. [Google Scholar] [CrossRef]
- Balk, B.; Dees, M.; Bender, K.; Luke, B. The differential processing of telomeres in response to increased telomeric transcription and RNA–DNA hybrid accumulation. RNA Biol. 2014, 11, 95–100. [Google Scholar] [CrossRef]
- Feretzaki, M.; Pospisilova, M.; Valador Fernandes, R.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 2020, 587, 303–308. [Google Scholar] [CrossRef]
- Porro, A.; Feuerhahn, S.; Reichenbach, P.; Lingner, J. Molecular dissection of telomeric repeat-containing RNA biogenesis unveils the presence of distinct and multiple regulatory pathways. Mol. Cell. Biol. 2010, 30, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G. Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018, 28, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, L.; Wagner, T.; Luke, B. Telomere Interacting Proteins and TERRA Regulation. Front. Genet. 2022, 13, 872636. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Voon, H.P.J.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; Sharpe, J.A.; Sloane-Stanley, J.A. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; De Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef]
- Eustermann, S.; Yang, J.-C.; Law, M.J.; Amos, R.; Chapman, L.M.; Jelinska, C.; Garrick, D.; Clynes, D.; Gibbons, R.J.; Rhodes, D. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 2011, 18, 777–782. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.-M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X. Distinct factors control histone variant H3. 3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.-M.; Stadler, S.C.; Allis, C.D. Daxx is an H3. 3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; De Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709. [Google Scholar] [CrossRef]
- Kannan, K.; Inagaki, A.; Silber, J.; Gorovets, D.; Zhang, J.; Kastenhuber, E.R.; Heguy, A.; Petrini, J.H.; Chan, T.A.; Huse, J.T. Whole exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 2012, 3, 1194. [Google Scholar] [CrossRef]
- Cheung, N.-K.V.; Zhang, J.; Lu, C.; Parker, M.; Bahrami, A.; Tickoo, S.K.; Heguy, A.; Pappo, A.S.; Federico, S.; Dalton, J. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012, 307, 1062–1071. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M. Driver mutations in histone H3. 3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Lee, J.-C.; Jeng, Y.-M.; Liau, J.-Y.; Tsai, J.-H.; Hsu, H.-H.; Yang, C.-Y. Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1064–1073. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.-M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Bower, K.; Napier, C.E.; Cole, S.L.; Dagg, R.A.; Lau, L.M.; Duncan, E.L.; Moy, E.L.; Reddel, R.R. Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of alternative lengthening of telomeres. PLoS ONE 2012, 7, e50062. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; De Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Chu, H.-P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.-g.; Wei, C.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA antagonizes ATRX and protects telomeres. Cell 2017, 170, 86–101.e116. [Google Scholar] [CrossRef]
- Yan, Q.; Wulfridge, P.; Doherty, J.; Fernandez-Luna, J.L.; Real, P.J.; Tang, H.-Y.; Sarma, K. Proximity labeling identifies a repertoire of site-specific R-loop modulators. Nat. Commun. 2022, 13, 53. [Google Scholar] [CrossRef]
- Petti, E.; Buemi, V.; Zappone, A.; Schillaci, O.; Broccia, P.V.; Dinami, R.; Matteoni, S.; Benetti, R.; Schoeftner, S. SFPQ and NONO suppress RNA: DNA-hybrid-related telomere instability. Nat. Commun. 2019, 10, 1001. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Bond, C.S.; Fox, A.H. The DBHS proteins SFPQ, NONO and PSPC1: A multipurpose molecular scaffold. Nucleic Acids Res. 2016, 44, 3989–4004. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.M. BRCA1 in the DNA damage response and at telomeres. Front. Genet. 2013, 4, 85. [Google Scholar] [CrossRef]
- Ballal, R.D.; Saha, T.; Fan, S.; Haddad, B.R.; Rosen, E.M. BRCA1 localization to the telomere and its loss from the telomere in response to DNA damage. J. Biol. Chem. 2009, 284, 36083–36098. [Google Scholar] [CrossRef]
- Acharya, S.; Kaul, Z.; Gocha, A.S.; Martinez, A.R.; Harris, J.; Parvin, J.D.; Groden, J. Association of BLM and BRCA1 during telomere maintenance in ALT cells. PLoS ONE 2014, 9, e103819. [Google Scholar] [CrossRef]
- Pan, X.; Drosopoulos, W.C.; Sethi, L.; Madireddy, A.; Schildkraut, C.L.; Zhang, D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. USA 2017, 114, E5940–E5949. [Google Scholar] [CrossRef]
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef]
- Chiang, H.-C.; Zhang, X.; Li, J.; Zhao, X.; Chen, J.; Wang, H.T.; Jatoi, I.; Brenner, A.; Hu, Y.; Li, R. BRCA1-associated R-loop affects transcription and differentiation in breast luminal epithelial cells. Nucleic Acids Res. 2019, 47, 5086–5099. [Google Scholar] [CrossRef]
- Vohhodina, J.; Goehring, L.J.; Liu, B.; Kong, Q.; Botchkarev Jr, V.V.; Huynh, M.; Liu, Z.; Abderazzaq, F.O.; Clark, A.P.; Ficarro, S.B. BRCA1 binds TERRA RNA and suppresses R-Loop-based telomeric DNA damage. Nat. Commun. 2021, 12, 3542. [Google Scholar] [CrossRef]
- West, S.; Gromak, N.; Proudfoot, N.J. Human 5′→3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 2004, 432, 522–525. [Google Scholar] [CrossRef]
- Björkman, A.; Johansen, S.L.; Lin, L.; Schertzer, M.; Kanellis, D.C.; Katsori, A.-M.; Christensen, S.T.; Luo, Y.; Andersen, J.S.; Elsässer, S.J. Human RTEL1 associates with Poldip3 to facilitate responses to replication stress and R-loop resolution. Genes Dev. 2020, 34, 1065–1074. [Google Scholar] [CrossRef]
- Kotsantis, P.; Segura-Bayona, S.; Margalef, P.; Marzec, P.; Ruis, P.; Hewitt, G.; Bellelli, R.; Patel, H.; Goldstone, R.; Poetsch, A.R. RTEL1 regulates G4/R-loops to avert replication-transcription collisions. Cell Rep. 2020, 33, 108546. [Google Scholar] [CrossRef]
- Wu, W.; Bhowmick, R.; Vogel, I.; Özer, Ö.; Ghisays, F.; Thakur, R.S.; Sanchez de Leon, E.; Richter, P.H.; Ren, L.; Petrini, J.H. RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol. 2020, 27, 424–437. [Google Scholar] [CrossRef]
- Ghisays, F.; Garzia, A.; Wang, H.; Canasto-Chibuque, C.; Hohl, M.; Savage, S.A.; Tuschl, T.; Petrini, J.H. RTEL1 influences the abundance and localization of TERRA RNA. Nat. Commun. 2021, 12, 3016. [Google Scholar] [CrossRef]
- Yadav, T.; Zhang, J.-M.; Ouyang, J.; Leung, W.; Simoneau, A.; Zou, L. TERRA and RAD51AP1 promote alternative lengthening of telomeres through an R-to D-loop switch. Mol. Cell 2022, 82, 3985–4000.e3984. [Google Scholar] [CrossRef]
- Kaminski, N.; Wondisford, A.R.; Kwon, Y.; Lynskey, M.L.; Bhargava, R.; Barroso-González, J.; García-Expósito, L.; He, B.; Xu, M.; Mellacheruvu, D. RAD51AP1 regulates ALT-HDR through chromatin-directed homeostasis of TERRA. Mol. Cell 2022, 82, 4001–4017.e4007. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, C.; Ma, W.; Huang, J.; Zhao, Y.; Liu, H. METTL3-mediated m6A modification stabilizes TERRA and maintains telomere stability. Nucleic Acids Res. 2022, 50, 11619–11634. [Google Scholar] [CrossRef]
- Shiromoto, Y.; Sakurai, M.; Minakuchi, M.; Ariyoshi, K.; Nishikura, K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 2021, 12, 1654. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing—Immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.R.; Slack, F.J. ADAR1 and its implications in cancer development and treatment. Trends Genet. 2022, 38, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Thomis, D.C.; Hans, S.L.; Samuel, C.E. Mechanism of interferon action: Double-stranded RNA-specific adenosine deaminase from human cells is inducible by alpha and gamma interferons. Virology 1995, 210, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef]
- Marión, R.M.; Montero, J.J.; Lopez de Silanes, I.; Grana-Castro, O.; Martinez, P.; Schoeftner, S.; Palacios-Fabrega, J.A.; Blasco, M.A. TERRA regulate the transcriptional landscape of pluripotent cells through TRF1-dependent recruitment of PRC2. eLife 2019, 8, e44656. [Google Scholar] [CrossRef]
- Porreca, R.M.; Herrera-Moyano, E.; Skourti, E.; Law, P.P.; Gonzalez Franco, R.; Montoya, A.; Faull, P.; Kramer, H.; Vannier, J.-B. TRF1 averts chromatin remodelling, recombination and replication dependent-break induced replication at mouse telomeres. eLife 2020, 9, e49817. [Google Scholar] [CrossRef]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 2009, 35, 403–413. [Google Scholar] [CrossRef]
- Lee, Y.W.; Arora, R.; Wischnewski, H.; Azzalin, C.M. TRF1 participates in chromosome end protection by averting TRF2-dependent telomeric R loops. Nat. Struct. Mol. Biol. 2018, 25, 147–153. [Google Scholar] [CrossRef]
- Mei, Y.; Deng, Z.; Vladimirova, O.; Gulve, N.; Johnson, F.B.; Drosopoulos, W.C.; Schildkraut, C.L.; Lieberman, P.M. TERRA G-quadruplex RNA interaction with TRF2 GAR domain is required for telomere integrity. Sci. Rep. 2021, 11, 3509. [Google Scholar] [CrossRef]
- Nie, X.; Xiao, D.; Ge, Y.; Xie, Y.; Zhou, H.; Zheng, T.; Li, X.; Liu, H.; Huang, H.; Zhao, Y. TRF2 recruits nucleolar protein TCOF1 to coordinate telomere transcription and replication. Cell Death Differ. 2021, 28, 1062–1075. [Google Scholar] [CrossRef]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef]
- Zhang, J.; Walter, J.C. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014, 19, 135–142. [Google Scholar] [CrossRef]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi anemia pathway in cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Li, D.; Hou, K.; Zhang, K.; Jia, S. Regulation of replication stress in alternative lengthening of telomeres by fanconi anaemia protein. Genes 2022, 13, 180. [Google Scholar] [CrossRef]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Martinez, D.L.; Liang, C.-C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef]
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252. [Google Scholar] [CrossRef]
- Silva, B.; Pentz, R.; Figueira, A.M.; Arora, R.; Lee, Y.W.; Hodson, C.; Wischnewski, H.; Deans, A.J.; Azzalin, C.M. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019, 10, 2253. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 19110. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef]
- Bakker, S.T.; van de Vrugt, H.J.; Rooimans, M.A.; Oostra, A.B.; Steltenpool, J.; Delzenne-Goette, E.; van der Wal, A.; van der Valk, M.; Joenje, H.; te Riele, H. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 2009, 18, 3484–3495. [Google Scholar] [CrossRef] [PubMed]
- Ishino, S.; Yamagami, T.; Kitamura, M.; Kodera, N.; Mori, T.; Sugiyama, S.; Ando, T.; Goda, N.; Tenno, T.; Hiroaki, H. Multiple interactions of the intrinsically disordered region between the helicase and nuclease domains of the archaeal Hef protein. J. Biol. Chem. 2014, 289, 21627–21639. [Google Scholar] [CrossRef] [PubMed]
- Crismani, W.; Girard, C.; Froger, N.; Pradillo, M.; Santos, J.L.; Chelysheva, L.; Copenhaver, G.P.; Horlow, C.; Mercier, R. FANCM limits meiotic crossovers. Science 2012, 336, 1588–1590. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ijpma, A.; Greider, C.W. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol. Cell. Biol. 2001, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Martínez, L.; Öztürk, M.; Butter, F.; Luke, B. Npl3 stabilizes R-loops at telomeres to prevent accelerated replicative senescence. EMBO Rep. 2020, 21, e49087. [Google Scholar] [CrossRef]
- Li, B. Keeping balance between genetic stability and plasticity at the telomere and subtelomere of trypanosoma brucei. Front. Cell Dev. Biol. 2021, 9, 699639. [Google Scholar] [CrossRef]
- Apte, M.S.; Cooper, J.P. Life and cancer without telomerase: ALT and other strategies for making sure ends (don’t) meet. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 57–73. [Google Scholar] [CrossRef]
- Ng, L.J.; Cropley, J.E.; Pickett, H.A.; Reddel, R.R.; Suter, C.M. Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res. 2009, 37, 1152–1159. [Google Scholar] [CrossRef]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londoño-Vallejo, A.; Decottignies, A. Alternative lengthening of telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef]
- Ouyang, J.; Yadav, T.; Zhang, J.-M.; Yang, H.; Rheinbay, E.; Guo, H.; Haber, D.A.; Lan, L.; Zou, L. RNA transcripts stimulate homologous recombination by forming DR-loops. Nature 2021, 594, 283–288. [Google Scholar] [CrossRef]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 30. [Google Scholar] [CrossRef]
- Vukic, M.; Daxinger, L. DNA methylation in disease: Immunodeficiency, Centromeric instability, Facial anomalies syndrome. Essays Biochem. 2019, 63, 773–783. [Google Scholar]
- Toubiana, S.; Selig, S. Human subtelomeric DNA methylation: Regulation and roles in telomere function. Curr. Opin. Genet. Dev. 2020, 60, 9–16. [Google Scholar] [CrossRef]
- Weemaes, C.M.; Van Tol, M.J.; Wang, J.; van Ostaijen-Ten Dam, M.M.; Van Eggermond, M.C.; Thijssen, P.E.; Aytekin, C.; Brunetti-Pierri, N.; Van Der Burg, M.; Graham Davies, E. Heterogeneous clinical presentation in ICF syndrome: Correlation with underlying gene defects. Eur. J. Hum. Genet. 2013, 21, 1219–1225. [Google Scholar] [CrossRef]
- Nergadze, S.G.; Farnung, B.O.; Wischnewski, H.; Khoriauli, L.; Vitelli, V.; Chawla, R.; Giulotto, E.; Azzalin, C.M. CpG-island promoters drive transcription of human telomeres. RNA 2009, 15, 2186–2194. [Google Scholar] [CrossRef]
- Deng, Z.; Campbell, A.E.; Lieberman, P.M. TERRA, CpG methylation, and telomere heterochromatin: Lessons from ICF syndrome cells. Cell Cycle 2010, 9, 69–74. [Google Scholar] [CrossRef]
- Yehezkel, S.; Segev, Y.; Viegas-Pequignot, E.; Skorecki, K.; Selig, S. Hypomethylation of subtelomeric regions in ICF syndrome is associated with abnormally short telomeres and enhanced transcription from telomeric regions. Hum. Mol. Genet. 2008, 17, 2776–2789. [Google Scholar] [CrossRef]
- Sagie, S.; Ellran, E.; Katzir, H.; Shaked, R.; Yehezkel, S.; Laevsky, I.; Ghanayim, A.; Geiger, D.; Tzukerman, M.; Selig, S. Induced pluripotent stem cells as a model for telomeric abnormalities in ICF type I syndrome. Hum. Mol. Genet. 2014, 23, 3629–3640. [Google Scholar] [CrossRef]
- Yehezkel, S.; Shaked, R.; Sagie, S.; Berkovitz, R.; Shachar-Bener, H.; Segev, Y.; Selig, S. Characterization and rescue of telomeric abnormalities in ICF syndrome type I fibroblasts. Front. Oncol. 2013, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Sagie, S.; Toubiana, S.; Hartono, S.R.; Katzir, H.; Tzur-Gilat, A.; Havazelet, S.; Francastel, C.; Velasco, G.; Chédin, F.; Selig, S. Telomeres in ICF syndrome cells are vulnerable to DNA damage due to elevated DNA: RNA hybrids. Nat. Commun. 2017, 8, 14015. [Google Scholar] [CrossRef] [PubMed]
- Sagie, S.; Edni, O.; Weinberg, J.; Toubiana, S.; Kozlovski, T.; Frostig, T.; Katzin, N.; Bar-Am, I.; Selig, S. Non-random length distribution of individual telomeres in immunodeficiency, centromeric instability and facial anomalies syndrome, type I. Hum. Mol. Genet. 2017, 26, 4244–4256. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Telomeres: Influencing the rate of aging. Ann. N. Y. Acad. Sci. 1998, 854, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kawanishi, S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999, 453, 365–368. [Google Scholar] [CrossRef]
- Lonkar, P.; Dedon, P.C. Reactive species and DNA damage in chronic inflammation: Reconciling chemical mechanisms and biological fates. Int. J. Cancer 2011, 128, 1999–2009. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Sun, C.; Wang, K.; Stock, A.J.; Gong, Y.; Demarest, T.G.; Yang, B.; Giri, N.; Harrington, L.; Alter, B.P.; Savage, S.A. Re-equilibration of imbalanced NAD metabolism ameliorates the impact of telomere dysfunction. EMBO J. 2020, 39, e103420. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef]
- Westin, E.R.; Aykin-Burns, N.; Buckingham, E.M.; Spitz, D.R.; Goldman, F.D.; Klingelhutz, A.J. The p53/p21WAF/CIP pathway mediates oxidative stress and senescence in dyskeratosis congenita cells with telomerase insufficiency. Antioxid. Redox Signal. 2011, 14, 985–997. [Google Scholar] [CrossRef]
- Wang, Z.; Rhee, D.B.; Lu, J.; Bohr, C.T.; Zhou, F.; Vallabhaneni, H.; de Souza-Pinto, N.C.; Liu, Y. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010, 6, e1000951. [Google Scholar] [CrossRef]
- von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol. Cell 2019, 75, 117–130.e116. [Google Scholar] [CrossRef]
- Tan, J.; Duan, M.; Yadav, T.; Phoon, L.; Wang, X.; Zhang, J.-M.; Zou, L.; Lan, L. An R-loop-initiated CSB–RAD52–POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Res. 2020, 48, 1285–1300. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; Liu, Y. R-Loops at Chromosome Ends: From Formation, Regulation, and Cellular Consequence. Cancers 2023, 15, 2178. https://doi.org/10.3390/cancers15072178
Gong Y, Liu Y. R-Loops at Chromosome Ends: From Formation, Regulation, and Cellular Consequence. Cancers. 2023; 15(7):2178. https://doi.org/10.3390/cancers15072178
Chicago/Turabian StyleGong, Yi, and Yie Liu. 2023. "R-Loops at Chromosome Ends: From Formation, Regulation, and Cellular Consequence" Cancers 15, no. 7: 2178. https://doi.org/10.3390/cancers15072178
APA StyleGong, Y., & Liu, Y. (2023). R-Loops at Chromosome Ends: From Formation, Regulation, and Cellular Consequence. Cancers, 15(7), 2178. https://doi.org/10.3390/cancers15072178