

Targeting the Molecular and Immunologic Features of Leiomyosarcoma
 and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Current Standard of Care Treatment for LMS
2.1. uLMS-Specific Considerations
2.2. ST-LMS-Specific Considerations
3. Emerging Role of Immunotherapy in the Treatment of LMS
4. Molecular Landscape of LMS
4.1. Complex Karyotype
4.2. Molecular Subtypes of LMS
4.3. TP53
4.4. RB1
4.5. PTEN Deletion and PI3k/AKt/mOR Pathway
4.6. DNA Damage Response
4.7. Whole-Genome Doubling
4.8. Alternative Lengthening of Telomeres
5. Immune Landscape of LMS
5.1. Tertiary Lymphoid Structures
5.2. PD-L1
5.3. Tumor-Infiltrating Lymphocytes
5.4. Macrophages
5.5. Association of Molecular Subtypes with Immune Microenvironment
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Katz, D.; Palmerini, E.; Pollack, S.M. More Than 50 Subtypes of Soft Tissue Sarcoma: Paving the Path for Histology-Driven Treatments. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 925–938. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Neyman, N.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2010; National Cancer Institute: Bethesda, MD, USA, 2010.
- George, S.; Serrano, C.; Hensley, M.L.; Ray-Coquard, I. Soft Tissue and Uterine Leiomyosarcoma. J. Clin. Oncol. 2018, 36, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Amant, F.; Coosemans, A.; Debiec-Rychter, M.; Timmerman, D.; Vergote, I. Clinical management of uterine sarcomas. Lancet Oncol. 2009, 10, 1188–1198. [Google Scholar] [CrossRef]
- Tropé, C.G.; Abeler, V.M.; Kristensen, G.B. Diagnosis and treatment of sarcoma of the uterus. A review. Acta Oncol. 2012, 51, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; O’Connell, F.; Fletcher, C.D.M. Dedifferentiated leiomyosarcoma: Clinicopathological analysis of 18 cases. Histopathology 2011, 59, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Demicco, E.G.; Boland, G.M.; Savannah, K.J.B.; Lusby, K.; Young, E.D.; Ingram, D.; Watson, K.L.; Bailey, M.; Guo, X.; Hornick, J.L.; et al. Progressive loss of myogenic differentiation in leiomyosarcoma has prognostic value. Histopathology 2015, 66, 627–638. [Google Scholar] [CrossRef]
- Miettinen, M.; Fetsch, J.F. Evaluation of biological potential of smooth muscle tumours. Histopathology 2006, 48, 97–105. [Google Scholar] [CrossRef]
- Massi, D.; Beltrami, G.; Mela, M.; Pertici, M.; Capanna, R.; Franchi, A. Prognostic factors in soft tissue leiomyosarcoma of the extremities: A retrospective analysis of 42 cases. Eur. J. Surg. Oncol. (EJSO) 2004, 30, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, P.; Rydholm, A.; Willén, H.; Åkerman, M.; Baldetorp, B.; Fernö, M. Soft tissue leiomyosarcoma. A population-based epidemiologic and prognostic study of 48 patients, including cellular dna content. Cancer 1992, 70, 114–119. [Google Scholar] [CrossRef]
- Pautier, P.; Floquet, A.; Chevreau, C.; Penel, N.; Guillemet, C.; Delcambre, C.; Cupissol, D.; Selle, F.; Isambert, N.; Piperno-Neumann, S.; et al. A single-arm multicentre phase II trial of doxorubicin in combination with trabectedin in the first-line treatment for leiomyosarcoma with long-term follow-up and impact of cytoreductive surgery. ESMO Open 2021, 6, 100209. [Google Scholar] [CrossRef]
- Pautier, P.; Italiano, A.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Firmin, N.; Boudou-Rouquette, P.; Bertucci, F.; Balleyguier, C.; Lebrun-Ly, V.; et al. Doxorubicin alone versus doxorubicin with trabectedin followed by trabectedin alone as first-line therapy for metastatic or unresectable leiomyosarcoma (LMS-04): A randomised, multicentre, open-label phase 3 trial. Lancet Oncol. 2022, 23, 1044–1054. [Google Scholar] [CrossRef]
- Van Glabbeke, M.; Van Oosterom, A.T.; Oosterhuis, J.W.; Mouridsen, H.; Crowther, D.; Somers, R.; Verweij, J.; Santoro, A.; Buesa, J.; Tursz, T. Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: An analysis of 2,185 patients treated with anthracycline-containing first-line regimens—A European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J. Clin. Oncol. 1999, 17, 150–157. [Google Scholar]
- Savina, M.; Le Cesne, A.; Blay, J.-Y.; Ray-Coquard, I.; Mir, O.; Toulmonde, M.; Cousin, S.; Terrier, P.; Ranchere-Vince, D.; Meeus, P.; et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: The METASARC observational study. BMC Med. 2017, 15, 78. [Google Scholar] [CrossRef]
- Roland, C.L.; Boland, G.M.; Demicco, E.G.; Lusby, K.; Ingram, D.R.; May, C.D.; Kivlin, C.M.; Watson, K.L.; Al Sannaa, G.A.; Wang, W.-L.; et al. Clinical Observations and Molecular Variables of Primary Vascular Leiomyosarcoma. JAMA Surg. 2016, 151, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Giuntoli, R.L., 2nd; Metzinger, D.S.; DiMarco, C.S.; Cha, S.S.; Sloan, J.A.; Keeney, G.L.; Gostout, B.S. Retrospective review of 208 patients with leiomyosarcoma of the uterus: Prognostic indicators, surgical management, and adjuvant therapy. Gynecol. Oncol. 2003, 89, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Seagle, B.-L.L.; Sobecki-Rausch, J.; Strohl, A.E.; Shilpi, A.; Grace, A.; Shahabi, S. Prognosis and treatment of uterine leiomyosarcoma: A National Cancer Database study. Gynecol. Oncol. 2017, 145, 61–70. [Google Scholar] [CrossRef]
- Hensley, M.L.; Wathen, J.K.; Maki, R.G.; Araujo, D.M.; Sutton, G.; Priebat, D.A.; George, S.; Soslow, R.A.; Baker, L.H. Adjuvant therapy for high-grade, uterus-limited leiomyosarcoma: Results of a phase 2 trial (SARC 005). Cancer 2013, 119, 1555–1561. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Soibinet, P.; Penel, N.; Bompas, E.; Duffaud, F.; Stoeckle, E.; Mir, O.; Adam, J.; Chevreau, C.; Bonvalot, S.; et al. Improved survival using specialized multidisciplinary board in sarcoma patients. Ann. Oncol. 2017, 28, 2852–2859. [Google Scholar] [CrossRef]
- Blay, J.Y.; Honore, C.; Stoeckle, E.; Meeus, P.; Jafari, M.; Gouin, F.; Anract, P.; Ferron, G.; Rochwerger, A.; Ropars, M.; et al. Surgery in reference centers improves survival of sarcoma patients: A nationwide study. Ann. Oncol. 2019, 30, 1407. [Google Scholar] [CrossRef] [PubMed]
- Bonvalot, S.; Gaignard, E.; Stoeckle, E.; Meeus, P.; Decanter, G.; Carrere, S.; Honore, C.; Delhorme, J.B.; Fau, M.; Tzanis, D.; et al. Survival Benefit of the Surgical Management of Retroperitoneal Sarcoma in a Reference Center: A Nationwide Study of the French Sarcoma Group from the NetSarc Database. Ann. Surg. Oncol. 2019, 26, 2286–2293. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Soft Tissue Sarcoma (Version 2.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- National Comprehensive Cancer Network. Uterine Neoplasms (Version 2.2022); National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2022. [Google Scholar]
- ESMO/European Sarcoma Network Working Group. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow. Ann. Oncol. 2012, 23, vii92–vii99. [Google Scholar] [CrossRef] [PubMed]
- Picci, P. Adjuvant chemotherapy for extremity soft-tissue sarcomas in adults. Curr. Oncol. Rep. 2000, 2, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Antman, K.; Crowley, J.; Balcerzak, S.P.; Rivkin, S.E.; Weiss, G.R.; Elias, A.; Natale, R.B.; Cooper, R.M.; Barlogie, B.; Trump, D.L. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J. Clin. Oncol. 1993, 11, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Maurel, J.; López-Pousa, A.; Peñas, R.D.L.; Fra, J.; Martín, J.; Cruz, J.; Casado, A.; Poveda, A.; Martínez-Trufero, J.; Balañá, C.; et al. Efficacy of Sequential High-Dose Doxorubicin and Ifosfamide Compared With Standard-Dose Doxorubicin in Patients With Advanced Soft Tissue Sarcoma: An Open-Label Randomized Phase II Study of the Spanish Group for Research on Sarcomas. J. Clin. Oncol. 2009, 27, 1893–1898. [Google Scholar] [CrossRef]
- Sutton, G.; Blessing, J.A.; Malfetano, J.H. Ifosfamide and Doxorubicin in the Treatment of Advanced Leiomyosarcomas of the Uterus: A Gynecologic Oncology Group Study. Gynecol. Oncol. 1996, 62, 226–229. [Google Scholar] [CrossRef]
- Hensley, M.L.; Maki, R.G.; Venkatraman, E.; Geller, G.; Lovegren, M.; Aghajanian, C.; Sabbatini, P.; Tong, W.; Barakat, R.; Spriggs, D.R. Gemcitabine and Docetaxel in Patients With Unresectable Leiomyosarcoma: Results of a Phase II Trial. J. Clin. Oncol. 2002, 20, 2824–2831. [Google Scholar] [CrossRef]
- Maki, R.G.; Wathen, J.K.; Patel, S.R.; Priebat, D.A.; Okuno, S.H.; Samuels, B.; Fanucchi, M.; Harmon, D.C.; Schuetze, S.M.; Reinke, D.; et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: Results of sarcoma alliance for research through collaboration study 002 [corrected]. J. Clin. Oncol. 2007, 25, 2755–2763. [Google Scholar] [CrossRef]
- Le Cesne, A.; Blay, J.-Y.; Judson, I.; Van Oosterom, A.; Verweij, J.; Radford, J.; Lorigan, P.; Rodenhuis, S.; Ray-Coquard, I.; Bonvalot, S.; et al. Phase II Study of ET-743 in Advanced Soft Tissue Sarcomas: A European Organisation for the Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group Trial. J. Clin. Oncol. 2005, 23, 576–584. [Google Scholar] [CrossRef]
- Demetri, G.D.; Chawla, S.P.; von Mehren, M.; Ritch, P.; Baker, L.H.; Blay, J.Y.; Hande, K.R.; Keohan, M.L.; Samuels, B.L.; Schuetze, S.; et al. Efficacy and Safety of Trabectedin in Patients With Advanced or Metastatic Liposarcoma or Leiomyosarcoma After Failure of Prior Anthracyclines and Ifosfamide: Results of a Randomized Phase II Study of Two Different Schedules. J. Clin. Oncol. 2009, 27, 4188–4196. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schöffski, P.; Collin, F.; Pandite, L.; Marreaud, S.; De Brauwer, A.; et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J. Clin. Oncol. 2009, 27, 3126–3132. [Google Scholar]
- Van Der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Bogani, G.; Cliby, W.A.; Aletti, G.D. Impact of morcellation on survival outcomes of patients with unexpected uterine leiomyosarcoma: A systematic review and meta-analysis. Gynecol. Oncol. 2015, 137, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.; Mangioni, C.; Malmström, H.; Scarfone, G.; Poveda, A.; Pecorelli, S.; Tateo, S.; Franchi, M.; Jobsen, J.; Coens, C.; et al. Phase III randomised study to evaluate the role of adjuvant pelvic radiotherapy in the treatment of uterine sarcomas stages I and II: An European Organisation for Research and Treatment of Cancer Gynaecological Cancer Group Study (protocol 55874). Eur. J. Cancer 2008, 44, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Denschlag, D.; Ackermann, S.; Battista, M.J.; Cremer, W.; Egerer, G.; Fehr, M.; Follmann, M.; Haase, H.; Harter, P.; Hettmer, S.; et al. Sarcoma of the Uterus. Guideline of the DGGG, OEGGG and SGGG (S2k-Level, AWMF Registry No. 015/074, April 2021). Geburtshilfe Frauenheilkd 2022, 82, 1337–1367. [Google Scholar] [CrossRef]
- Bogani, G.; Fucà, G.; Maltese, G.; Ditto, A.; Martinelli, F.; Signorelli, M.; Chiappa, V.; Scaffa, C.; Sabatucci, I.; Lecce, F.; et al. Efficacy of adjuvant chemotherapy in early stage uterine leiomyosarcoma: A systematic review and meta-analysis. Gynecol. Oncol. 2016, 143, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.H.; Shim, S.-H.; Chang, M.; Choi, A.Y.; Kang, G.G.; Lee, S.J.; Kim, S.-N. Effect of adjuvant therapy on the risk of recurrence in early-stage leiomyosarcoma: A meta-analysis. Gynecol. Oncol. 2019, 154, 638–650. [Google Scholar] [CrossRef]
- Pautier, P.; Floquet, A.; Gladieff, L.; Bompas, E.; Ray-Coquard, I.; Piperno-Neumann, S.; Selle, F.; Guillemet, C.; Weber, B.; Largillier, R.; et al. A randomized clinical trial of adjuvant chemotherapy with doxorubicin, ifosfamide, and cisplatin followed by radiotherapy versus radiotherapy alone in patients with localized uterine sarcomas (SARCGYN study): A study of the French Sarcoma Group. Ann. Oncol. 2013, 24, 1099–1104. [Google Scholar] [CrossRef]
- Shellenberger, T.D.; Sturgis, E.M. Sarcomas of the head and neck region. Curr. Oncol. Rep. 2009, 11, 135–142. [Google Scholar] [CrossRef]
- Anaya, D.A.; Lev, D.C.; Pollock, R. The role of surgical margin status in retroperitoneal sarcoma. J. Surg. Oncol. 2008, 98, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Pierie, J.-P.; Betensky, R.; Choudry, U.; Willett, C.; Souba, W.; Ott, M. Outcomes in a series of 103 retroperitoneal sarcomas. Eur. J. Surg. Oncol. (EJSO) 2006, 32, 1235–1241. [Google Scholar] [CrossRef]
- Hassan, I.; Park, S.Z.; Donohue, J.H.; Nagorney, D.M.; Kay, P.A.; Nasciemento, A.G.; Schleck, C.D.; Ilstrup, D.M. Operative management of primary retroperitoneal sarcomas: A reappraisal of an institutional experience. Ann Surg 2004, 239, 244–250. [Google Scholar] [CrossRef]
- Albertsmeier, M.; Rauch, A.; Roeder, F.; Hasenhütl, S.; Pratschke, S.; Kirschneck, M.; Gronchi, A.; Jebsen, N.L.; Cassier, P.A.; Sargos, P.; et al. External Beam Radiation Therapy for Resectable Soft Tissue Sarcoma: A Systematic Review and Meta-Analysis. Ann. Surg. Oncol. 2018, 25, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Bonvalot, S.; Gronchi, A.; Le Péchoux, C.; Swallow, C.J.; Strauss, D.; Meeus, P.; van Coevorden, F.; Stoldt, S.; Stoeckle, E.; Rutkowski, P.; et al. Preoperative radiotherapy plus surgery versus surgery alone for patients with primary retroperitoneal sarcoma (EORTC-62092: STRASS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2020, 21, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with Single Agent Nivolumab for Advanced Leiomyosarcoma of the Uterus: Results of a Phase 2 Study. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Soto, L.S.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Liu, J.; Liang, D.; Zhao, M.; Yu, W.; Chen, P. Nivolumab plus ipilimumab versus nivolumab in individuals with treatment-naive programmed death-ligand 1 positive metastatic soft tissue sarcomas: A multicentre retrospective study. BMC Cancer 2021, 21, 108. [Google Scholar] [CrossRef]
- Saerens, M.; Brusselaers, N.; Rottey, S.; Decruyenaere, A.; Creytens, D.; Lapeire, L. Immune checkpoint inhibitors in treatment of soft-tissue sarcoma: A systematic review and meta-analysis. Eur. J. Cancer 2021, 152, 165–182. [Google Scholar] [CrossRef]
- Italiano, A.; Bellera, C.; D’Angelo, S. PD1/PD-L1 targeting in advanced soft-tissue sarcomas: A pooled analysis of phase II trials. J. Hematol. Oncol. 2020, 13, 55. [Google Scholar] [CrossRef]
- Tan, Z.; Wang, X.; Liu, J.; Fan, Z.; Gao, T.; Bai, C.; Xue, R.; Li, S.; Zhang, L. Efficacy of PD-1 inhibitors combined with pegylated liposomal doxorubicin and dacarbazine compared with liposomal doxorubicin and dacarbazine in advanced leiomyosarcoma patients: A retrospective, single-institutional cohort study. Ann. Transl. Med. 2022, 10, 1000. [Google Scholar] [CrossRef]
- Pollack, S.M.; Redman, M.W.; Baker, K.K.; Wagner, M.J.; Schroeder, B.A.; Loggers, E.T.; Trieselmann, K.; Copeland, V.C.; Zhang, S.; Black, G. Assessment of Doxorubicin and Pembrolizumab in Patients With Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol. 2020, 6, 1778–1782. [Google Scholar] [CrossRef]
- Livingston, M.B.; Jagosky, M.H.; Robinson, M.M.; Ahrens, W.A.; Benbow, J.H.; Farhangfar, C.J.; Foureau, D.M.; Maxwell, D.M.; Baldrige, E.A.; Begic, X.; et al. Phase II Study of Pembrolizumab in Combination with Doxorubicin in Metastatic and Unresectable Soft-Tissue Sarcoma. Clin. Cancer Res. 2021, 27, 6424–6431. [Google Scholar] [CrossRef] [PubMed]
- Martin-Broto, J.; Hindi, N.; Grignani, G.; Martinez-Trufero, J.; Redondo, A.; Valverde, C.; Stacchiotti, S.; Lopez-Pousa, A.; D’Ambrosio, L.; Gutierrez, A.; et al. Nivolumab and sunitinib combination in advanced soft tissue sarcomas: A multicenter, single-arm, phase Ib/II trial. J. Immunother. Cancer 2020, 8, e001561. [Google Scholar] [CrossRef] [PubMed]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Guo, X.; Zhuang, R.; Zhang, C.; Wang, Z.; Shen, F.; Wang, Y.; Liu, W.; Zhang, Y.; Lu, W.; et al. Activity of PD-1 Inhibitor Combined With Anti-Angiogenic Therapy in Advanced Sarcoma: A Single-Center Retrospective Analysis. Front. Mol. Biosci. 2021, 8, 747650. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Williams, H.R.; Brzezinska, B.N.; Gaidis, A.; Patel, B.; Munroe, J.; White, J.; Rungruang, B. Use of pembrolizumab in MSI-high uterine leiomyosarcoma; a case report and review of the literature. Gynecol. Oncol. Rep. 2021, 35, 100701. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef]
- Heine, A.; Kristiansen, G.; Schild, H.H.; Brossart, P. Successful treatment of refractory leiomyosarcoma with the PD-1 inhibitor nivolumab. Ann. Oncol. 2016, 27, 1813–1814. [Google Scholar] [CrossRef]
- Wagner, M.J.; Zhang, Y.; Cranmer, L.D.; Loggers, E.T.; Black, G.; McDonnell, S.; Maxwell, S.; Johnson, R.; Moore, R.; Hermida de Viveiros, P.; et al. A Phase 1/2 Trial Combining Avelumab and Trabectedin for Advanced Liposarcoma and Leiomyosarcoma. Clin Cancer Res 2022, 28, 2306–2312. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.D.; Babichev, Y.; Fuligni, F.; Comitani, F.; Layeghifard, M.; Venier, R.E.; Dentro, S.C.; Maheshwari, A.; Guram, S.; Wunker, C.; et al. Lineage-defined leiomyosarcoma subtypes emerge years before diagnosis and determine patient survival. Nat. Commun. 2021, 12, 4496. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.J.; McLellan, M.D.; Bailey, M.H.; Miller, C.A.; Appelbaum, E.L.; Cordes, M.G.; Fronick, C.C.; Fulton, L.A.; Fulton, R.S.; Mardis, E.R.; et al. Cancer Genome Atlas Research, Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar]
- Beck, A.H.; Lee, C.-H.; Witten, D.M.; Gleason, B.C.; Edris, B.; Espinosa, I.; Zhu, S.; Li, R.; Montgomery, K.D.; Marinelli, R.J.; et al. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene 2010, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Lagarde, P.; Brulard, C.; Terrier, P.; Laë, M.; Marques, B.; Ranchere-Vince, D.; Michels, J.-J.; Trassard, M.; Cioffi, A.; et al. Genetic Profiling Identifies Two Classes of Soft-Tissue Leiomyosarcomas with Distinct Clinical Characteristics. Clin. Cancer Res. 2013, 19, 1190–1196. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Liu, W.; Tong, H.; Zhang, C.; Zhuang, R.; Guo, H.; Lv, C.; Yang, H.; Lin, Q.; Guo, X.; Wang, Z.; et al. Integrated genomic and transcriptomic analysis revealed mutation patterns of de-differentiated liposarcoma and leiomyosarcoma. BMC Cancer 2020, 20, 1035. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Mani, D.R.; Krug, K.; Zhang, B.; Satpathy, S.; Clauser, K.R.; Ding, L.; Ellis, M.; Gillette, M.A.; Carr, S.A. Cancer proteogenomics: Current impact and future prospects. Nat. Rev. Cancer 2022, 22, 298–313. [Google Scholar] [CrossRef]
- Harbers, L.; Agostini, F.; Nicos, M.; Poddighe, D.; Bienko, M.; Crosetto, N. Somatic Copy Number Alterations in Human Cancers: An Analysis of Publicly Available Data From The Cancer Genome Atlas. Front. Oncol. 2021, 11, 700568. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.-H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e673. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, aaf8399. [Google Scholar] [CrossRef]
- Guo, X.; Jo, V.Y.; Mills, A.M.; Zhu, S.X.; Lee, C.-H.; Espinosa, I.; Nucci, M.R.; Varma, S.; Forgó, E.; Hastie, T.; et al. Clinically Relevant Molecular Subtypes in Leiomyosarcoma. Clin. Cancer Res. 2015, 21, 3501–3511. [Google Scholar] [CrossRef]
- Pérot, G.; Derré, J.; Coindre, J.-M.; Tirode, F.; Lucchesi, C.; Mariani, O.; Gibault, L.; Guillou, L.; Terrier, P.; Aurias, A. Strong Smooth Muscle Differentiation Is Dependent on Myocardin Gene Amplification in Most Human Retroperitoneal Leiomyosarcomas. Cancer Res 2009, 69, 2269–2278. [Google Scholar] [CrossRef]
- Ito, M.; Barys, L.; O’Reilly, T.; Young, S.; Gorbatcheva, B.; Monahan, J.; Zumstein-Mecker, S.; Choong, P.F.; Dickinson, I.; Crowe, P.; et al. Comprehensive Mapping of p53 Pathway Alterations Reveals an Apparent Role for Both SNP309 and MDM2 Amplification in Sarcomagenesis. Clin. Cancer Res. 2011, 17, 416–426. [Google Scholar] [CrossRef]
- Pérot, G.; Chibon, F.; Montero, A.; Lagarde, P.; de Thé, H.; Terrier, P.; Guillou, L.; Ranchère, D.; Coindre, J.-M.; Aurias, A. Constant p53 Pathway Inactivation in a Large Series of Soft Tissue Sarcomas with Complex Genetics. Am. J. Pathol. 2010, 177, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2018, 11, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A. and K.H. Vousden, p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Hensley, M.L.; Chavan, S.S.; Solit, D.B.; Murali, R.; Soslow, R.; Chiang, S.; Jungbluth, A.A.; Bandlamudi, C.; Srinivasan, P.; Tap, W.D.; et al. Genomic Landscape of Uterine Sarcomas Defined Through Prospective Clinical Sequencing. Clin. Cancer Res. 2020, 26, 3881–3888. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Schaefer, I.; Lundberg, M.Z.; Demicco, E.G.; Przybyl, J.; Matusiak, M.; Chibon, F.; Ingram, D.R.; Hornick, J.L.; Wang, W.; Bauer, S.; et al. Relationships between highly recurrent tumor suppressor alterations in 489 leiomyosarcomas. Cancer 2021, 127, 2666–2673. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Chaikovsky, A.C.; Sage, J. Beyond the Cell Cycle: Enhancing the Immune Surveillance of Tumors Via CDK4/6 Inhibition. Mol. Cancer Res. 2018, 16, 1454–1457. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Manzano, A.; Dong, W.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Yao, X.; Deshpande, A.; Zaidi, S.; Guglielmi, A.; et al. Integrated mutational landscape analysis of uterine leiomyosarcomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2025182118. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Jang, H.J.; Truong, C.Y.; Lo, E.M.; Holmes, H.M.; Ramos, D.; Ramineni, M.; Lee, J.S.; Wang, D.Y.; Pietropaolo, M.; Ripley, R.T.; et al. Inhibition of Cyclin Dependent Kinase 4/6 Overcomes Primary Resistance to Programmed Cell Death 1 Blockade in Malignant Mesothelioma. Ann. Thorac. Surg. 2022, 114, 1842–1852. [Google Scholar] [CrossRef]
- Lee, E.K.; Esselen, K.M.; Kolin, D.L.; Lee, L.J.; Matulonis, U.A.; Konstantinopoulos, P.A. Combined CDK4/6 and PD-1 Inhibition in Refractory SMARCA4-Deficient Small-Cell Carcinoma of the Ovary, Hypercalcemic Type. JCO Precis Oncol. 2020, 4, 736–742. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef]
- Hutcheson, J.; Bourgo, R.J.; Balaji, U.; Ertel, A.; Witkiewicz, A.K.; Knudsen, E.S. Retinoblastoma protein potentiates the innate immune response in hepatocytes: Significance for hepatocellular carcinoma. Hepatology 2014, 60, 1231–1240. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef]
- Movva, S.; Wen, W.; Chen, W.; Millis, S.Z.; Gatalica, Z.; Reddy, S.; von Mehren, M.; Van Tine, B.A. Multi-platform profiling of over 2000 sarcomas: Identification of biomarkers and novel therapeutic targets. Oncotarget 2015, 6, 12234–12247. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Suzuki, A.; De La Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative Regulation of PKB/Akt-Dependent Cell Survival by the Tumor Suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Rao, U.N.; Jasani, S.; Khanna, V.; Yaw, K.; Surti, U. Loss of DNA copy number of 10q is associated with aggressive behavior of leiomyosarcomas: A comparative genomic hybridization study. Cancer Genet. Cytogenet. 2005, 161, 20–27. [Google Scholar] [CrossRef]
- Zhang, Q.; Ubago, J.; Li, L.; Guo, H.; Liu, Y.; Qiang, W.; Kim, J.J.; Kong, B.; Wei, J.-J. Molecular analyses of 6 different types of uterine smooth muscle tumors: Emphasis in atypical leiomyoma. Cancer 2014, 120, 3165–3177. [Google Scholar] [CrossRef]
- Knight, D.A.; Ngiow, S.F.; Li, M.; Parmenter, T.; Mok, S.; Cass, A.; Haynes, N.M.; Kinross, K.; Yagita, H.; Koya, R.C.; et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J. Clin. Investig. 2013, 123, 1371–1381. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res 2010, 70, 6171–6180. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Vyse, S.; Thway, K.; Huang, P.H.; Jones, R.L. Next-generation sequencing for the management of sarcomas with no known driver mutations. Curr. Opin. Oncol. 2021, 33, 315–322. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Jonsson, P.; Seier, K.; Qin, L.-X.; Chi, P.; Dickson, M.; Gounder, M.; Kelly, C.; Keohan, M.L.; Nacev, B.; et al. Clinical Outcome of Leiomyosarcomas With Somatic Alteration in Homologous Recombination Pathway Genes. JCO Precis. Oncol. 2020, 4, 1350–1360. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Huang, J.-L.; Chang, Y.-T.; Hong, Z.-Y.; Lin, C.-S. Targeting DNA Damage Response and Immune Checkpoint for Anticancer Therapy. Int. J. Mol. Sci. 2022, 23, 3238. [Google Scholar] [CrossRef]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLOS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT Cells Is Characterized by Free Telomeric Circles and Heterogeneous t-Loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef] [PubMed]
- Nabetani, A.; Ishikawa, F. Unusual Telomeric DNAs in Human Telomerase-Negative Immortalized Cells. Mol. Cell. Biol. 2009, 29, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.-Y.; Tsai, J.-H.; Jeng, Y.-M.; Lee, J.-C.; Hsu, H.-H.; Yang, C.-Y. Leiomyosarcoma With Alternative Lengthening of Telomeres Is Associated With Aggressive Histologic Features, Loss of ATRX Expression, and Poor Clinical Outcome. Am. J. Surg. Pathol. 2015, 39, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Onitake, Y.; Hiyama, E.; Kamei, N.; Yamaoka, H.; Sueda, T.; Hiyama, K. Telomere biology in neuroblastoma: Telomere binding proteins and alternative strengthening of telomeres. J. Pediatr. Surg. 2009, 44, 2258–2266. [Google Scholar] [CrossRef]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef]
- Macha, S.J.; Koneru, B.; Burrow, T.A.; Zhu, C.; Savitski, D.; Rahman, R.L.; Ronaghan, C.A.; Nance, J.; McCoy, K.; Eslinger, C.; et al. Alternative Lengthening of Telomeres in Cancer Confers a Vulnerability to Reactivation of p53 Function. Cancer Res 2022, 82, 3345–3358. [Google Scholar] [CrossRef]
- Degtjarik, O.; Golovenko, D.; Diskin-Posner, Y.; Abrahmsén, L.; Rozenberg, H.; Shakked, Z. Structural basis of reactivation of oncogenic p53 mutants by a small molecule: Methylene quinuclidinone (MQ). Nat. Commun. 2021, 12, 7057. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. Correction: APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2019, 10, 439. [Google Scholar] [CrossRef]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin. Cancer Res. 2020, 26, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Bessede, A.; Pulido, M.; Bompas, E.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Bertucci, F.; Toulmonde, M.; Bellera, C.; et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: A phase 2 PEMBROSARC trial cohort. Nat. Med. 2022, 28, 1199–1206. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.-X.; Carvajal, R.D.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Schwartz, G.K.; et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef]
- Kostine, M.; Bruijn, I.H.B.-D.; Cleven, A.H.G.; Vervat, C.; Corver, W.E.; Schilham, M.W.; Van Beelen, E.; Van Boven, H.; Haas, R.L.; Italiano, A.; et al. Increased infiltration of M2-macrophages, T-cells and PD-L1 expression in high grade leiomyosarcomas supports immunotherapeutic strategies. Oncoimmunology 2018, 7, e1386828. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.R.; Moon, Y.J.; Kwon, K.S.; Bae, J.S.; Wagle, S.; Kim, K.M.; Park, H.S.; Lee, H.; Moon, W.S.; Chung, M.J.; et al. Tumor Infiltrating PD1-Positive Lymphocytes and the Expression of PD-L1 Predict Poor Prognosis of Soft Tissue Sarcomas. PLoS ONE 2013, 8, e82870. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Perrot, D.; Leroux, A.; Collin, F.; Le Cesne, A.; Coindre, J.-M.; Blay, J.-Y.; Birnbaum, D.; Mamessier, E. PDL1 expression is a poor-prognosis factor in soft-tissue sarcomas. Oncoimmunology 2017, 6, e1278100. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Datar, I.; Mani, N.; Henick, B.S.; Wurtz, A.; Kaftan, E.; Herbst, R.S.; Rimm, D.L.; Gettinger, S.N.; Politi, K.A.; Schalper, K.A. Measurement of PD-1, TIM-3 and LAG-3 protein in non-small cell lung carcinomas (NSCLCs) with acquired resistance to PD-1 axis blockers. J. Clin. Oncol. 2017, 35, e14611. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Setsu, N.; Gao, D.; Blay, J.-Y.; Thomas, D.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Expression of lymphocyte immunoregulatory biomarkers in bone and soft-tissue sarcomas. Mod. Pathol. 2019, 32, 1772–1785. [Google Scholar] [CrossRef]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) Modulates the Ability of CD4 T-cells to Be Suppressed In Vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef]
- Workman, C.J.; Cauley, L.S.; Kim, I.-J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A.A. Lymphocyte Activation Gene-3 (CD223) Regulates the Size of the Expanding T Cell Population Following Antigen Activation In Vivo. J. Immunol. 2004, 172, 5450–5455. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.N.; Postow, M.; Burton, E.M.; Tetzlaff, M.T.; Ross, M.I.; Torres-Cabala, C.; Glitza, I.C.; Duan, F.; Milton, D.R.; Busam, K.; et al. Neoadjuvant relatlimab and nivolumab in resectable melanoma. Nature 2022, 611, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.; Fang, Z.; Guan, Y.; Xiao, W.; Xu, B.; Zhao, J.; Chen, H.; Zhang, X.; Zeng, M.; Liang, Y.; et al. LAG-3 expression on tumor-infiltrating T cells in soft tissue sarcoma correlates with poor survival. Cancer Biol. Med. 2019, 16, 331–340. [Google Scholar]
- Iwasaki, T.; Kohashi, K.; Toda, Y.; Ishihara, S.; Yamada, Y.; Oda, Y. Association of PD-L1 and IDO1 expression with JAK–STAT pathway activation in soft-tissue leiomyosarcoma. J. Cancer Res. Clin. Oncol. 2021, 147, 1451–1463. [Google Scholar] [CrossRef]
- A Study of BMS-986205 Alone and in Combination with Nivolumab in Chinese Patients with Advanced Malignant Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03792750 (accessed on 15 November 2022).
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Duhen, R.; Ballesteros-Merino, C.; Frye, A.K.; Tran, E.; Rajamanickam, V.; Chang, S.-C.; Koguchi, Y.; Bifulco, C.B.; Bernard, B.; Leidner, R.S.; et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nat. Commun. 2021, 12, 1047. [Google Scholar] [CrossRef]
- Melake, M.; Smith, H.; Mansfield, D.; Davies, E.; Dillon, M.; Wilkins, A.; Patin, E.; Pedersen, M.; Buus, R.; Melcher, A.; et al. OX40 and 4-1BB delineate distinct immune profiles in sarcoma. Oncoimmunology 2022, 11, 2066050. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Cichocki, F.; Zhang, B.; Yingst, A.; Spellman, S.R.; Cooley, S.; Verneris, M.R.; Blazar, B.R.; Miller, J.S. Adaptive NK Cells with Low TIGIT Expression Are Inherently Resistant to Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5696–5706. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Judge, S.J.; Darrow, M.A.; Thorpe, S.W.; Gingrich, A.A.; O’Donnell, E.F.; Bellini, A.R.; Sturgill, I.R.; Vick, L.V.; Dunai, C.; Stoffel, K.M.; et al. Analysis of tumor-infiltrating NK and T cells highlights IL-15 stimulation and TIGIT blockade as a combination immunotherapy strategy for soft tissue sarcomas. J. Immunother. Cancer 2020, 8, e001355. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Dirkx, A.E.M.; Egbrink, M.G.A.O.; Wagstaff, J.; Griffioen, A.W. Monocyte/macrophage infiltration in tumors: Modulators of angiogenesis. J. Leukoc. Biol. 2006, 80, 1183–1196. [Google Scholar] [CrossRef]
- Hamada, I.; Kato, M.; Yamasaki, T.; Iwabuchi, K.; Watanabe, T.; Yamada, T.; Itoyama, S.; Ito, H.; Okada, K. Clinical effects of tumor-associated macrophages and dendritic cells on renal cell carcinoma. Anticancer. Res. 2002, 22, 4281–4284. [Google Scholar]
- Hanada, T.; Nakagawa, M.; Emoto, A.; Nomura, T.; Nasu, N.; Nomura, Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int. J. Urol. 2000, 7, 263–269. [Google Scholar] [CrossRef]
- Koide, N.; Nishio, A.; Sato, T.; Sugiyama, A.; Miyagawa, S.I. Significance of macrophage chemoattractant protein-1 expression and macrophage infiltration in squamous cell carcinoma of the esophagus. Am. J. Gastroenterol. 2004, 99, 1667–1674. [Google Scholar] [CrossRef]
- Leek, R.D.; Landers, R.J.; Harris, A.; Lewis, C.E. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br. J. Cancer 1999, 79, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Espinosa, I.; Vrijaldenhoven, S.; Subramanian, S.; Montgomery, K.D.; Zhu, S.; Marinelli, R.J.; Peterse, J.L.; Poulin, N.; Nielsen, T.O.; et al. Prognostic Significance of Macrophage Infiltration in Leiomyosarcomas. Clin. Cancer Res. 2008, 14, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Yamao, T.; Noguchi, T.; Takeuchi, O.; Nishiyama, U.; Morita, H.; Hagiwara, T.; Akahori, H.; Kato, T.; Inagaki, K.; Okazawa, H.; et al. Negative Regulation of Platelet Clearance and of the Macrophage Phagocytic Response by the Transmembrane Glycoprotein SHPS-1. J. Biol. Chem. 2002, 277, 39833–39839. [Google Scholar] [CrossRef] [PubMed]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.-Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Ozaniak, A.; Smetanova, J.; Bartolini, R.; Rataj, M.; Capkova, L.; Hacek, J.; Fialova, M.; Krupickova, L.; Striz, I.; Lischke, R.; et al. A novel anti-CD47-targeted blockade promotes immune activation in human soft tissue sarcoma but does not potentiate anti-PD-1 blockade. J. Cancer Res. Clin. Oncol. 2022, 1–13. [Google Scholar] [CrossRef]



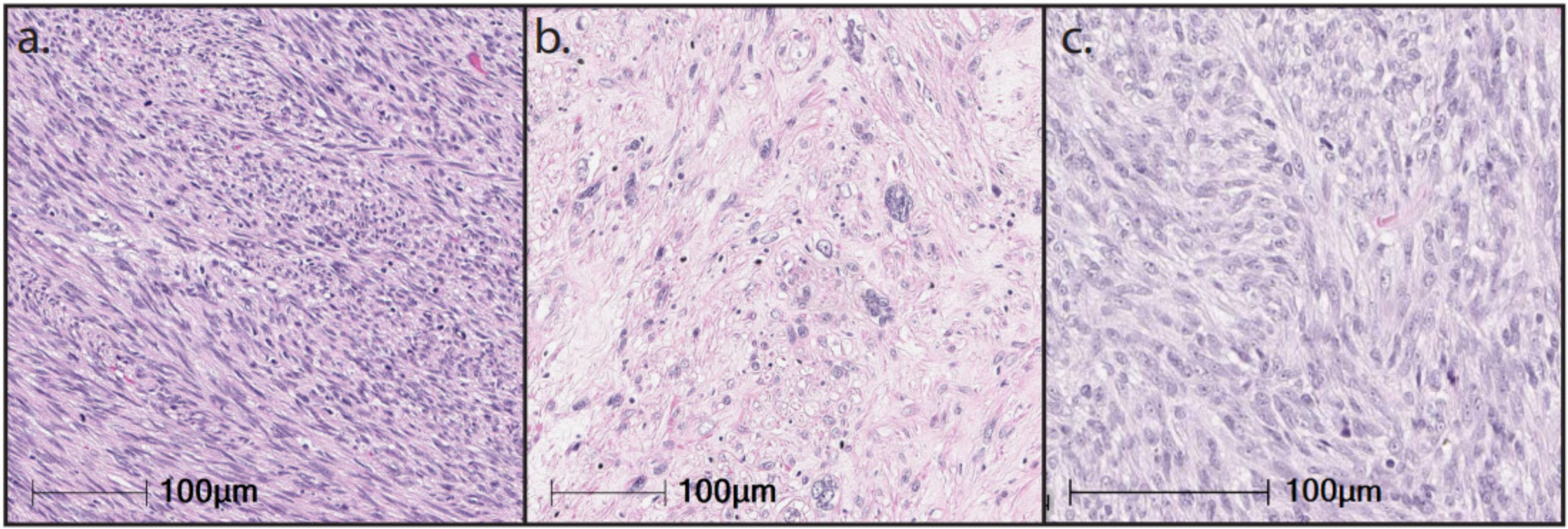{kind=link}
{kind=link}
| Type of LMS | Molecule Tested | Target | Design | ORR | DCR (ORR + SD) | PFS and OS Duration, Median (Range) | Reference |
|---|---|---|---|---|---|---|---|
| uLMS | Nivolumab | Anti-PD1 | Phase II | 0% | 8.3% | PFS: 1.8 months (0.8-unknown) OS: not met; 4 of 12 died during the 100-day study follow-up period because of progression | [48] |
| Pembrolizumab | Anti-PD1 | Retrospective case study | 100% | 100% | NA | [61] | |
| Pembrolizumab | Anti-PD1 | Retrospective case study | 100% | 100% | NA | [62] | |
| ST-LMS | Pembrolizumab | Anti-PD1 | Phase II | 0% | 60% | * PFS: 15 weeks (8–21) * OS: 49 weeks (24–73) | [49] |
| Ipilimumab plus nivolumab | Anti-PD1 and anti-CTLA-4 | Retrospective | 13% | NR | PFS: 4.1 months (3.2–4.5) OS: 12.2 months (6.1–13.7) | [52] | |
| Nivolumab | Anti-PD1 | Retrospective case study | 100% | 100% | NA | [63] | |
| Both | Iplimumab plus nivolumab | Anti-PD1 and anti-CTLA-4 | Phase II | 14.3% | NR | PFS: 4.1 months (2.6–4.7) OS: 14.3 months (9.6-nr) | [51] |
| Pembro plus axitinib | Anti-PD1 and VEGF inhibitor | Phase II | * 25% | 53.1% | * PFS: 4.7 months (3–9.4) * OS: 18.7 months (12-nr) | [59] | |
| Pembrolizumab plus doxorubicin | Anti-PD1 | Phase II | 40% | 100% | * PFS: 5.7 months (4.1–8.9) * OS: 17.0 months (9.9-nr) | [57] | |
| Pembrolizumab plus doxorubicin | Anti-PD1 | Phase I/II | 20% | 50% | * PFS: 8.1 months (7.6–10.8) * OS: 27.6 months (18.7-nr) | [56] | |
| Durvalumab plus tremelimumab | Anti-PDL1 and anti-CTLA-4 | Phase II | [50] | ||||
| Avelumab plus trabectedin | Anti-PDL1 | Phase I/II | * 13% | * 43% | * PFS: 8.3 months Study was halted as it did not meet the primary objective response rate endpoint | [64] | |
| PD-1 inhibitors (pembrolizumab/toripalimab/sintilimab) plus standard chemotherapy | Anti-PD1 | Retrospective cohort study | 17.1% | 73.2% | PFS: 8.8 months (4.57–13.0) OS: not reached; no difference in OS was observed for chemotherapy alone and chemotherapy plus PD-1 | [55] | |
| PD-1/PDL1 inhibitors plus TKI | Anti-PD1/PDL1 and VEGF inhibitor | Retrospective cohort study | 0% | 50% | * PFS: 11.74 months | [60] |
| LMS Subtype | Prognosis | Mutational Burden | Cellular Lineage | Molecular Characteristics | Gene Expression | Immune Microenvironment |
|---|---|---|---|---|---|---|
| Well-differentiated ST-LMS | Better | Low | Vascular and digestive smooth muscle | MYOCD amplification and upregulated muscle-associated transcripts TP53 mutations | Enriched: PDGFRA, LRRC15, IGF1R | Inflammatory NK cell signature |
| Dedifferentiated ST-LMS | Poor | Vascular smooth muscle | DMD deletion Reduced markers of muscle differentiation PTEN loss, overexpression of Akt pathway RB1 and TP53 mutations | Enriched: ACTA1, SYNM, LMO1 | Higher immune infiltration overall, dominated by M2-Macrophages Higher leukocyte count overexpression | |
| uLMS | Poor | Gynecologic (uterine, vaginal, fallopian tube) smooth muscle | DMD expression inhibition PTEN loss, overexpression of Akt pathway TP53 mutations RB1 fusion and loss of function | Enriched: ESR1, PGR, EMX2 | M2 macrophage-dominant |
| Primary Intervention | Secondary Intervention | NCT | Tumor Type | Phase | Status | |
|---|---|---|---|---|---|---|
| Immunotherapy | ||||||
| Durvalumab | Tremelimumab | NCT02815995 | Advanced/metastatic STS | II | Active, not recruiting | |
| Nivolumab | Ipilimumab | NCT02428192 | uLMS | II | Active, not recruiting | |
| Itacitinib | NCT03670069 | Advanced/metastatic STS | I | Recruiting | ||
| LMP1/2 CTLs | NCT01956084 | ST-LMS | I | Active, not recruiting | ||
| Nivolumab | NCT03241745 | Uterine sarcoma | II | Active, not recruiting | ||
| Tabelecleucel | NCT04554914 | ST-LMS | II | Recruiting | ||
| Talimogene laherparepvec | NCT02923778 | ST-LMS | II | Recruiting | ||
| Immunotherapy + anti-angiogenics | ||||||
| Durvalumab | Olaparib, cediranib | NCT03851614 | ST-LMS | II | Active, not recruiting | |
| Nivolumab | Sunitinib, epirubicin | NCT03277924 | Advanced/metastatic STS | I/II | Recruiting | |
| PD-1 inhibition | BA3011 | NCT03425279 | ST-LMS | I/II | Recruiting | |
| Pembrolizumab | Eribulin | NCT03899805 | ST-LMS | II | Active, not recruiting | |
| Immunotherapy + chemotherapy | ||||||
| APX005M | Doxorubicin | NCT03719430 | Advanced/metastatic STS | II | Recruiting | |
| Cabozantinib | Temozolomide | NCT04200443 | ST-LMS, uLMS | II | Recruiting | |
| Nivolumab | Rucaparib | NCT04624178 | ST-LMS | II | Active, not recruiting | |
| Nivolumab | BO-112 | NCT04420975 | ST-LMS | I | Active, not recruiting | |
| OR2805 | Cemiplimab, docetaxel | NCT05094804 | Advanced/metastatic STS | I/II | Recruiting | |
| Pembrolizumab | Gemcitabine | NCT03123276 | ST-LMS | I/II | Active, not recruiting | |
| Pembrolizumab | Cyclophosphamide | NCT02406781 | ST-LMS | II | Active, not recruiting | |
| Ribociclib | Doxorubicin | NCT03009201 | Advanced/metastatic STS | I | Active, not recruiting | |
| TTI-621 | Doxorubicin | NCT04996004 | ST-LMS | II | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cope, B.M.; Traweek, R.S.; Lazcano, R.; Keung, E.Z.; Lazar, A.J.; Roland, C.L.; Nassif, E.F. Targeting the Molecular and Immunologic Features of Leiomyosarcoma. Cancers 2023, 15, 2099. https://doi.org/10.3390/cancers15072099
Cope BM, Traweek RS, Lazcano R, Keung EZ, Lazar AJ, Roland CL, Nassif EF. Targeting the Molecular and Immunologic Features of Leiomyosarcoma. Cancers. 2023; 15(7):2099. https://doi.org/10.3390/cancers15072099
Chicago/Turabian StyleCope, Brandon M., Raymond S. Traweek, Rossana Lazcano, Emily Z. Keung, Alexander J. Lazar, Christina L. Roland, and Elise F. Nassif. 2023. "Targeting the Molecular and Immunologic Features of Leiomyosarcoma" Cancers 15, no. 7: 2099. https://doi.org/10.3390/cancers15072099
APA StyleCope, B. M., Traweek, R. S., Lazcano, R., Keung, E. Z., Lazar, A. J., Roland, C. L., & Nassif, E. F. (2023). Targeting the Molecular and Immunologic Features of Leiomyosarcoma. Cancers, 15(7), 2099. https://doi.org/10.3390/cancers15072099