

Moyamoya Vasculopathy in Neurofibromatosis Type 1 Pediatric Patients: The Role of Rare Variants of RNF213
 , ,
, ,  , , , , , , , , and add
Show full author list
, , , , , , , , and add
Show full author list
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Controls
2.2. DNA Extraction
2.3. Next-Generation Sequencing (NGS)
2.4. Multiplex Ligation-Dependent Probe Amplification (MLPA)
2.5. RNA Extraction, RT-PCR and Sequencing of NF1 Gene
2.6. Statistical Analysis
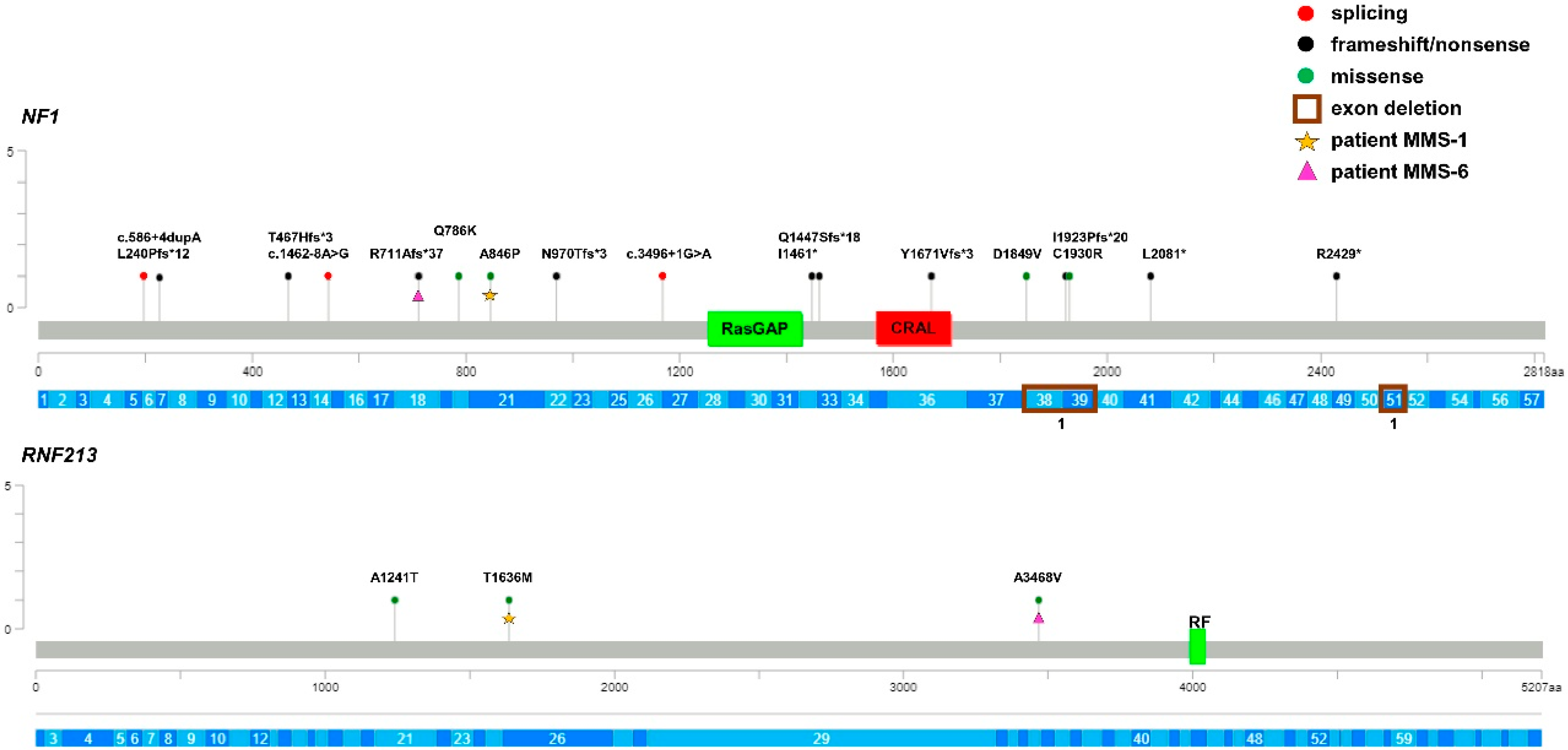
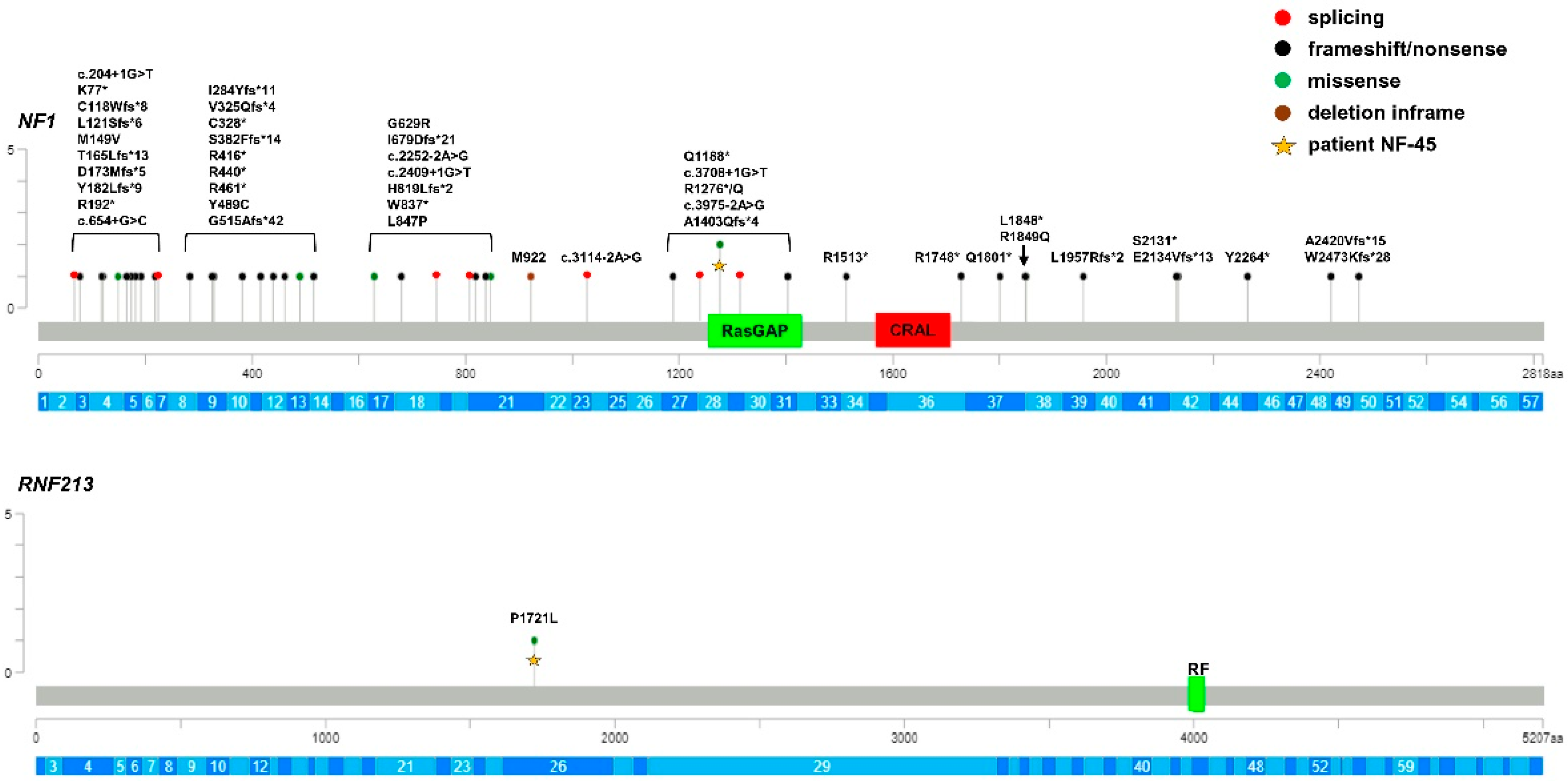
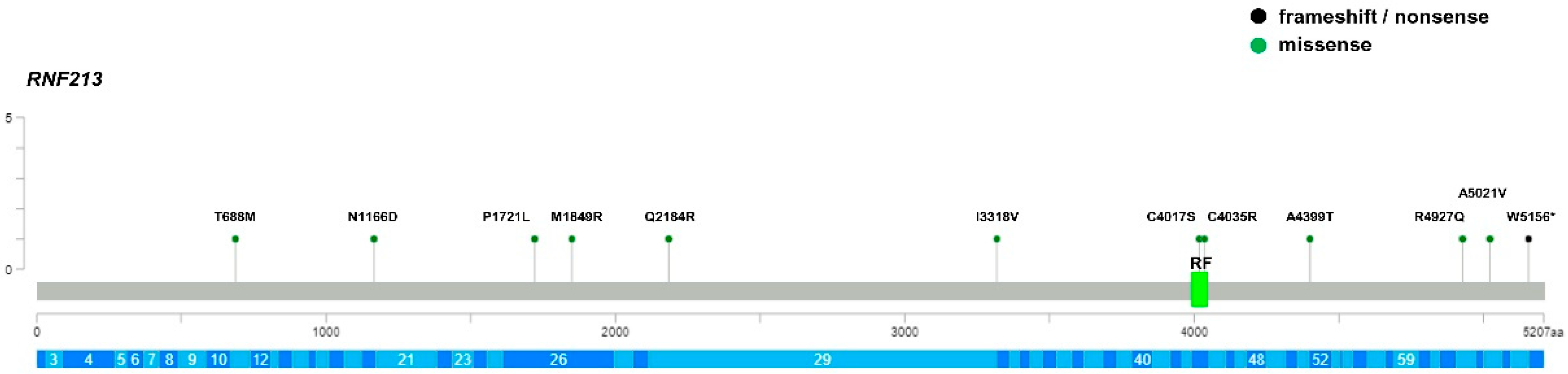
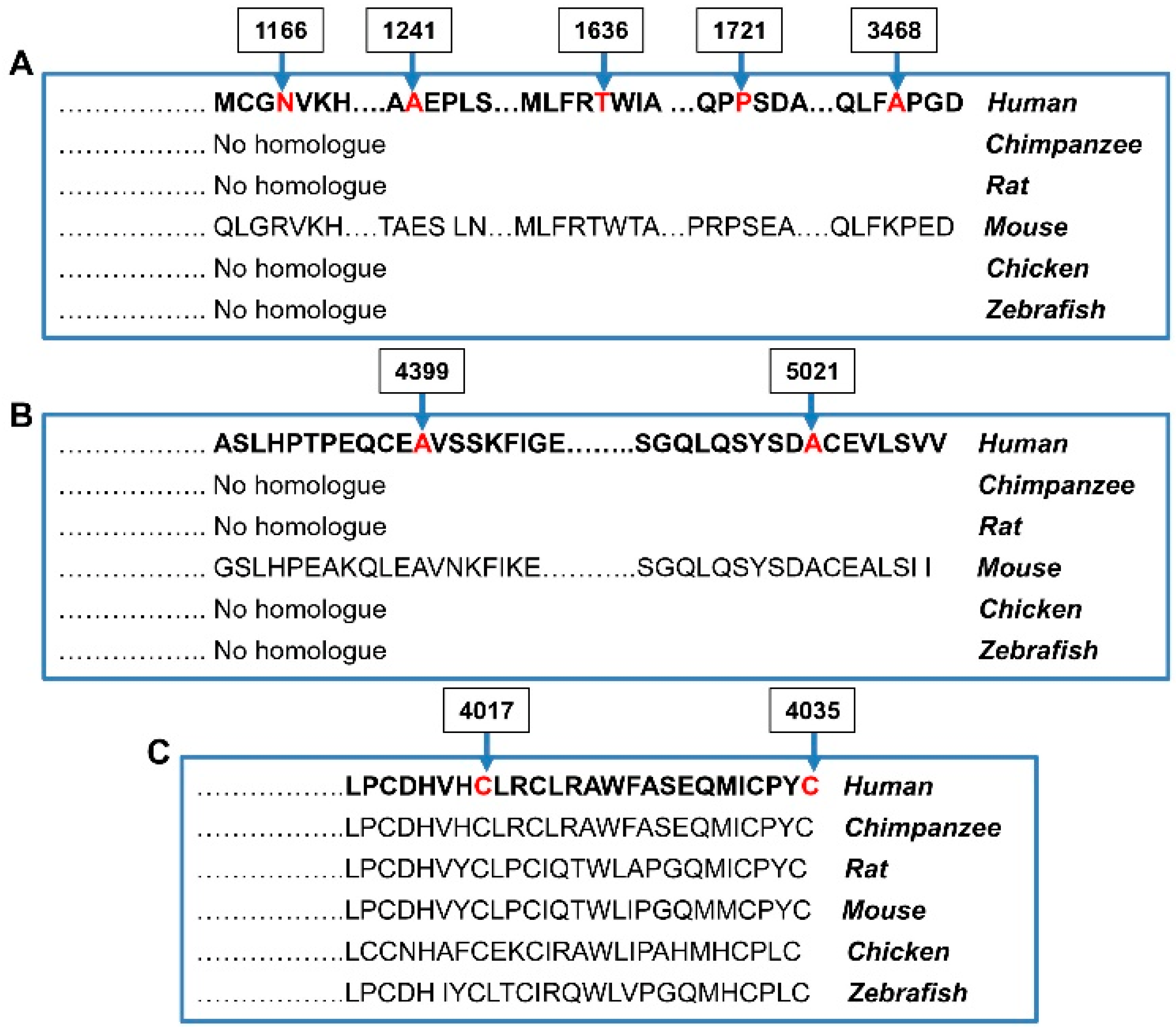
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cairns, A.G.; North, K.N. Cerebrovascular dysplasia in neurofibromatosis type 1. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1165–1170. [Google Scholar] [CrossRef]
- Rea, D.; Brandsema, J.F.; Armstrong, D.; Parkin, P.C.; deVeber, G.; MacGregor, D.; Logan, W.J.; Askalan, R. Cerebral arteriopathy in children with neurofibromatosis type 1. Pediatrics 2009, 124, 476–483. [Google Scholar]
- Ghosh, P.S.; Rothner, A.D.; Emch, T.M.; Friedman, N.R.; Moodley, M. Cerebral vasculopathy in children with neurofibromatosis type 1. J. Child. Neurol. 2013, 28, 95–101. [Google Scholar] [PubMed]
- Duat-Rodríguez, A.; Carceller Lechon, F.; Lopez Pino, M.A.; Rodríguez Fernandez, C.; Gonzalez-Gutierrez-Solana, L. Neurofibromatosis type 1 associated with moyamoya syndrome in children. Pediatr. Neurol. 2014, 50, 96–98. [Google Scholar] [CrossRef]
- Suzuki, J.; Takaku, A. Cerebrovascular “Moyamoya” disease: Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 1969, 20, 288–289. [Google Scholar] [PubMed]
- Kuroda, S.; Houkin, K. Moyamoya disease: Current concepts and future perspectives. Lancet Neurol. 2008, 7, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.M.; Smith, E.R. Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [PubMed]
- Nakamura, H.; Sato, K.; Yoshimura, S.; Hayashi, Y.; Izumo, T.; Tokunaga, Y. Moyamoya disease associated with Graves’ disease and Down syndrome: A case report and literature Review. J. Stroke Cerebrovasc Dis. 2021, 30, 105414. [Google Scholar] [CrossRef]
- Newman, S.; Boulter, J.H.; Malcolm, J.G.; Pradilla, I.; Pradilla, G. Outcomes in patients with Moyamoya syndrome and Sickle Cell disease: A systematic review. World Neurosurg. 2020, 135, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhang, H.; Liu, S.; Liu, D.; Peng, P.; Hao, F.; Yuan, F.; Liu, Y.; Sheng, F.; Zhang, L.; et al. Long-term outcomes of moyamoya disease versus atherosclerosis-associated moyamoya vasculopathy using high-resolution MR vessel wall imaging. J. Neurol. Neurosurg. Psychiatry 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Mertens, R.; Graupera, M.; Gerhardt, H.; Bersano, A.; Tournier-Lasserve, E.; Mensah, M.A.; Mundlos, S.; Vajkoczy, P. The genetic basis of Moyamoya Disease. Transl. Stroke Res. 2022, 13, 25–45. [Google Scholar]
- Guey, S.; Tournier-Lasserve, E.; Hervé, D.; Kossorotoff, M. Moyamoya disease and syndromes: From genetics to clinical management. Appl. Clin. Genet. 2015, 16, 49–68. [Google Scholar]
- Hirano, Y.; Miyawaki, S.; Imai, H.; Hongo, H.; Teranishi, Y.; Dofuku, S.; Ishigami, D.; Ohara, K.; Koizumi, S.; Ono, H.; et al. Differences in clinical features among different onset patterns in moyamoya disease. J. Clin. Med. 2021, 10, 2815. [Google Scholar]
- Scala, M.; Fiaschi, P.; Capra, V.; Garrè, M.L.; Tortora, D.; Ravegnani, M.; Pavanello, M. When and why is surgical revascularization indicated for the treatment of moyamoya syndrome in patients with RASopathies? A systematic review of the literature and a single institute experience. Childs Nerv. Syst. 2018, 34, 1311–1323. [Google Scholar]
- Scala, M.; Fiaschi, P.; Cama, A.; Consales, A.; Piatelli, G.; Giannelli, F.; Barra, S.; Satragno, C.; Pacetti, M.; Secci, F.; et al. Radiation-Induced Moyamoya Syndrome in Children with Brain Tumors: Case Series and Literature Review. World Neurosurg. 2020, 135, 118–129. [Google Scholar] [PubMed]
- Fiaschi, P.; Scala, M.; Piatelli, G.; Tortora, D.; Secci, F.; Cama, A.; Pavanello, M. Limits and pitfalls of indirect revascularization in moyamoya disease and syndrome. Neurosurg. Rev. 2021, 44, 1877–1887. [Google Scholar] [PubMed]
- Tortora, D.; Scavetta, C.; Rebella, G.; Bertamino, M.; Scala, M.; Giacomini, T.; Morana, G.; Pavanello, M.; Rossi, A.; Severino, M. Spatial coefficient of variation applied to arterial spin labeling MRI may contribute to predict surgical revascularization outcomes in pediatric moyamoya vasculopathy. Neuroradiology 2020, 62, 1003–1015. [Google Scholar] [PubMed]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Yu, A.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar]
- Miyatake, S.; Miyake, N.; Touho, H.; Nishimura-Tadaki, A.; Kondo, Y.; Okada, I.; Tsurusaki, Y.; Doi, H.; Sakai, H.; Saitsu, H.; et al. Homozygous c.14576G>A variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology 2012, 78, 803–810. [Google Scholar]
- Koizumi, A.; Kobayashi, H.; Hitomi, T.; Harada, K.H.; Habu, T.; Youssefian, S. A new horizon of moyamoya disease and associated health risks explored through RNF213. Environ. Health Prev. Med. 2016, 21, 55–70. [Google Scholar] [PubMed]
- Kim, D.-K.; Jang, S.H.; Chang, S.A.; Park, T.K. Systemic vasculopathy associated with an RNF213 p.Arg4810Lys variant in moyamoya disease: A review. Precis. Future Med. 2022, 6, 155–160. [Google Scholar] [CrossRef]
- Romanisio, G.; Chelleri, C.; Scala, M.; Piccolo, G.; Carlini, B.; Gatti, L.; Capra, V.; Zara, F.; Bersano, A.; Pavanello, M.; et al. RNF213 variant in a patient with Legius syndrome associated with moyamoya syndrome. Mol. Genet. Genom. Med. 2021, 9, e1669. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef]
- Mineharu, Y.; Miyamoto, S. RNF213 and GUCY1A3 in Moyamoya Disease: Key Regulators of Metabolism, Inflammation, and Vascular Stability. Front Neurol. 2021, 12, 687088. [Google Scholar] [PubMed]
- Keylock, A.; Hong, Y.; Saunders, D.; Omoyinmi, E.; Mulhern, C.; Roebuck, D.; Brogan, P.; Ganesan, V.; Eleftheriou, D. Moyamoya-like cerebrovascular disease in a child with a novel mutation in myosin heavy chain 11. Neurology 2018, 90, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yang, W.Z.; Zhang, H.T.; Ye, T.; Duan, L. Clinical characteristics and long-term outcomes of moyamoya syndrome associated with neurofibromatosis type 1. J. Clin. Neurosci. 2015, 22, 286–290. [Google Scholar]
- Santoro, C.; Di Rocco, F.; Kossorotoff, M.; Zerah, M.; Boddaert, N.; Calmon, R.; Vidaud, D.; Cirillo, M.; Cinalli, G.; Mirone, G.; et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am. J. Med. Genet. Part A 2017, 173, 1521–1530. [Google Scholar] [CrossRef]
- Xu, J.; Ismat, F.A.; Wang, T.; Yang, J.; Epstein, J.A. NF1 regulates a Ras-dependent vascular smooth muscle proliferative injury response. Circulation 2007, 116, 2148–2156. [Google Scholar] [PubMed]
- Bessler, W.K.; Hudson, F.Z.; Zhang, H.; Harris, V.; Wang, Y.; Mund, J.A.; Downing, B.; Ingram, D.A., Jr.; Case, J.; Fulton, D.J.; et al. Neurofibromin is a novel regulator of Ras-induced reactive oxygen species production in mice and humans. Free Radic. Biol. Med. 2016, 97, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Norton, K.K.; Xu, J.; Gutmann, D.H. Expression of the neurofibromatosis I gene product, neurofibromin, in blood vessel endothelial cells and smooth muscle. Neurobiol. Dis. 1995, 2, 13–21. [Google Scholar] [PubMed]
- Ozerdem, U. Targeting neovascular pericytes in neurofibromatosis type 1. Angiogenesis 2004, 7, 307–311. [Google Scholar]
- Munchhof, A.M.; Li, F.; White, H.A.; Mead, L.E.; Krier, T.R.; Fenoglio, A.; Li, X.; Yuan, J.; Yang, F.C.; Ingram, D.A. Neurofibroma-associated growth factors activate a distinct signaling network to alter the function of neurofibromin-deficient endothelial cells. Hum. Mol. Genet. 2006, 15, 1858–1869. [Google Scholar] [PubMed]
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Scala, M.; Schiavetti, I.; Madia, F.; Chelleri, C.; Piccolo, G.; Accogli, A.; Riva, A.; Salpietro, V.; Bocciardi, R.; Morcaldi, G.; et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study. Cancers 2021, 13, 1879. [Google Scholar] [PubMed]
- Guey, S.; Kraemer, M.; Hervé, D.; Ludwig, T.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.-C.; Choi, S.; Broseus, L.; Callebaut, I.; et al. RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar]
- Liao, X.; Deng, J.; Dai, W.; Zhang, T.; Yan, J. Rare variants of RNF213 and moyamoya/non-moyamoya intracranial artery stenosis/occlusion disease risk: A meta-analysis and systematic review. Environ. Health Prev. Med. 2017, 22, 75. [Google Scholar]
- Li, F.; Munchhof, A.M.; White, H.A.; Mead, L.E.; Krier, T.R.; Fenoglio, A.; Chen, S.; Wu, X.; Cai, S.; Yang, F.-C.; et al. Neurofibromin is a novel regulator of RAS-induced signals in primary vascular smooth muscle cells. Hum. Mol. Genet. 2006, 15, 1921–1930. [Google Scholar] [PubMed]
- Scholz, B.; Korn, C.; Wojtarowicz, J.; Boutros, M.; Niehrs, C.; Hellmut, A. Endothelial RSPO3 controls vascular stability and pruning through non-canonical WNT/Ca2+/NFAT signaling. Developmental. Cell 2016, 36, 79–93. [Google Scholar]
- Phi, J.H.; Choi, J.W.; Seon, M.-W.; Kim, T. Association between moyamoya syndrome and the RNF213 c.14576G>A variant in patients with neurofibromatosis Type 1. J. Neurosurg. Pediatr. 2016, 17, 717–722. [Google Scholar]
- Santoro, C.; Giugliano, T.; Kraemer, M.; Torella, A.; Schwitalla, J.-C.; Cirillo, M.; Melis, D.; Berlit, P.; Nigro, V.; Perrotta, S.; et al. Whole exome sequencing identifies MRVI1 as a susceptibility gene for moyamoya syndrome in neurofibromatosis type 1. PLoS ONE 2018, 13, e0200446. [Google Scholar]
- Bajaj, A.; Li, Q.; Zheng, Q.; Pumiglia, K. Loss of NF1 expression in human endothelial cells promotes autonomous proliferation and altered vascular morphogenesis. PLoS ONE 2012, 7, e49222. [Google Scholar]
- Wang, D.; Uhrin, P.; Mocan, A.; Waltenberger, B.; Breuss, J.M.; Tewari, D.; Mihaly-Bison, J.; Huminiecki, Ł.; Starzyński, R.R.; Tzvetkov, N.T.; et al. Vascular smooth muscle cell proliferation as a therapeutic target. Part 1: Molecular targets and pathways. Biotechnol. Adv. 2018, 36, 1586–1607. [Google Scholar] [PubMed]
- Chen, J.; Zhou, Y.; Liu, S.; Li, C.J. Biomechanical signal communication in vascular smooth muscle cells. Cell Commun. Signal. 2020, 14, 357–376. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall (n = 92) | MMS (n = 20) | MMD (n = 25) | NF1 (n = 47) | p-Value | p-Value Pairwise Comparisons | ||
|---|---|---|---|---|---|---|---|
| Sex | Females | 48 (52.2%) | 12 (60.0%) | 15 (60.0%) | 21 (44.7%) | 0.34 ^ | |
| Males | 44 (47.8%) | 8 (40.0%) | 10 (40.0%) | 26 (55.3%) | |||
| Age | years | 9.3 ± 5.02 | 11.2 ± 4.84 | 7.4 ± 3.33 | 9.5 (±5.55) | 0.036 ^^* | MMD/NF1: 0.29 MMD/MMS: 0.032 * NF1/MMS: 0.56 |
| Diagnosis age | years | 5.5 ± 2.10 | 6.7 ± 1.93 | 4.6 ± 1.04 | 5.4 (±2.38) | 0.003 ^^* | MMD/NF1: 0.18 MMD/MMS: 0.002 * NF1/MMS: 0.10 |
| Familial cases | 6 | 2 | - | 4 |
| Patient | Sex | Genotype (cDNA and Protein Change) | ACMG/ClinVar/LOVD Prediction | gnomAD/ DGV Frequency | Biogear Frequency |
|---|---|---|---|---|---|
| MMS-1 | F | NF1 c.2536G>C (p.Ala846Pro) RNF213 c.4907C>T (p.Thr1636Met) | P/P/P VUS/-/- | - 2.6 × 10−5 | - - |
| MMS-2 | M | NF1 c.7285C>T (p.Arg2429 *) RNF213 WT | P/P/P | - | - |
| MMS-3 | F | NF1 c.4381delA (p.Ile1461 *) RNF213 WT | LP/-/- | - | - |
| MMS-4 | F | NF1 c.5483A>T (p.Asp1849Val) RNF213 WT | P/C/VUS | - | - |
| MMS-5 | F | NF1 c.4276_4282delinsAG (p.Gln1447Serfs * 18) RNF213 WT | LP/-/- | - | - |
| MMS-6 | F | NF1 c.2131delC (p.Arg711Alafs * 37) RNF213 c.10403C>T (p.Ala3468Val) | P/P/- VUS/-/- | - 1.3 × 10−4 | - 7 × 10−4 |
| MMS7 | F | NF1 c.3496+1G>A RNF213WT | P/P/- | - | - |
| MMS-8 | F | NF1 c.2909delA (p.Asn970Thrfs * 3) RNF213 WT | LP/-/- | - | - |
| MMS-9 | M | NF1 c.1399dupA (p.Thr467Hisfs * 3) RNF213 WT | P/P/- | - | - |
| MMS-10 | M | NF1 c.5725T>C (p.Cys1930Arg) RNF213 WT | P/C/P | - | - |
| MMS-11 | F | NF1 c.1642-8A>G RNF213 WT | P/P/- | - | - |
| MMS-12 | M | NF1c.7553_7575del (exon 51 del) RNF213 WT | LP/-/- | - | - |
| MMS-13 | F | NF1 c5547_5943del (exons 38–39 del) RNF213 WT | P/P/- - | Very rare in DGV - | - |
| MMS-14 | F | NF1 WT ^ RNF213 c.3721G>A (p.Ala1241Thr) | - VUS/-/- | - 1.9 × 10−5 | - 3 × 10−4 |
| MMS-15 | M | NF1 c.6179T>G (p.Leu2081 *) RNF213 WT | LP/-/- | - | - |
| MMS-16 | F | NF1 c.2356C>A (p.Gln786Lys) RNF213 WT | LP/VUS/- | - | - |
| MMS-17 | M | NF1 c.610dupC (p.Leu204Profs * 12) RNF213 WT | P/LP/- | - | - |
| MMS-18 ‡ | M | NF1 c.586+4dupA RNF213 WT | VUS/VUS/- | - | - |
| MMS-19 | F | NF1 c.5010dupG (p.Tyr1671Valfs * 3) RNF213 WT | LP/-/- | - | - |
| MMS-20 ‡ | M | NF1 c.5767dupC (p.Ile1923Profs * 20) RNF213 WT | P/-/- | - | - |
| Patient | Sex | Genotype (cDNA and Protein Change) | ACMG/ClinVar/LOVD Prediction | gnomAD Frequency | Biogear Frequency |
|---|---|---|---|---|---|
| NF-1 | M | NF1 c.1885G>A (p.Gly629Arg) RNF213 WT | P/P/P | - | - |
| NF-2 | M | NF1 c.5242C>T (p.Arg1748 *) RNF213 WT | P/P/P | - | - |
| NF-3 | M | NF1 c.2970_2972del (p.Met992del) RNF213 WT | P/P/P | 8.2 × 10−6 | - |
| NF-4 | M | NF1 c.545del (p.Tyr182Leufs * 9) RNF213 WT | LP/-/- | - | - |
| NF-5 | M | NF1 c.493del (p.Thr165Leufs * 13) RNF213 WT | LP/-/- | - | - |
| NF-6 | M | NF1 c.1544_1545del (p.Gly515Alafs * 42) RNF213 WT | LP/-/- | - | - |
| NF-7 | M | NF1 c.973_974del (p.Val325Glnfs * 4) RNF213 WT | LP/-/- | - | - |
| NF-8 | F | NF1 c.360dup (p.Leu121Serfs * 6) RNF213 WT | LP/-/- | - | - |
| NF-9 | F | NF1 c.1144dup (p.Ser382Phefs * 14) RNF213 WT | LP/-/- | - | - |
| NF-10 | F | NF1 c.5870del (p.Leu1957Argfs * 2) RNF213 WT | LP/-/- | - | - |
| NF-11 | F | NF1 c.6391_6394del (p.Ser2131 *) RNF213 WT | LP/-/- | - | - |
| NF-12 ‡ | M | NF1 c.850del (p.Ile284Tyrfs * 11) RNF213 WT | LP/-/- | - | - |
| NF-13 ‡ | M | NF1 c.656del (p.Ala219Aspfs * 6) RNF213 WT | LP/-/- | - | - |
| NF-14 | M | NF1 c.5401C>T (p.Gln1801 *) RNF213 WT | P/P/P | - | - |
| NF-15 | F | NF1 c.229A>T (p.Lys77 *) RNF213 WT | LP/-/- | - | - |
| NF-16 | M | NF1 c.3562C>T (p.Gln1188 *) RNF213 WT | P/P/P | - | - |
| NF-17 | F | NF1 c.2033dup (p.Ile679Aspfs * 21) RNF213 WT | P/P/P | - | - |
| NF-18 | M | NF1 c.3114-2A>G RNF213 WT | P/LP/P | - | - |
| NF-19 | M | NF1 c.1381C>T (p.Arg461 *) RNF213 WT | P/P/P | - | - |
| NF-20 ‡ | F | NF1 c.354_356delinsG (p.Cys118Trpfs * 8) RNF213 WT | LP/-/- | - | - |
| NF-21 | M | NF1 c.3975-2A>G RNF213 WT | P/P/P | - | - |
| NF-22 | F | NF1 c.5546G>A (p.Arg1849Gln) RNF213 WT | P/P/P | - | - |
| NF-23 | F | NF1 c.4537C>T (p.Arg1513 *) RNF213 WT | P/C/P | 6 × 10−6 | - |
| NF-24 | F | NF1 c.574C>T (p.Arg192 *) RNF213 WT | P/C/P | 1 × 10−5 | - |
| NF-25 | M | NF1 c.7259-1_7259del (p.Ala2420Valfs * 15) RNF213 WT | LP/-/- | - | - |
| NF-26 | F | NF1 c.2511G>A (p.Trp837 *) RNF213 WT | P/P/P | - | - |
| NF-27 | F | NF1 c.3826C>T (p.Arg1276 *) RNF213 WT | P/P/P | - | - |
| NF-28 | M | NF1 c.2409+1G>T RNF213 WT | P/P/P | - | - |
| NF-29 | M | NF1 c.6401_6402del (p.Glu2134Valfs * 13) RNF213 WT | LP/-/- | - | - |
| NF-30 | M | NF1 c.1246C>T (p.Arg416 *) RNF213 WT | P/P/P | 8 × 10−6 | - |
| NF-31 | M | NF1 c.2540T>C (p.Leu847Pro) RNF213 WT | P/C/P | - | - |
| NF-32 ‡ | F | NF1 c.3708+1G>T RNF213 WT | P/LP/P | - | - |
| NF-33 | F | NF1 c.4206del (p.Ala1403Glnfs * 4) RNF213 WT | LP/-/- | - | - |
| NF-34 | M | NF1 c.983_984del (p.Cys328 *) RNF213 WT | P/P/P | - | - |
| NF-35 | M | NF1 c.516delT (p.Asp173Metfs * 5) RNF213 WT | LP/-/- | - | - |
| NF-36 | M | NF1 c.2540T>C (p.Leu847Pro) RNF213 WT | P/C/P | - | - |
| NF-37 | F | NF1 c.2456del (p.His819Leufs * 2) RNF213 WT | LP/-/- | - | |
| NF-38 | F | NF1 c.6792C>A (p.Tyr2264 *) RNF213 WT | P/P/P | - | - |
| NF-39 | M | NF1 c.1318C>T (p.Arg440 *) RNF213 WT | P/P/P | - | 1 × 10−4 |
| NF-40 | M | NF1 c.7417_7425delinsAAGGT(Trp2473Lysfs * 28) RNF213 WT | LP/-/- | - | - |
| NF-41 | F | NF1 c.2252-2A>G RNF213 WT | P/C/P | - | - |
| NF-42 | F | NF1 c.204+1G>T RNF213 WT | P/P/P | 6 × 10−6 | - |
| NF-43 | F | NF1 c.3445A>G (p.Met1149Val) RNF213 WT | P/C/P | 6 × 10−6 | - |
| NF-44 | F | NF1 c.1466A>G (p.Tyr489Cys) RNF213 WT | P/P/P | 6 × 10−6 | - |
| NF-45 | M | NF1 c.3827G>A (p.Arg1276Gln) RNF213 c.5162C>T (p.Pro1721Leu) | P/P/P VUS/-/- | 1 × 10−5 1 × 10−3 | - 2.7 × 10−3 |
| NF-46 | F | NF1 c.5543T>G (p.Leu1848 *) RNF213 WT | P/P/P | - | - |
| NF-47 | M | NF1 c.654+1G>C RNF213 WT | LP/-/P | - | - |
| Patient | Sex | RNF213 Genotype | Protein Domain | ACMG/ClinVar/LOVD Prediction | gnomAD Frequency | Biogear Frequency |
|---|---|---|---|---|---|---|
| MMD-1 | F | c.5546T>G (pMet1849Arg) | N-term | VUS/-/- | - | - |
| MMD-2 † | F | c.15062C>T (p.Ala5021Val) | C-term | VUS/B/- | 1.5 × 10−4 | - |
| MMD-3 ‡ | M | c.10443G>A (p.Ala3481=) | B/-/- | - | 7 × 10−4 | |
| MMD-4 | F | c.4146T>G (p.Thr1382=) | B/B/- | 8 × 10−3 | 5 × 10−3 | |
| MMD-5 | M | WT | - | |||
| MMD-6 | M | c.12050G>C (p.Cys4017Ser) | RF | VUS/-/- | - | - |
| MMD-7 | F | c.5162C>T (p.Pro1721Leu) | N-term | VUS/VUS/- | 1 × 10−3 | 1 × 10−3 |
| MMD-8 | F | WT | - | |||
| MMD-9 | F | WT | - | |||
| MMD-10 | M | WT | - | |||
| MMD-11 | M | c.12103T>C (p.Cys4035Arg) | RF | VUS/-/- | - | - |
| MMD-12 | M | WT | - | |||
| MMD-13 | F | WT | - | |||
| MMD-14 | F | c.9952A>G (p.Ile3318Val) | N-term | B/B/- | 1 × 10−3 | 1 × 10−2 |
| MMD-15 | F | c.2063C>T (p.Thr688Met) | N-term | B/B/- | 1 × 10−3 | 3 × 10−4 |
| MMD-16 | F | c.3496A>G (p.Asn1166Asp) | N-term | VUS/-/- | - | - |
| MMD-17 | M | WT | - | |||
| MMD-18 | F | WT | - | |||
| MMD-19 | M | c.6042C>T (p.Asn2014=) c.15467G>A (p.Trp5156 *) | - C-term | B/B/- LP/-/- | 1 × 10−3 - | 3 × 10−3 - |
| MMD-20 | F | c.14780G>A (p.Arg4927Gln) | C-term | VUS/-/- | 5 × 10−5 | - |
| MMD-21 | F | WT | - | |||
| MMD-22 | F | WT | - | |||
| MMD-23 | M | WT | - | |||
| MMD-24 | F | c.6551A>G (p.Gln2184Arg) | N-term | B/LB/- | 2 × 10−3 | - |
| MMD-25 | M | c.13195G>A (p.Ala4399Thr) | C-term | B/P/- | 8 × 10−3 | 1 × 10−3 |
| Carrier Frequency | IRR (95%CI); p-Value | |||||
|---|---|---|---|---|---|---|
| Domain | MMS (n = 20) | MMD (n = 25) | Biogear (n = 2504) | gnomAD (n = 141,456) | MMS | MMD |
| N-term | 3 (15%) | 3 (12%) | 282 (11%) | NA | 1.33 (0.273–3.932); p = 0.58 * NA ** | 1.07 (0.22–3.15); p = 0.85 * NA ** |
| RING finger | 0 | 2 (8%) | 8 (0.3%) | 227 (0.1%) | 0.00 (0.00–73.05); p = 0.80 * 0.00 (0.00–115.88); p = 0.86 ** | 25.04(2.59–125.47); p = 0.004 * 49.85 (6.00–182.17); p < 0.001 ** |
| C-term | 0 | 4 (16%) | 61 (2.3%) | 1373 (0.9%) | 0.00 (0.00–7.80); p = 0.49 * 0.00 (0.00–19.03); p = 0.66 ** | 6.57(1.73–17.69); p = 0.004 * 16.48 (4.49–42.30); p < 0.001 ** |
| RING finger + C-term | 0 | 6 (24%) | 69 (2.7%) | 1600 (1%) | 0.00 (0.00–6.88); p = 0.46 * 0.00 (0.00–19.03); p = 0.66 ** | 8.71(3.09–19.94); p < 0.001 * 21.22 (7.78–46.29); p < 0.001 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ognibene, M.; Scala, M.; Iacomino, M.; Schiavetti, I.; Madia, F.; Traverso, M.; Guerrisi, S.; Di Duca, M.; Caroli, F.; Baldassari, S.; et al. Moyamoya Vasculopathy in Neurofibromatosis Type 1 Pediatric Patients: The Role of Rare Variants of RNF213. Cancers 2023, 15, 1916. https://doi.org/10.3390/cancers15061916
Ognibene M, Scala M, Iacomino M, Schiavetti I, Madia F, Traverso M, Guerrisi S, Di Duca M, Caroli F, Baldassari S, et al. Moyamoya Vasculopathy in Neurofibromatosis Type 1 Pediatric Patients: The Role of Rare Variants of RNF213. Cancers. 2023; 15(6):1916. https://doi.org/10.3390/cancers15061916
Chicago/Turabian StyleOgnibene, Marzia, Marcello Scala, Michele Iacomino, Irene Schiavetti, Francesca Madia, Monica Traverso, Sara Guerrisi, Marco Di Duca, Francesco Caroli, Simona Baldassari, and et al. 2023. "Moyamoya Vasculopathy in Neurofibromatosis Type 1 Pediatric Patients: The Role of Rare Variants of RNF213" Cancers 15, no. 6: 1916. https://doi.org/10.3390/cancers15061916
APA StyleOgnibene, M., Scala, M., Iacomino, M., Schiavetti, I., Madia, F., Traverso, M., Guerrisi, S., Di Duca, M., Caroli, F., Baldassari, S., Tappino, B., Romano, F., Uva, P., Vozzi, D., Chelleri, C., Piatelli, G., Diana, M. C., Zara, F., Capra, V., ... De Marco, P. (2023). Moyamoya Vasculopathy in Neurofibromatosis Type 1 Pediatric Patients: The Role of Rare Variants of RNF213. Cancers, 15(6), 1916. https://doi.org/10.3390/cancers15061916