
Inhibition of IDH3α Enhanced the Efficacy of Chemoimmunotherapy by Regulating Acidic Tumor Microenvironments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis
2.2. Cell Line Culture and Drug Treatment
2.3. Construction of Stable Cell Lines
2.4. CCK8 Assay and EdU Assay
2.5. Colony Formation Assays
2.6. Cell Invasion Assay and Wound Healing Assay
2.7. Flow Cytometry
2.8. RNA-seq
2.9. qPCR
2.10. Western Blotting
2.11. Quantification of Glucose Uptake, L-lactate, NAD+/NADH, and ROS
2.12. Chemosensitivity Assay
2.13. In Vivo Experiments
2.14. HE and Immunohistochemistry Assays
2.15. Immunofluorescence
2.16. Statistical Analysis
3. Results
3.1. Upregulation of IDH3α Is Associated with Poor Patient Survival in UCC and LUAD
3.2. Downregulation of IDH3α Exhibited Tumor-Suppressive Functions in Cancer Cell Lines
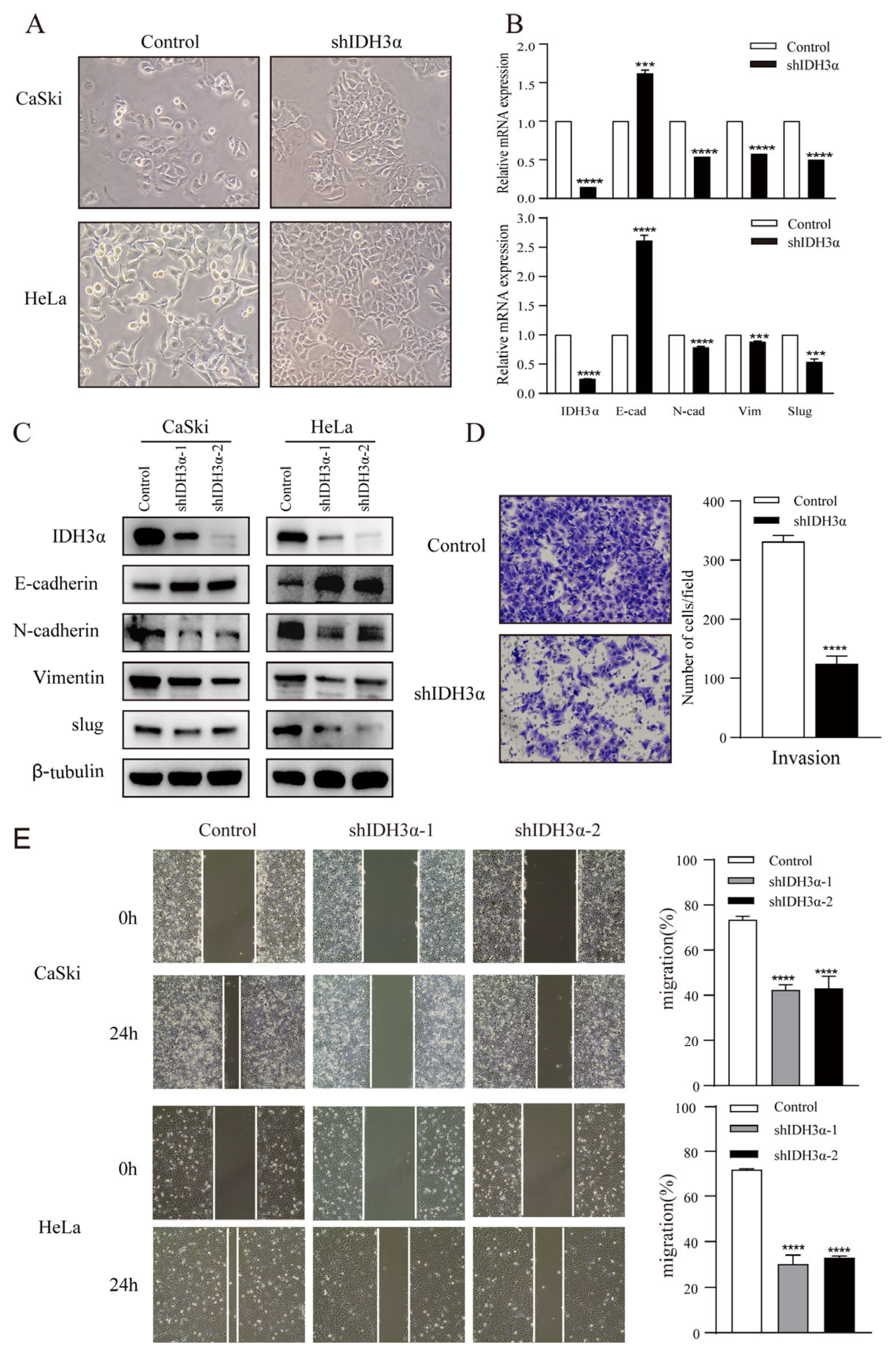
3.3. IDH3α Regulates the EMT in UCC
3.4. IDH3α Controls Intracellular Redox Status and Glycolysis Ability
3.5. Downregulation of IDH3α Expression Increased Sensitivity to Cisplatin
3.6. IDH3α Knockdown Inhibited UCC Growth In Vivo and Increased CD8+ T Cell Infiltration Proportion after Cisplatin Treatment
3.7. IDH3α Knockdown Enhanced Chemoimmunotherapy Sensitivity by Activating cGAS–STING Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bhattacharjee, R.; Dey, T.; Kumar, L.; Kar, S.; Sarkar, R.; Ghorai, M.; Malik, S.; Jha, N.K.; Vellingiri, B.; Kesari, K.K.; et al. Cellular landscaping of cisplatin resistance in cervical cancer. Biomed. Pharmacother. 2022, 153, 113345. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Aisner, D.L.; Wood, D.E.; Akerley, W.; Bauman, J.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; Dobelbower, M.; et al. NCCN Guidelines Insights: Non–Small Cell Lung Cancer, Version 5.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 807–821. [Google Scholar] [CrossRef]
- Mutlu, L.; Tymon-Rosario, J.; Harold, J.; Menderes, G. Targeted treatment options for the management of metastatic/persistent and recurrent cervical cancer. Expert Rev. Anticancer. Ther. 2022, 22, 633–645. [Google Scholar] [CrossRef] [PubMed]
- van Luijk, I.F.; Smith, S.M.; Ojeda, M.C.M.; Oei, A.L.; Kenter, G.G.; Jordanova, E.S. A Review of the Effects of Cervical Cancer Standard Treatment on Immune Parameters in Peripheral Blood, Tumor Draining Lymph Nodes, and Local Tumor Microenvironment. J. Clin. Med. 2022, 11, 2277. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef]
- Hönigova, K.; Navratil, J.; Peltanova, B.; Polanska, H.H.; Raudenska, M.; Masarik, M. Metabolic tricks of cancer cells. Biochim. Biophys. Acta (BBA) Rev. Cancer 2022, 1877, 188705. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef]
- Zeng, P.; Lu, W.; Tian, J.; Qiao, S.; Li, J.; Glorieux, C.; Wen, S.; Zhang, H.; Li, Y.; Huang, P. Reductive TCA cycle catalyzed by wild-type IDH2 promotes acute myeloid leukemia and is a metabolic vulnerability for potential targeted therapy. J. Hematol. Oncol. 2022, 15, 30. [Google Scholar] [CrossRef]
- Liu, X.; Qiao, Y.; Ting, X.; Si, W. Isocitrate dehydrogenase 3A, a rate-limiting enzyme of the TCA cycle, promotes hepatocellular carcinoma migration and invasion through regulation of MTA1, a core component of the NuRD complex. Am. J. Cancer Res. 2020, 10, 3212–3229. [Google Scholar]
- Zeng, L.; Morinibu, A.; Kobayashi, M.; Zhu, Y.; Wang, X.; Goto, Y.; Yeom, C.J.; Zhao, T.; Hirota, K.; Shinomiya, K.; et al. Aberrant IDH3α expression promotes malignant tumor growth by inducing HIF-1-mediated metabolic reprogramming and angiogenesis. Oncogene 2015, 34, 4758–4766. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vanpouille-Box, C.; Bakhoum, S.F.; Demaria, S. SnapShot: CGAS-STING Signaling. Cell 2018, 173, 276.e1. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.; Chen, X.; Corrales, L.; Kline, D.E.; Dubensky, T.W.; Duttagupta, P.; Kortylewski, M.; Kline, J. STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep. 2016, 15, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, D.; Kurbatov, V.; Wang, Q.; Wang, Y.; Fang, D.; Wu, L.; Bosenberg, M.; Muzumdar, M.D.; Khan, S.; et al. 5-Fluorouracil efficacy requires anti-tumor immunity triggered by cancer-cell-intrinsic STING. EMBO J. 2021, 40, e106065. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Xu, W.; Li, J.; Liu, Y.; Zeng, X.; Zhong, M.; Zhu, Y. The Multi-Omics Analysis of Key Genes Regulating EGFR-TKI Resistance, Immune Infiltration, SCLC Transformation in EGFR-Mutant NSCLC. J. Inflamm. Res. 2022, 15, 649–667. [Google Scholar] [CrossRef]
- Zeng, X.; Zhong, M.; Yang, Y.; Wang, Z.; Zhu, Y. Down-regulation of RCC1 sensitizes immunotherapy by up-regulating PD-L1 via p27 kip1 /CDK4 axis in non-small cell lung cancer. J. Cell Mol. Med. 2021, 25, 4136–4147. [Google Scholar] [CrossRef]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef]
- Rawat, S.G.; Tiwari, R.K.; Jaiswara, P.K.; Gupta, V.K.; Sonker, P.; Vishvakarma, N.K.; Kumar, S.; Pathak, C.; Gautam, V.; Kumar, A. Phosphodiesterase 5 inhibitor sildenafil potentiates the antitumor activity of cisplatin by ROS-mediated apoptosis: A role of deregulated glucose metabolism. Apoptosis 2022, 27, 606–618. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Pol, J.; Levesque, S.; Petrazzuolo, A.; Pfirschke, C.; Engblom, C.; Rickelt, S.; Yamazaki, T.; Iribarren, K.; et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 2019, 10, 1486. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Paul, M.S.; Ohashi, P.S. The Roles of CD8+ T Cell Subsets in Antitumor Immunity. Trends Cell Biol. 2020, 30, 695–704. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Li, B.; Simon, M.C. Moonlighting functions of metabolic enzymes and metabolites in cancer. Mol. Cell 2021, 81, 3760–3774. [Google Scholar] [CrossRef]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, A.; Chandel, N.S.; et al. IDH3α regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019, 5, eaat0456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, S.; Chen, Y.; Yang, F.; Liu, S. LDH-A promotes epithelial–mesenchymal transition by upregulating ZEB2 in intestinal-type gastric cancer. OncoTargets Ther. 2018, 11, 2363–2373. [Google Scholar] [CrossRef]
- Blaha, C.S.; Ramakrishnan, G.; Jeon, S.-M.; Nogueira, V.; Rho, H.; Kang, S.; Bhaskar, P.; Terry, A.R.; Aissa, A.F.; Frolov, M.V.; et al. A non-catalytic scaffolding activity of hexokinase 2 contributes to EMT and metastasis. Nat. Commun. 2022, 13, 899. [Google Scholar] [CrossRef]
- Sun, Q.; Yuan, M.; Wang, H.; Zhang, X.; Zhang, R.; Wang, H.; Chen, X.; Zhu, M.; Liu, S.; Wu, J. PKM2 Is the Target of a Multi-Herb-Combined Decoction During the Inhibition of Gastric Cancer Progression. Front. Oncol. 2021, 11, 767116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Zhang, M.; Cong, Q.; Zhang, M.-X.; Zhang, M.Y.; Lu, Y.-Y.; Xu, C.-J. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int. J. Biochem. Cell Biol. 2018, 95, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Wu, J.; Guo, L.; Xu, Y.; Liu, L.; Lu, J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int. J. Oncol. 2017, 50, 1848–1856. [Google Scholar] [CrossRef]
- Dai, Y.; Liu, Y.; Li, J.; Jin, M.; Yang, H.; Huang, G. Shikonin inhibited glycolysis and sensitized cisplatin treatment in non-small cell lung cancer cells via the exosomal pyruvate kinase M2 pathway. Bioengineered 2022, 13, 13906–13918. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Queckborner, S.; Turnbull, A.; Michie, C.O.; Williams, A.R.W.; Rye, T.; Gourley, C.; Langdon, S.P. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer 2018, 18, 636. [Google Scholar] [CrossRef] [PubMed]
- Baczewska, M.; Supruniuk, E.; Bojczuk, K.; Guzik, P.; Milewska, P.; Konończuk, K.; Dobroch, J.; Chabowski, A.; Knapp, P. Energy Substrate Transporters in High-Grade Ovarian Cancer: Gene Expression and Clinical Implications. Int. J. Mol. Sci. 2022, 23, 8968. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Azmi, A.S.; Mohammad, R.M. Deregulated transcription factors and poor clinical outcomes in cancer patients. Semin. Cancer Biol. 2022, 86, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Kalantari, M.; Mohammadinejad, R.; Javaheri, T.; Sethi, G. Association of the Epithelial–Mesenchymal Transition (EMT) with Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 4002. [Google Scholar] [CrossRef]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 81. [Google Scholar] [CrossRef]
- Du, M.; Yu, T.; Zhan, Q.; Li, H.; Zou, Y.; Geng, M.; Meng, T.; Xie, Z. Development of a novel lactate dehydrogenase A inhibitor with potent antitumor activity and immune activation. Cancer Sci. 2022, 113, 2974–2985. [Google Scholar] [CrossRef]
- Baggiolini, M. Chemokines in pathology and medicine. J. Intern. Med. 2001, 250, 91–104. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, Y. CXCL11 Signaling in the Tumor Microenvironment. Single Mol. Single Cell Seq. 2021, 1302, 41–50. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, C.; Ding, J.; Wu, H.; Cheng, W.; Li, M.; Zhu, R.; Li, H. Altered T lymphocyte subtypes and cytokine profiles in follicular fluid associated with diminished ovary reserve. Am. J. Reprod. Immunol. 2022, 87, e13522. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Masi, T.; Goff, M.R.; Karlstad, M.D.; Collier, J.J. NF-κB and STAT1 control CXCL1 and CXCL2 gene transcription. Am. J. Physiol. Metab. 2014, 306, E131–E149. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Kosaka, A.; Yajima, Y.; Yasuda, S.; Ohara, M.; Ohara, K.; Harabuchi, S.; Hayashi, R.; Funakoshi, H.; Ueda, J.; et al. A critical role of STING-triggered tumor-migrating neutrophils for anti-tumor effect of intratumoral cGAMP treatment. Cancer Immunol. Immunother. 2021, 70, 2301–2312. [Google Scholar] [CrossRef]
- Leca, J.; Fortin, J.; Mak, T.W. Illuminating the cross-talk between tumor metabolism and immunity in IDH-mutated cancers. Curr. Opin. Biotechnol. 2021, 68, 181–185. [Google Scholar] [CrossRef]
- Yang, Q.; Hao, J.; Chi, M.; Wang, Y.; Li, J.; Huang, J.; Zhang, J.; Zhang, M.; Lu, J.; Zhou, S.; et al. D2HGDH-mediated D2HG catabolism enhances the anti-tumor activities of CAR-T cells in an immunosuppressive microenvironment. Mol. Ther. 2022, 30, 1188–1200. [Google Scholar] [CrossRef]
- Lv, H.; Lv, G.; Chen, C.; Zong, Q.; Jiang, G.; Ye, D.; Cui, X.; He, Y.; Xiang, W.; Han, Q.; et al. NAD+ Metabolism Maintains Inducible PD-L1 Expression to Drive Tumor Immune Evasion. Cell Metab. 2020, 33, 110–127.e5. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Song, Y.; Dai, X.; Xu, W.; Li, M.; Zhu, Y. Inhibition of IDH3α Enhanced the Efficacy of Chemoimmunotherapy by Regulating Acidic Tumor Microenvironments. Cancers 2023, 15, 1802. https://doi.org/10.3390/cancers15061802
Zhang L, Song Y, Dai X, Xu W, Li M, Zhu Y. Inhibition of IDH3α Enhanced the Efficacy of Chemoimmunotherapy by Regulating Acidic Tumor Microenvironments. Cancers. 2023; 15(6):1802. https://doi.org/10.3390/cancers15061802
Chicago/Turabian StyleZhang, Lingling, Yang Song, Xiaoyan Dai, Wenwen Xu, Mengxia Li, and Yuxi Zhu. 2023. "Inhibition of IDH3α Enhanced the Efficacy of Chemoimmunotherapy by Regulating Acidic Tumor Microenvironments" Cancers 15, no. 6: 1802. https://doi.org/10.3390/cancers15061802
APA StyleZhang, L., Song, Y., Dai, X., Xu, W., Li, M., & Zhu, Y. (2023). Inhibition of IDH3α Enhanced the Efficacy of Chemoimmunotherapy by Regulating Acidic Tumor Microenvironments. Cancers, 15(6), 1802. https://doi.org/10.3390/cancers15061802