
Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives

 and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Molecular Landscape
2.1. Carcinogenesis and Drivers
2.2. Immunology of HCC
2.3. Molecular and Immune HCC Classes
3. First-Line Treatment
3.1. Tyrosine Kinase Inhibitors
3.2. Immune Checkpoint Inhibitors and Combination Regimens
3.3. Other Treatment Options under Investigation
4. Second-Line and Beyond
4.1. Tyrosine Kinase Inhibitors
4.2. Immune Checkpoint Inhibitors
4.3. Other Immune Checkpoint Inhibitors under Investigation
5. Sequencing Treatment
6. Biomarkers
6.1. Biomarkers for Immunotherapy
6.1.1. PD-L1 Expression
6.1.2. Tumor Mutational Burden and Microsatellite Instability
6.1.3. Other Possible Biomarkers: Circulating Tumor Cells, Gut Microbiota and WNT/β-Catenin Signaling
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D. Cancer statistics. ACS J. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- White, D.L.; Thrift, A.P.; Kanwal, F.; Davila, J.; El-Serag, H.B. Incidence of Hepatocellular Carcinoma in All 50 United States, From 2000 Through 2012. Gastroenterology 2017, 152, 812–820.e815. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: A meta-analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef]
- Chen, C.J.; Liang, K.Y.; Chang, A.S.; Chang, Y.C.; Lu, S.N.; Liaw, Y.F.; Chang, W.Y.; Sheen, M.C.; Lin, T.M. Effects of hepatitis B virus, alcohol drinking, cigarette smoking and familial tendency on hepatocellular carcinoma. Hepatology 1991, 13, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Di Bisceglie, A.M.; Bruix, J.; Kramer, B.S.; Lencioni, R.; Zhu, A.X.; Sherman, M.; Schwartz, M.; Lotze, M.; Talwalkar, J.; et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J. Natl. Cancer Inst. 2008, 100, 698–711. [Google Scholar] [CrossRef]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e1224. [Google Scholar] [CrossRef]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef] [PubMed]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef]
- Llovet, J.M.; Pinyol, R.; Kelley, R.K.; El-Khoueiry, A.; Reeves, H.L.; Wang, X.W.; Gores, G.J.; Villanueva, A. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat. Cancer 2022, 3, 386–401. [Google Scholar] [CrossRef]
- Nault, J.C.; Calderaro, J.; di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e1323. [CrossRef]
- Giraud, J.; Chalopin, D.; Blanc, J.F.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.Z.; Liu, Z.W.; Zhang, J.Y.; Yang, Y.P.; Tien, P.; Wang, F.S. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Sarobe, P. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Solé, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Montironi, C.; Castet, F. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2023, 72, 129–140. [Google Scholar] [CrossRef]
- Ikeda, M.; Mitsunaga, S.; Ohno, I.; Hashimoto, Y.; Takahashi, H.; Watanabe, K.; Umemoto, K.; Okusaka, T. Systemic Chemotherapy for Advanced Hepatocellular Carcinoma: Past, Present, and Future. Diseases 2015, 3, 360–381. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Rimassa, L.; Cheng, A.L.; Kaseb, A.; Qin, S.; Zhu, A.X.; Chan, S.L.; Melkadze, T.; Sukeepaisarnjaroen, W.; Breder, V.; et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 995–1008. [Google Scholar] [CrossRef]
- Kudo, M. Durvalumab plus tremelimumab in unresectable hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2022, 11, 592–596. [Google Scholar] [CrossRef]
- Qin, S.; Finn, R.S.; Kudo, M.; Meyer, T.; Vogel, A.; Ducreux, M.; Macarulla, T.M.; Tomasello, G.; Boisserie, F.; Hou, J.; et al. RATIONALE 301 study: Tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Future Oncol. 2019, 15, 1811–1822. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Matilla, A.; Santoro, A.; Melero, I.; Gracián, A.C.; Acosta-Rivera, M.; Choo, S.P.; El-Khoueiry, A.B.; Kuromatsu, R.; El-Rayes, B.; et al. CheckMate 040 cohort 5: A phase I/II study of nivolumab in patients with advanced hepatocellular carcinoma and Child-Pugh B cirrhosis. J. Hepatol. 2021, 75, 600–609. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Lyu, N.; Wang, X.; Li, J.B.; Lai, J.F.; Chen, Q.F.; Li, S.L. Arterial Chemotherapy of Oxaliplatin Plus Fluorouracil Versus Sorafenib in Advanced Hepatocellular Carcinoma: A Biomolecular Exploratory, Randomized, Phase III Trial (FOHAIC-1). J. Clin. Oncol. 2022, 40, 468–480. [Google Scholar] [CrossRef]
- Finn, R.S.; Kudo, M.; Merle, P.; Meyer, T.; Qin, S.; Ikeda, M.; Xu, R.; Edeline, J.; Ryoo, B.Y.; Ren, Z.; et al. LBA34 Primary results from the phase III LEAP-002 study: Lenvatinib plus pembrolizumab versus lenvatinib as first-line (1L) therapy for advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2022, 33, S1401. [Google Scholar] [CrossRef]
- Qin, S.; Chan, L.S.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. LBA35 Camrelizumab (C) plus rivoceranib (R) vs. sorafenib (S) as first-line therapy for unresectable hepatocellular carcinoma (uHCC): A randomized, phase III trial. Ann. Oncol. 2022, 33, S1401–S1402. [Google Scholar] [CrossRef]
- Peng, Z.; Fan, W.; Zhu, B. Lenvatinib Combined With Transarterial Chemoembolization as First-Line Treatment for Advanced Hepatocellular Carcinoma: A Phase III, Randomized Clinical Trial (LAUNCH). J. Clin. Oncol. 2023, 41, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K. Antitumor activity of lenvatinib (e7080): An angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Gordan, J.D.; Kennedy, E.B. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 4317–4345. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Tai, D.; Chan, S.L.; Huang, Y.H.; Choo, S.P.; Hsu, C.; Cheung, T.T.; Lin, S.M.; Yong, W.P.; Lee, J.; et al. Systemic Treatment of Advanced Unresectable Hepatocellular Carcinoma after First-Line Therapy: Expert Recommendations from Hong Kong, Singapore, and Taiwan. Liver Cancer 2022, 11, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; Toni, E.N.D.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Breedis, C.; Young, G. The blood supply of neoplasms in the liver. Am. J. Pathol. 1954, 30, 969–977. [Google Scholar]
- Ueshima, K.; Ogasawara, S.; Ikeda, M.; Yasui, Y.; Terashima, T.; Yamashita, T.; Obi, S.; Sato, S.; Aikata, H.; Ohmura, T.; et al. Hepatic Arterial Infusion Chemotherapy versus Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Liver Cancer 2020, 9, 583–595. [Google Scholar] [CrossRef]
- Lyu, N.; Kong, Y.; Mu, L.; Lin, Y.; Li, J.; Liu, Y.; Zhang, Z.; Zheng, L.; Deng, H.; Li, S.; et al. Hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin vs. sorafenib for advanced hepatocellular carcinoma. J. Hepatol. 2018, 69, 60–69. [Google Scholar] [CrossRef]
- Lyu, N.; Lin, Y.; Kong, Y.; Zhang, Z.; Liu, L.; Zheng, L.; Mu, L.; Wang, J.; Li, X.; Pan, T.; et al. FOXAI: A phase II trial evaluating the efficacy and safety of hepatic arterial infusion of oxaliplatin plus fluorouracil/leucovorin for advanced hepatocellular carcinoma. Gut 2018, 67, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Park, J.W.; Kim, J.H.; An, M.; Kong, S.Y.; Nam, B.H.; Choi, J.I.; Kim, H.B.; Lee, W.J.; Kim, C.M. Association between increment of serum VEGF level and prognosis after transcatheter arterial chemoembolization in hepatocellular carcinoma patients. Cancer Sci. 2008, 99, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Ryoo, B.Y.; Merle, P.; Park, J.W.; Bolondi, L.; Chan, S.L.; Lim, H.Y.; Baron, A.D.; Parnis, F.; Knox, J.; et al. Second-line cabozantinib after sorafenib treatment for advanced hepatocellular carcinoma: A subgroup analysis of the phase 3 CELESTIAL trial. ESMO Open 2020, 5, e000714. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Decaens, T.; Raoul, J.L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.K.; Assenat, E.; Lim, H.Y.; Boige, V.; et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: Results from the randomized phase III BRISK-PS study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3509–3516. [Google Scholar] [CrossRef]
- Qin, S.; Li, Q.; Gu, S.; Chen, X.; Lin, L.; Wang, Z.; Xu, A.; Chen, X.; Zhou, C.; Ren, Z.; et al. Apatinib as second-line or later therapy in patients with advanced hepatocellular carcinoma (AHELP): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 559–568. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chen, Z.; Fang, W. Pembrolizumab Versus Placebo as Second-Line Therapy in Patients From Asia With Advanced Hepatocellular Carcinoma: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2023, 41, 1434–1443. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 Antibody SHR-1210 Combined with Apatinib for Advanced Hepatocellular Carcinoma, Gastric, or Esophagogastric Junction Cancer: An Open-label, Dose Escalation and Expansion Study. Clin. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef] [PubMed]
- He, A.R.; Weiss, G.J.; Falchook, G.; Yee, N.S.; Gil-Martin, M.; Shahda, S.; Moreno, V.; Brana, I.; Crittenden, M.; Formenti, S.; et al. Cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with advanced or metastatic hepatocellular carcinoma (HCC): Data from an expansion cohort (EC) in a phase I study. Ann. Oncol. 2018, 29, x26. [Google Scholar] [CrossRef]
- Shen, L.; Guo, J.; Zhang, Q.; Pan, H.; Yuan, Y.; Bai, Y.; Liu, T.; Zhou, Q.; Zhao, J.; Shu, Y.; et al. Tislelizumab in Chinese patients with advanced solid tumors: An open-label, non-comparative, phase 1/2 study. J. Immunother. Cancer 2020, 8, e000437. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Ramjeesingh, R.; Liu, D.; Tam, V.C.; Knox, J.J.; Card, P.B.; Meyers, B.M. Optimizing Survival and the Changing Landscape of Targeted Therapy for Intermediate and Advanced Hepatocellular Carcinoma: A Systematic Review. J. Natl. Cancer Inst. 2021, 113, 123–136. [Google Scholar] [CrossRef]
- Storandt, M.H.; Gile, J.J. Cabozantinib Following Immunotherapy in Patients with Advanced Hepatocellular Carcinoma. Cancers 2022, 14, 5173. [Google Scholar] [CrossRef]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef]
- Zhang, D.; Liao, X. Pan-TRK Immunohistochemistry and NTRK Gene Fusions in Primary Carcinomas of the Liver. Appl. Immunohistochem. Mol. Morphol. AIMM 2022, 30, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Teufel, M.; Seidel, H.; Köchert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers Associated With Response to Regorafenib in Patients With Hepatocellular Carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Kang, Y.K.; Yen, C.J.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Motomura, K.; Ohno, I.; et al. Serum alpha-fetoprotein and clinical outcomes in patients with advanced hepatocellular carcinoma treated with ramucirumab. Br. J. Cancer 2021, 124, 1388–1397. [Google Scholar] [CrossRef]
- Girardi, D.M.; Pacífico, J.P.M.; Guedes de Amorim, F.P.L.; Dos Santos Fernandes, G.; Teixeira, M.C.; Pereira, A.A.L. Immunotherapy and Targeted Therapy for Hepatocellular Carcinoma: A Literature Review and Treatment Perspectives. Pharmaceuticals 2020, 14, 28. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.L.; Yau, T.; Furuse, J.; Park, J.W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Hou, M.-M.; Rau, K.-M.; Kang, Y.-K.; Lee, J.-S.; Pan, H.; Yuan, Y.; Yu, C.; Zhang, Y.; Ma, X.; Wu, X.; et al. 77 Association between programmed death-ligand 1 (PD-L1) expression and gene signatures of response or resistance to tislelizumab monotherapy in hepatocellular carcinoma (HCC). J. ImmunoTherapy Cancer 2020, 8, A47–A48. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Abou-Alfa Ghassan, K.; Lau, G.; Kudo, M.; Chan Stephen, L.; Kelley Robin, K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni Enrico, N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070. [Google Scholar] [CrossRef]
- He, Y.; Lu, M.; Che, J.; Chu, Q.; Zhang, P.; Chen, Y. Biomarkers and Future Perspectives for Hepatocellular Carcinoma Immunotherapy. Front. Oncol. 2021, 11, 716844. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef] [PubMed]
- von Felden, J.; Garcia-Lezana, T.; Schulze, K.; Losic, B.; Villanueva, A. Liquid biopsy in the clinical management of hepatocellular carcinoma. Gut 2020, 69, 2025–2034. [Google Scholar] [CrossRef]
- Winograd, P.; Hou, S.; Court, C.M. Hepatocellular Carcinoma-Circulating Tumor Cells Expressing PD-L1 Are Prognostic and Potentially Associated With Response to Checkpoint Inhibitors. Hepatol. Commun. 2020, 4, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ying, J. Gut Microbiota and Immunotherapy. Front. Microbiol. 2022, 13, 945887. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Gao, J.; Cui, Q.; Wang, Q. Strategies to Improve the Antitumor Effect of Immunotherapy for Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 783236. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Galle, P.R. The Current Landscape of Clinical Trials for Systemic Treatment of HCC. Cancers 2021, 13, 1962. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zhang, J.; Li, A.; Niu, M.; Yan, Y.; Jiao, Y.; Luo, S.; Zhou, P.; Wu, K. The construction, expression, and enhanced anti-tumor activity of YM101: A bispecific antibody simultaneously targeting TGF-β and PD-L1. J. Hematol. Oncol. 2021, 14, 27. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Joshi, J.J.; Coffey, H.; Corcoran, E.; Tsai, J.; Huang, C.L.; Ichikawa, K.; Prajapati, S.; Hao, M.H.; Bailey, S.; Wu, J.; et al. H3B-6527 Is a Potent and Selective Inhibitor of FGFR4 in FGF19-Driven Hepatocellular Carcinoma. Cancer Res. 2017, 77, 6999–7013. [Google Scholar] [CrossRef] [PubMed]


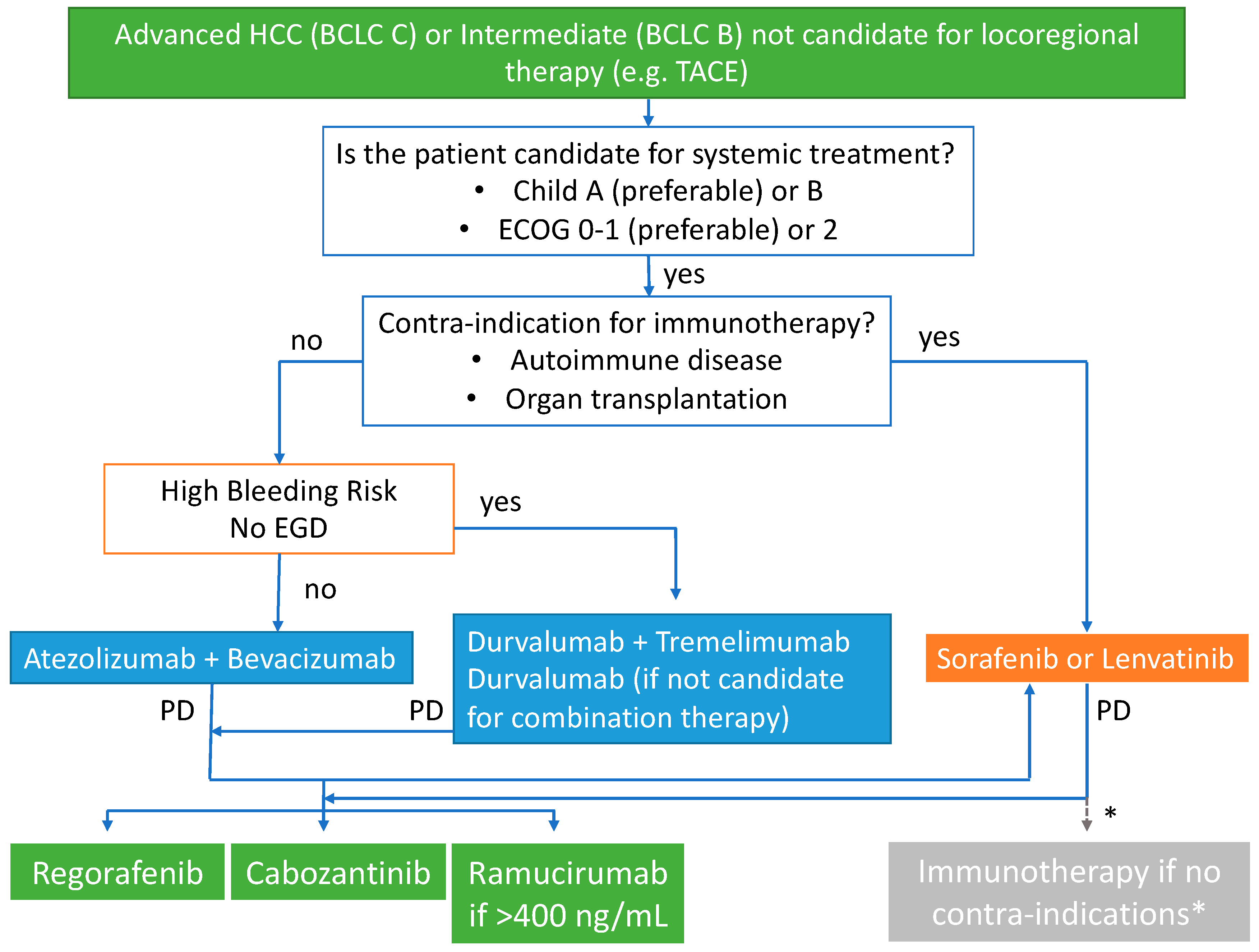{kind=link}
| Study (Year) | Phase | N | Population | Geographical Region | Drug | Median Overall Survival | Median Progression-Free Survival | Objective Response Rate |
|---|---|---|---|---|---|---|---|---|
| REFLECT trial (2018) [31] | III noninferiority | 954 | Unresectable HCC and no prior systemic therapy (99% Child-Turcotte-Pugh class A) | 29% (white); 69% (Asian); 2% (other) | Lenvatinib vs. sorafenib | 13.6 mo for lenvatinib vs. 12.3 mo for sorafenib (HR: 0.92, 95% CI: 0.79–1.06) | 7.4 mo for lenvatinib vs. 3.7 mo for sorafenib (HR: 0.66; p < 0.0001) | 24.1% for lenvatinib vs. 9.2% for sorafenib (p < 0.0001) |
| IMbrave 150 trial (2021) [32,33] | III | 336 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 40% (Asians, excluding Japan); 60% (rest of the world) | Atezolizumab-bevacizumab vs. sorafenib | 19.2 mo for atezolizumab-bevacizumab vs. 13.4 mo for sorafenib (HR: 0.66; p < 0.001) | 6.9 mo for atezolizumab-bevacizumab vs. 4.3 mo for sorafenib (HR: 0.65; p < 0.001) | 30% to atezolizumab-bevacizumab vs. 11% to sorafenib |
| COSMIC-321 trial (2022) [34] | III | 837 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 29.3% (Asians); 70.7% (Other) | Cabozantinib-atezolizumab vs. sorafenib | 15.4 mo for cabozantinib-atezolizumab vs. 15.5 mo for sorafenib (HR: 0.90, p = 0.44) | 6.8 mo for cabozantinib-atezolizumab vs. 4.2 mo for sorafenib (HR: 0.63, p = 0.0012) | 13% to cabozantinib-atezolizumab vs. 6% to sorafenib |
| HIMALAYA trial (2022) [35] | III | 1171 | Unresectable HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 40.9% (Asians, excluding Japan); 59.1% (rest of the world) | Durvalumab-tremelimumab or durvalumab vs. sorafenib | 16.43 mo for STRIDE vs. 13.77 for sorafenib (HR: 0.78; p = 0.0035) | 3.78 mo for STRIDE and 3.65 mo for durvalumab vs. 4.07 for sorafenib (HR: 0.90; p = 0.0035 and HR: 1.02, p = 0.0674) | 20.1% to STRIDE, 17% to durvalumab vs. 5.1 to sorafenib |
| RATIONALE-301 trial (2022) [36] | III | 674 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 63.1% (Asians, excluding Japan); 11.4 (Japan); 25.5% (rest of the world) | Tislelizumab vs. sorafenib | 15.9 mo for tislelizumab vs. 14.1 mo for sorafenib (HR: 0.8) | 2.2 mo for tislelizumab vs. 3.6 mo for sorafenib (HR: 1.1) | 14.3% to tislelizumab vs. 5.4% to sorafenib |
| CheckMate 459 (2019) [37] | III | 743 | Unresectable Child-Pugh A HCC naïve to systemic treatment | 40% (Asian); 60% (United States, Canada or Europe) | Nivolumab vs. sorafenib | 16.4 mo for nivolumab vs. 14.7 mo for sorafenib (HR: 0.85; p = 0.0752) | 3.7 mo for nivolumab vs. 3.8 mo for sorafenib | 15% for nivolumab and 7% to sorafenib |
| CheckMate-040: cohort B (2021) [38] | I/II | 49 | Unresectable or metastatic HCC, Child-Pugh liver function score B, with or without prior systemic therapy | 55% (Asian); 41% (white); 2% (black); 2% (other) | Nivolumab single arm | 9.8 mo for sorafenib naïve patients and 7.4 mo for previously treated patients | 3.4 mo for sorafenib naïve patients and 2.2 mo for previously treated patients | 12% |
| KEYNOTE-524 trial (2022) [39] | Ib | 104 | Unresectable or metastatic HCC, Child-Pugh liver function score < 7, and no prior systemic therapy | 51% (white); 28% (Asian); 2% (black); 5% (other); 14% (missing) | Lenvatinib-pembrolizumab single arm | 22 mo | 9.3 mo per mRECIST; 8.6 per RECIST v1.1 | 46% per mRECIST; 36% per RECIST v1.1 |
| FOHAIC-1 (2021) [40] | III | 262 | Locally advanced or unresectable HCC with or without extrahepatic oligometastasis, Child-Pugh liver function score ≤ 7 | HAIC (FOLFOX) vs. sorafenib | 13.9 mo for HAIC vs. 8.2 mo for sorafenib (HR: 0.408, p < 0.001) | 7.8 mo for HAIC vs. 4.3 mo for sorafenib (HR: 0.451, p < 0.001) | 31.5% to HAIC and 1.5% to sorafenib per RECIST; 35.4% to HAIC and 5.3% to sorafenib per mRECIST (p < 0.001) | |
| LEAP-002 (2021) [41] | III | 794 | Primary treatment-naive HCC, non-amenable to curative therapy, Child-Pugh A | 30.7% (Asian without Japan) vs. 69.3% (western regions and Japan) | lenvantinib plus pembrolizumab vs. lenvantinib | 21.2 mo for lenvatinib and pembrolizumab vs. 19 mo for lenvatinib (HR: 0.84; p = 0.0227) | 8.2 mo for lenvatinib and pembrolizumab vs. 8.1 mo for lenvatinib (HR: 0.834; p = 0.0466) | 26.1% for lenvatinib and pembrolizumab and 17.5% for lenvatinib per RECIST 1.1; 40.6% for lenvatinib and pembrolizumab and 34.1% for lenvatinib per mRECIST |
| Qin, et al. (2022) [42] | III | 543 | Unresectable or metastatic HCC primary treatment naive, BCLC satage B, Child-Pugh A | 82.7% (Asian) vs. 17.3% (non-Asian) | Camrelizumab + rivoceranib vs. sorafenib | 22.1 mo for canrelizumab + rivoceranib vs. 15.2 mo for sorafenib (HR: 0.62; 95% CI: 0.49–0.80) | 5.6 mo for canrelizumab + rivoceranib vs. 3.7 mo for sorafenib (HR: 0.52; 95% CI: 0.41–0.65) | 25.4% for camrelizumab and rivoceranib and 5.9% for sorafenib per RECIST 1.1; 33.1% for camrelizumab and rivoceranib and 10% for sorafenib per mRECIST |
| LAUNCH (2022) [43] | 338 | Primary treatment-naive or initial recurrent advanced HCC after surgery without adjuvant treatment, Child-Pugh class A | 100% (Asian—China) | LEN-TACE vs. lenvatinib | 17.8 mo forLEN-TACE vs. 11.5 mo for lenvatinib (HR: 0.33; p < 0.001) | 10.6 mo forLEN-TACE vs. 6.4 mo for lenvatinib (HR: 0.36; p < 0.001) | 45.9% to LEN_TACE and 20.8% to lenvatinib per RECIST; 54.1% to LEN-TACE and 25% to lenvatinib per mRECIST (p < 0.001) |
| Study (Year) | Phase | N | Population | Drug | Median Overall Survival | Median Progression-Free Survival | Objective Response Rate |
|---|---|---|---|---|---|---|---|
| RESORCE trial (2017) [56] | III | 573 | Advanced HCC that progressed after first-line treatment with sorafenib, Child-Pugh A | Regorafenib vs. placebo | 10.6 mo for regorafenib vs. 7.8 mo for placebo (HR: 0.63; p < 0.0001) | 3.1 mo for regorafenib vs. 1.5 mo for placebo (HR: 0.46; p < 0.0001) | 11% for regorafenib vs. 4% for placebo (p = 0.0047) |
| CELESTIAL trial (2018) [57] | III | 707 | Advanced and progressing HCC and not worse than Child-Pugh A | Cabozantinib vs. placebo | 10.2 mo for cabozantinib vs. 8.0 mo for placebo (HR: 0.76; p = 0.005) | 5.2 mo for cabozantinib vs. 1.9 mo for placebo (HR: 0.44; p < 0.001) | 4% for cabozantinib vs. less than 1% for placebo (p = 0.009) |
| REACH trial (2015) [58] | III | 565 | Advanced HCC following first-line therapy with sorafenib and Child-Pugh A | Ramucirumab vs. placebo | 9.2 mo for ramucirumab vs. 7.6 mo for placebo (HR: 0.87; p = 0.14). | 2.8 mo for ramucirumab vs. 2.1 mo for placebo (HR 0.63; p < 0.0001) | 7% for ramucirumab vs. < 1% for placebo (p < 0.0001) |
| REACH-2 trial (2019) [59] | III | 292 | Advanced HCC, Child-Pugh class A, and serum AFP ≥ 400 ng/mL in patients who had disease progression under first-line sorafenib | Ramucirumab vs. placebo | 8.5 mo for ramucirumab vs. 7.3 mo for placebo (HR: 0.71; p = 0.0199 | 2.8 mo for ramucirumab vs. 1.6 mo for placebo (HR: 0.452; p < 0. 0001) | 5% for ramucirumab vs. 1% for placebo (p = 0.1697) |
| BRISK-PS study [60] | III | 395 | Advanced HCC who progressed on/after or were intolerant to sorafenib | Brivanib vs. placebo | 9.4 mo for brivanib vs. 8.2 mo for placebo (HR: 0.89; p = 3307) | 4.2 mo for brivanib vs. 2.7 mo for placebo (HR, 0.56; p < 0.001) | 10% for brivanib vs. 2% for placebo (odds ratio, 5.72). |
| Qiu Li et al. (2020) [61] | III | 393 | Advanced HCC after failure of sorafenib and oxaliplatin-based chemotherapy and Child-Pugh liver function class A or B ≤ 7 points | Apatinib vs. placebo | 8.7 mo for apatinib vs. 6.8 mo for placebo (HR: 0.785; p = 0.0476) | 4.5 mo for apatinib vs. 1.9 mo for placebo (HR: 0.471; p < 0.0001) | 10.7% for ramucirumab vs. 1.5% for placebo |
| CheckMate 040 (2020) [62] | I/II | 148 | Advanced HCC patients who were treatment-naive or received sorafenib previously | Nivolumab and ipilimumab | Arm A: 22.8 mo Arm B: 12.5 mo ArmC: 12.7 mo | Arm A: 32% Arm B: 27% Arm C: 29% | |
| KeyNote 240 (2019) [63] | III | 413 | Child-Pugh A HCC patients, after progression or intolerance to sorafenib | Pembrolizumab vs. BSC | NR | 13.8 months | 18.3 versus 4.4%. |
| KeyNote 394 (2022) [64] | III | 453 | Second-line therapy for previously treated advanced HCC | Pembrolizumab vs. placebo | 14.6 vs. 13.0 months | 2.6 vs. 2.3 months | 12.7% vs. 1.3%, |
| Kelley et al.. (2020) [65] | I/II | 332 | Advanced HCC patients who progressed on, were intolerant to or refused sorafenib | Durvalumab + tremelimumab | T300 + D: 18.7 mo Durvalumab: 13.6 mo Tremelimumab: 15.1 mo T75 + D: 11.3 mo | T300 + D: 2.17 mo Durvalumab: 2.07 mo Tremelimumab: 2.69 mo T75 + D: 1.87 mo | T300 + D: 24.0% Durvalumab: 106% Tremelimumab: 7.2% T75 + D: 9.5% |
| Xu et al. (2019) [66] | I | 18 | HCC patients, Child-Pugh A, and previously treated with sorafenib | SHR-1210 + apatinib | NR | 5.8 mo | 50% |
| Qin et al. (2020) [67] | II | 217 | Advanced HCC, Child-Pugh A or B7 after sorafenib failure or intolerance to first-line systemic therapy | Camrelizumab | 13.8 mo | 2.1 mo | 14.7% |
| He et al. (2018) [68] | Ib | 26 | Advanced HCC, Child-Pugh A after failure or intolerance to first-line systemic therapy | Cemiplimab | 3.7 mo | 19.2% | |
| Shen et al. (2020) [69] | I/II | 300 | Advanced or metastatic solid tumors, including HCC, in patients who have progressed since their last standard antitumor treatment, had no available (or refused) standard treatment, or become intolerant to treatment | Tislelizumab | immature for HCC | 4.0 mo |
| Drugs | Phase | Setting | Endpoint | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| SBRT followed by sintilimab vs. SBRT | II/III | Palliative—1st line | Primary: 24-week PFS rate Secondary: PFS; OS; ORR; DCR; DOR | NCT04167293 |
| Lenvatinib + pembrolizumab +TACE vs. TACE | III | Palliative—1st line | Primary: PFS; OS Secondary: ORR; DCR; DOR; TTP; AEs | NCT04246177 |
| Arm A: TACE + durvalumab; Arm B: TACE + durvalumab + bevacizumab; Arm C: TACE | III | Palliative—1st line | Primary: PFS (Arm B vs. Arm C) Secondary: PFS (Arm A vs. Arm C); OS | NCT03778957 |
| Nivolumab + Ipilimumab vs. sorafenib or lenvatinib | III | Palliative—1st line | Primary: OS Secondary: ORR; DOR; TTSD | NCT04039607 |
| Finotonlimab (anti PD1) + SCT510 (bavacizumab) vs. Sorafenib | II/III | Palliative—1st line | Primary: OS Secondary: ORR; PFS | NCT04560894 |
| Toripalimab + Lenvatinib vs. Lenvatinib | III | Palliative—1st line | Primary: OS, PFS Secondary: ORR; DOR; TTP | NCT04523493 |
| Nofazinlimab (CS1003) + Lenvatinib vs. Lenvatinib | III | Palliative—1st line | Primary: OS, PFS | NCT04194775 |
| Atezolizumab + Lenvatinib or Sorafenib vs. Lenvatinib or Sorafenib | III | Palliative—2nd line | Primary: OS Secondary: ORR; PFS; DOR; TTP | NCT04770896 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girardi, D.M.; Sousa, L.P.; Miranda, T.A.; Haum, F.N.C.; Pereira, G.C.B.; Pereira, A.A.L. Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives. Cancers 2023, 15, 1680. https://doi.org/10.3390/cancers15061680
Girardi DM, Sousa LP, Miranda TA, Haum FNC, Pereira GCB, Pereira AAL. Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives. Cancers. 2023; 15(6):1680. https://doi.org/10.3390/cancers15061680
Chicago/Turabian StyleGirardi, Daniel M., Lara P. Sousa, Thiago A. Miranda, Fernanda N. C. Haum, Gabriel C. B. Pereira, and Allan A. L. Pereira. 2023. "Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives" Cancers 15, no. 6: 1680. https://doi.org/10.3390/cancers15061680
APA StyleGirardi, D. M., Sousa, L. P., Miranda, T. A., Haum, F. N. C., Pereira, G. C. B., & Pereira, A. A. L. (2023). Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives. Cancers, 15(6), 1680. https://doi.org/10.3390/cancers15061680