
Increased IL-17A Serum Levels and Gastric Th17 Cells in Helicobacter pylori-Infected Patients with Gastric Premalignant Lesions

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. IL-17 Production by Gastric Mucosa T Cells
2.3. Characterization of Antigen Specificity and IL-17 Production by Gastric T Cell Clones Obtained from Helicobacter pylori Patients with Gastric Intestinal Metaplasia, and Gastric Dysplasia
2.4. Luminex Assay for IL-17A
2.5. Statistical Analyses
3. Results
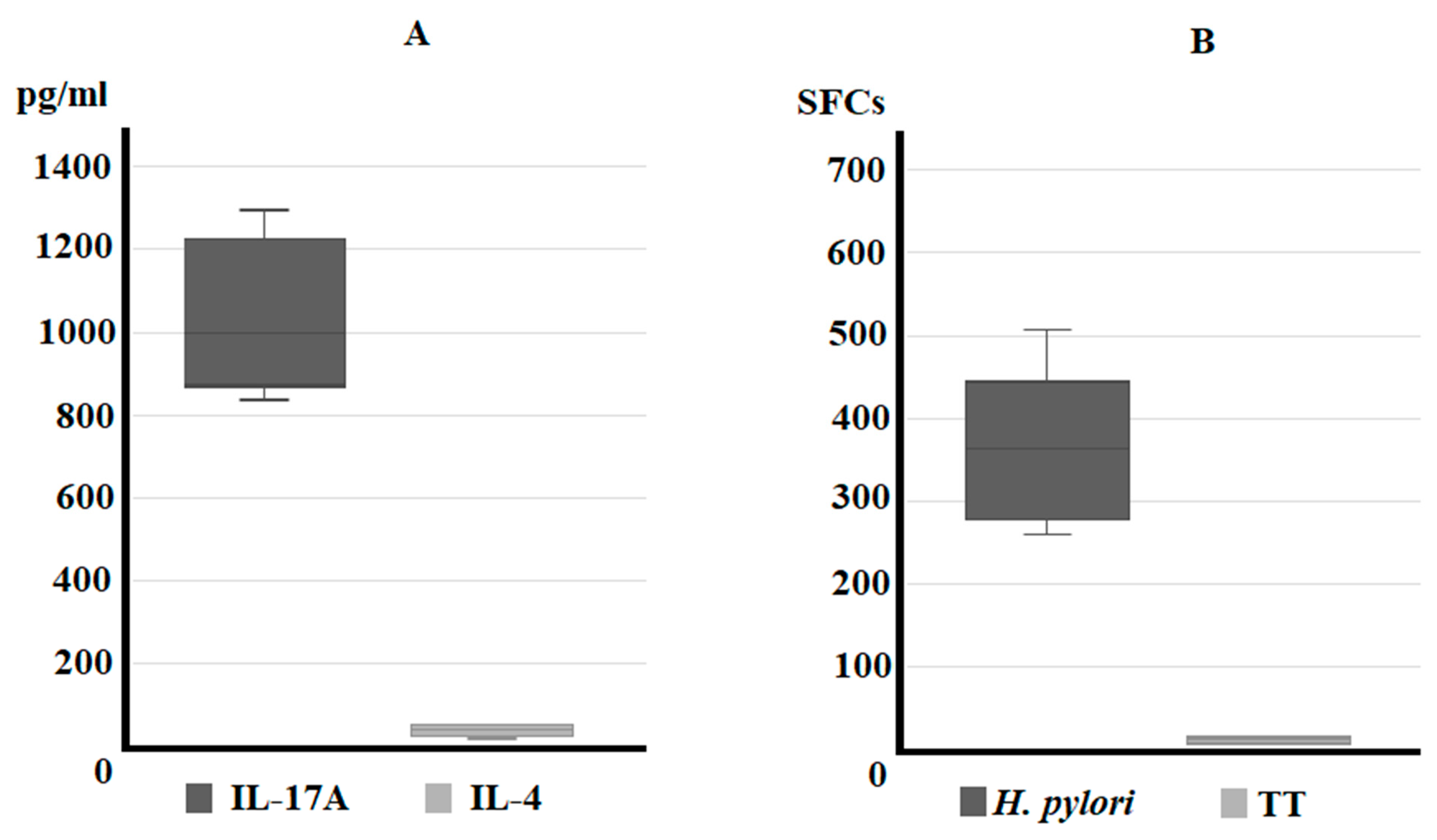
3.1. IL-17A Secretion by Gastric T Cells from Helicobacter pylori Patients with Gastric Intestinal Metaplasia and Dysplasia (IM/DYS)
3.2. Predominant T Helper 17 Phenotype of Helicobacter pylori-Specific CD4+ T Cell Clones Obtained from Helicobacter pylori Patients with Gastric Intestinal Metaplasia, and Gastric Dysplasia
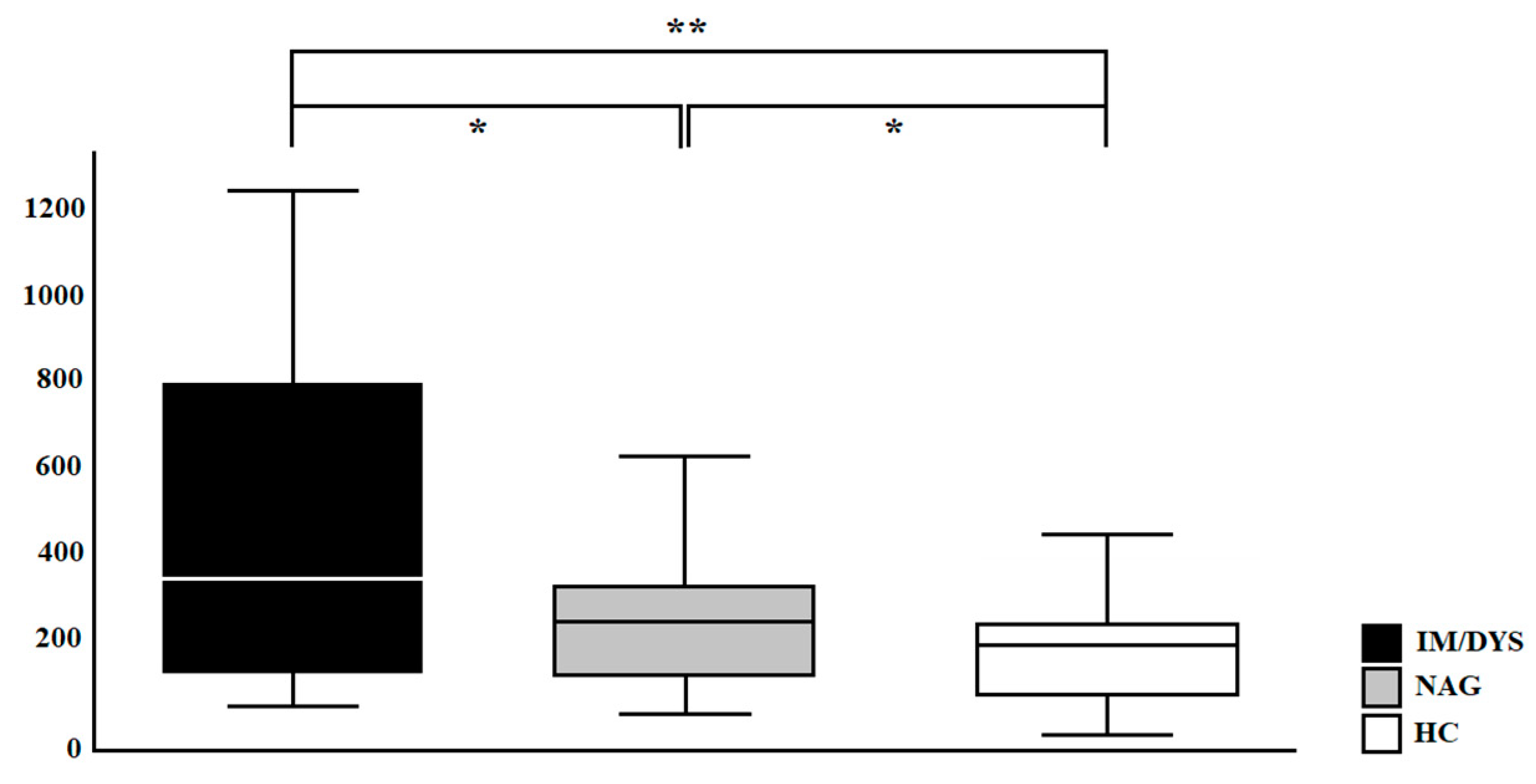
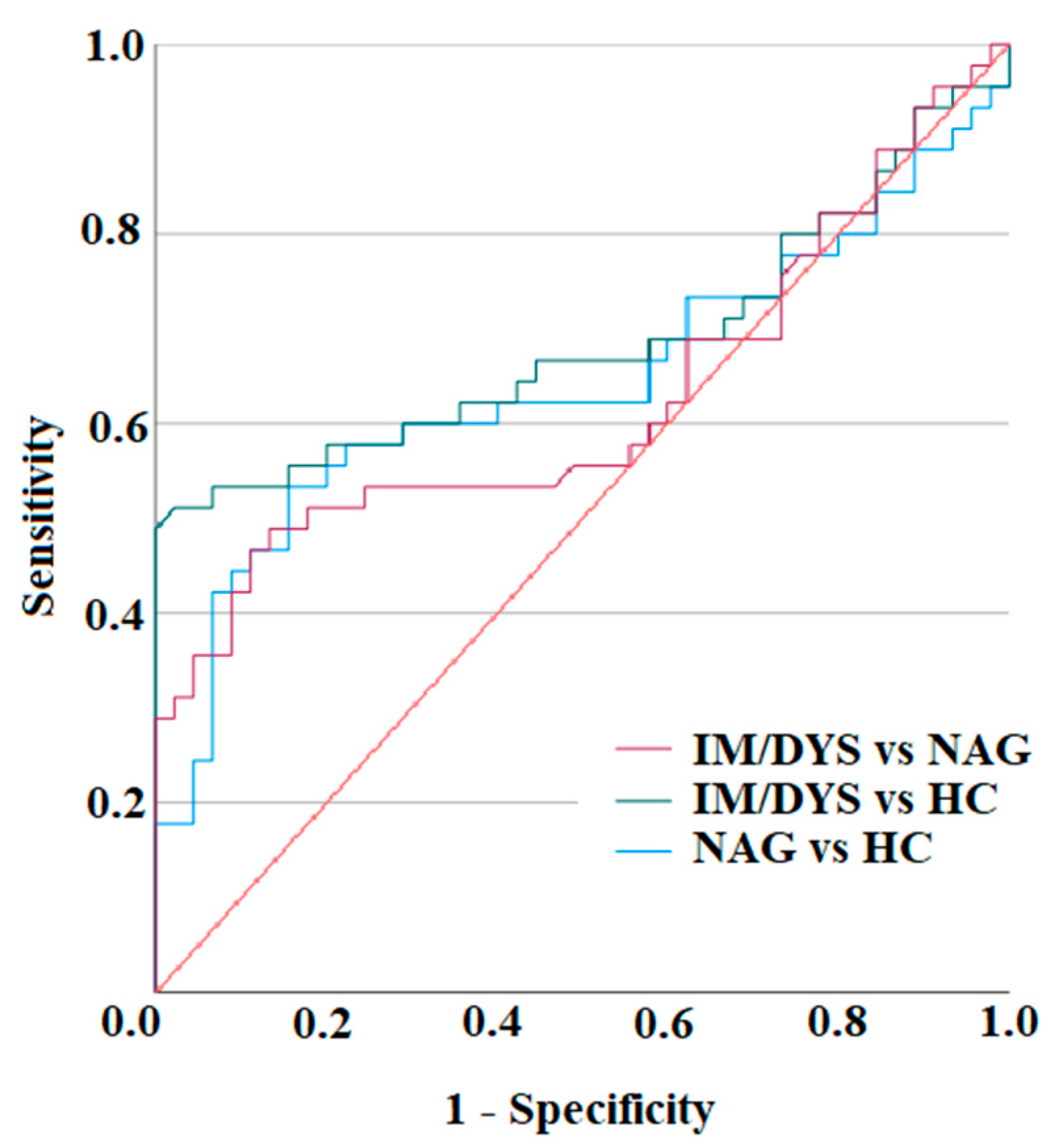
3.3. Serum IL-17A Levels Are Elevated in Sera of Helicobacter pylori Patients with Gastric Intestinal Metaplasia, and Gastric Dysplasia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marshall, B.; Warren, J. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 323, 1311–1315. [Google Scholar] [CrossRef]
- Robinson, K.; Atherton, J.C. The Spectrum of Helicobacter-Mediated Diseases. Annu. Rev. Pathol. 2021, 16, 123–144. [Google Scholar] [CrossRef]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Stemmermann, G.N.; Chyou, P.H.; Kato, I.; Perez-Perez, G.I.; Blaser, M.J. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 1991, 325, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef]
- Canzian, F.; Rizzato, C.; Stein, A.; Flores-Luna, L.; Camorlinga-Ponce, M.; Mendez-Tenorio, A.; Chen, W.; Kasamatsu, E.; Mercedes Bravo, M.; Torres, J.; et al. Phylogenetic origin of Helicobacter pylori pathogenicity island and risk of stomach cancer and high-grade premalignant gastric lesions. Eur. J. Cancer Prev. 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Wotherspoon, A.; Diss, T.; Pan, L.; Isaacson, P.; Doglioni, C.; Moschini, A.; De Boni, M. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993, 342, 575–577. [Google Scholar] [CrossRef]
- Floch, P.; Mégraud, F.; Lehours, P. Helicobacter pylori Strains and Gastric MALT Lymphoma. Toxins 2017, 9, 132. [Google Scholar] [CrossRef]
- Rossi, D.; Bertoni, F.; Zucca, E. Marginal-Zone Lymphomas. N. Engl. J. Med. 2022, 386, 568–581. [Google Scholar] [CrossRef]
- D’Elios, M.M.; Appelmelk, B.J.; Amedei, A.; Bergman, M.P.; Del Prete, G. Gastric autoimmunity: The role of Helicobacter pylori and molecular mimicry. Trends Mol. Med. 2004, 10, 316–323. [Google Scholar] [CrossRef]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241. [Google Scholar]
- Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kim, Y.I.; Kook, M.C.; Park, B.; Joo, J. Family history of gastric cancer and Helicobacter pylori treatment. N. Engl. J. Med. 2020, 382, 427–436. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Sipponen, P. Helicobacter pylori eradication for the prevention of gastric cancer. Helicobacter 2006, 11 (Suppl. S1), 52–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Dore, M.P.; Graham, D.Y. Diagnosis and Treatment of Helicobacter pylori Infection. Annu. Rev. Med. 2022, 73, 183–195. [Google Scholar] [CrossRef]
- Choi, I.J.; Kook, M.C.; Kim, Y.I.; Cho, S.J.; Lee, J.Y.; Kim, C.G.; Park, B.; Nam, B.H. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N. Engl. J. Med. 2018, 378, 1085–1095. [Google Scholar] [CrossRef]
- Kalali, B.; Mejías-Luque, R.; Javaheri, A.; Gerhard, M.H. pylori virulence factors: Influence on immune system and pathology. Mediators Inflamm. 2014, 2014, 426309. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.F.; Blaser, M.J. Mechanisms of disease: Inflammation and the origins of cancer. Nat. Clin. Pract. Oncol. 2005, 2, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Salio, M.; Galli, C.; Norais, N.; Rappuoli, R.; Lanzavecchia, A.; Montecucco, C. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J. Exp. Med. 1998, 187, 135–140. [Google Scholar] [CrossRef]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 2003, 301, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Boncristiano, M.; Paccani, S.R.; Barone, S.; Ulivieri, C.; Patrussi, L.; Ilver, D.; Amedei, A.; D’Elios, M.M.; Telford, J.L.; Baldari, C.T. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 2003, 198, 1887–1897. [Google Scholar] [CrossRef]
- Schmees, C.; Prinz, C.; Treptau, T.; Rad, R.; Hengst, L.; Voland, P.; Bauer, S.; Brenner, L.; Schmid, R.M.; Gerhard, M. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology 2007, 132, 1820–1833. [Google Scholar] [CrossRef]
- Mejías-Luque, R.; Gerhard, M. Immune Evasion Strategies and Persistence of Helicobacter pylori. Curr. Top. Microbiol. Immunol. 2017, 400, 53–71. [Google Scholar]
- Cadamuro, A.C.; Rossi, A.F.; Maniezzo, N.M.; Silva, A.E. Helicobacter pylori infection: Host immune response, implications on gene expression and microRNAs. World J. Gastroenterol. 2014, 20, 1424–1437. [Google Scholar] [CrossRef]
- Merrell, D.S.; Falkow, S. Frontal and stealth attack strategies in microbial pathogenesis. Nature 2004, 430, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Haenszel, W.; Cuello, C.; Tannenbaum, S.; Archer, M. A model for gastric cancer epidemiology. Lancet 1975, 2, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process–First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and epigenomic profiling of high-risk intestinal metaplasia reveals molecular determinants of progression to gastric cancer. Cancer Cell 2018, 33, 137–150.e5. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Corre, S.; Michel, V.; Le Luel, K.; Fernandes, J.; Ziveri, J.; Jouvion, G.; Danckaert, A.; Mouchet, N.; Da Silva Barreira, D.; et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut 2020, 69, 1582–1591. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, A.; Hu, C.; Zhou, Y.; Zhang, X.; Czajkowsky, D.M.; Li, J.; Cheng, S.; Shen, R.; Gu, J.; et al. Complex clonal mosaicism within microdissected intestinal metaplastic glands without concurrent gastric cancer. J. Med. Genet. 2016, 53, 643–646. [Google Scholar] [CrossRef]
- Touati, E.; Michel, V.; Thiberge, J.M.; Wuscher, N.; Huerre, M.; Labigne, A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003, 124, 1408–1419. [Google Scholar] [CrossRef]
- Dixon, B.R.E.A.; Hossain, R.; Patel, R.V.; Algood, H.M.S. Th17 Cells in Helicobacter pylori Infection: A Dichotomy of Help and Harm. Infect. Immun. 2019, 87, e00363-19. [Google Scholar] [CrossRef] [PubMed]
- Amedei, A.; Munari, F.; Bella, C.D.; Niccolai, E.; Benagiano, M.; Bencini, L.; Cianchi, F.; Farsi, M.; Emmi, G.; Zanotti, G.; et al. Helicobacter pylori secreted peptidyl prolyl cis, trans-isomerase drives Th17 inflammation in gastric adenocarcinoma. Intern. Emerg. Med. 2014, 9, 303–309. [Google Scholar] [CrossRef]
- Dewayani, A.; Fauzia, K.A.; Alfaray, R.I.; Waskito, L.A.; Doohan, D.; Rezkitha, Y.A.A.; Abdurachman, A.; Kobayashi, T.; I’tishom, R.; Yamaoka, Y.; et al. The Roles of IL-17, IL-21, and IL-23 in the Helicobacter pylori Infection and Gastrointesti-nal Inflammation: A Review. Toxins 2021, 13, 315. [Google Scholar] [CrossRef]
- Della Bella, C.; Antico, A.; Panozzo, M.P.; Capitani, N.; Petrone, L.; Benagiano, M.; D’Elios, S.; Sparano, C.; Azzurri, A.; Pratesi, S.; et al. Gastric Th17 Cells Specific for H+/K+-ATPase and Serum IL-17 Signature in Gastric Autoimmunity. Front. Immun. 2022, 13, 952674. [Google Scholar] [CrossRef] [PubMed]
- Capitani, N.; Codolo, G.; Vallese, F.; Minervini, G.; Grassi, A.; Cianchi, F.; Troilo, A.; Fischer, W.; Zanotti, G.; Baldari, C.T.; et al. The Lipoprotein HP1454 of Helicobacter pylori regulates T-Cell response by shaping T-cell receptor signalling. Cell Microbiol. 2019, 21, e13006. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, M.; Esmaeili, M.; Tashakoripour, M.; Mohagheghi, M.A.; Eshagh Hosseini, M.; Touati, E.; Vosough, M.; Mohammadi, M. Alteration in Serum Levels of Tumor Necrosis Factor Alpha is associated with Histopathologic Progression of Gastric Cancer. Iran Biomed. J. 2023, 27, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, C.; Antico, A.; Panozzo, M.P.; Capitani, N.; Benagiano, M.; Petrone, L.; Azzurri, A.; Pratesi, S.; D’Elios, S.; Cianchi, F.; et al. Elevated IL-19 serum levels in patients with pernicious anemia and autoimmune gastritis. Front. Immunol. 2022, 13, 887256. [Google Scholar] [CrossRef]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar] [CrossRef]
- Stolte, M.; Meining, A. The updated Sydney system: Classification and grading of gastritis as the basis of diagnosis and treatment. Can. J. Gastroenterol. 2001, 15, 591–598. [Google Scholar] [CrossRef]
- Rugge, M.; Pennelli, G.; Pilozzi, E.; Fassan, M.; Ingravallo, G.; Russo, V.M.; Di Mario, F. Gruppo Italiano Patologi Apparato Digerente (GIPAD); Società Italiana di Anatomia Patologica e Citopatologia Diagnostica/International Academy of Pathology, Italian division (SIAPEC/IAP). Gastritis: The histology report. Dig. Liver Dis. 2011, 43 (Suppl. S4), S373–S384. [Google Scholar] [CrossRef]
- Della Bella, C.; Corrà, A.; Mantengoli, E.; Galano, A.; Benagiano, M.; Bonciani, D.; Mariotti, E.B.; Pratesi, S.; Quintarelli, L.; Aimo, C.; et al. Skin IL-17A and IFN-γ production correlate with disease severity in patients with psoriasis and streptococcal infection. J. Investig. Dermatol. 2023, 12, S0022-202X(22)02899-8. [Google Scholar] [CrossRef]
- D’Elios, M.M.; Manghetti, M.; De Carli, M.; Costa, F.; Baldari, C.T.; Burroni, D.; Telford, J.L.; Romagnani, S.; Del Prete, G. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 1997, 158, 962–967. [Google Scholar] [CrossRef]
- D’Elios, M.M.; Amedei, A.; Manghetti, M.; Costa, F.; Baldari, C.T.; Quazi, A.S.; Telford, J.L.; Romagnani, S.; Del Prete, G. Impaired T-cell regulation of B-cell growth in Helicobacter pylori--related gastric low-grade MALT lymphoma. Gastroenterology 1999, 117, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, C.; Soluri, M.F.; Puccio, S.; Benagiano, M.; Grassi, A.; Bitetti, J.; Cianchi, F.; Sblattero, D.; Peano, C.; D’Elios, M.M. The Helicobacter pylori CagY Protein Drives Gastric Th1 and Th17 Inflammation and B Cell Proliferation in Gastric MALT Lymphoma. Int. J. Mol. Sci. 2021, 22, 9459. [Google Scholar] [CrossRef]
- D’Elios, M.M.; Manghetti, M.; Almerigogna, F.; Amedei, A.; Costa, F.; Burroni, D.; Baldari, C.T.; Romagnani, S.; Telford, J.L.; Del Prete, G. Different cytokine profile and antigen-specificity repertoire in Helicobacter pylori-specific T cell clones from the antrum of chronic gastritis patients with or without peptic ulcer. Eur. J. Immunol. 1997, 27, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Hornung, R.W.; Reed, L.D. Estimation of average concentration in the presence of non detectable. Values Appl. Occup. Environ. Hyg. 1990, 5, 46–51. [Google Scholar] [CrossRef]
- Swet, J.A. Measuring the accuracy of diagnostic systems. Science 1988, 240, 1285–1293. [Google Scholar] [CrossRef]
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Moyat, M.; Velin, D. Immune responses to Helicobacter pylori infection. World J. Gastroenterol. 2014, 20, 5583–5593. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Zhang, B.; Rong, G.; Wei, H.; Zhang, M.; Bi, J.; Ma, L.; Xue, X.; Wei, G.; Liu, X.; Fang, G. The prevalence of Th17 cells in patients with gastric cancer. Biochem. Biophys. Res. Commun. 2008, 374, 533–537. [Google Scholar] [CrossRef]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature 2001, 412, 99. [Google Scholar] [CrossRef]
- Luzza, F.; Parrello, T.; Monteleone, G.; Sebkova, L.; Romano, M.; Zarrilli, R.; Imeneo, M.; Pallone, F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J. Immunol. 2000, 165, 5332–5337. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, X.F.; Zhuang, Y.; Zhang, J.Y.; Liu, T.; Yin, Z.; Wu, C.; Mao, X.H.; Jia, K.R.; Wang, F.J.; et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J. Immunol. 2010, 184, 5121–5129. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, K.L.; Qin, L.; Ji, D.X.; Zhang, B.; Zheng, P.F.; Qin, Y.M. Notch signaling pathway regulates CD4+CD25+CD127dim/- regulatory T cells and T helper 17 cells function in gastric cancer patients. Biosci. Rep. 2019, 39, BSR20182044. [Google Scholar] [CrossRef] [PubMed]
- Suarez, G.; Romero-Gallo, J.; Piazuelo, M.B.; Sierra, J.C.; Delgado, A.G.; Washington, M.K.; Shah, S.C.; Wilson, K.T.; Peek, R.M., Jr. Nod1 imprints inflammatory and carcinogenic responses toward the gastric pathogen Helicobacter pylori. Cancer Res. 2019, 79, 1600–1611. [Google Scholar] [CrossRef]
- Nguyen, P.M.; Putoczki, T.L. Could the inhibition of IL-17 or IL-18 be a potential therapeutic opportunity for gastric cancer? Cytokine 2019, 118, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhu, S.; Dong, Q.; Zhang, S.; Ma, J.M.; Zhou, C. Expression of Th17/Treg related molecules in gastric cancer tissues. Turk. J. Gastroenterol. 2018, 29, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yue, R.; Zhao, P.; Yu, X.; Li, J.; Ma, G.; Tang, J.; Zhang, L.; Feng, L.; Sun, L.; et al. Proinflammatory follicular helper T cells promote immunoglobulin G secretion, suppress regulatory B cell development, and correlate with worse clinical outcomes in gastric cancer. Tumour. Biol. 2017, 39, 1010428317705747. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IM/DYS (N = 45) | NAG (N = 45) | HC (N = 45) | ||
|---|---|---|---|---|
| Age | <60 | 20 (44%) | 21 (47%) | 24 (53%) |
| ≥60 | 25 (56%) | 24 (53%) | 21 (47%) | |
| Sex | Female | 21 (47%) | 22 (49%) | 23 (51%) |
| Male | 24 (53%) | 23 (51%) | 22 (49%) | |
| Ethnicity | Caucasian | 45 (100%) | 45 (100%) | 45 (100%) |
| BMI (kg/m2) mean ± SD | 23.64 ± 2.61 | 23.67 ± 2.89 | 23.69 ± 2.77 | |
| Smoking habits | Never | 34 (75%) | 35 (77%) | 38 (84%) |
| Ever | 11 (25%) | 11 (23%) | 7 (16%) | |
| Alcohol use | Never/rare drinker | 31 (68%) | 32 (71%) | 37 (81%) |
| Drinker | 14 (32%) | 13 (29%) | 8 (19%) | |
| Family history of GC | No | 42 (93%) | 43 (95%) | 45 (100%) |
| Yes | 3 (7%) | 2 (5%) | 0 (0%) | |
| Pernicious anemia | No | 45 (100%) | 45 (100%) | 45 (100%) |
| Yes | 0 (0%) | 0 (0%) | 0 (0%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Bella, C.; D’Elios, S.; Coletta, S.; Benagiano, M.; Azzurri, A.; Cianchi, F.; de Bernard, M.; D’Elios, M.M. Increased IL-17A Serum Levels and Gastric Th17 Cells in Helicobacter pylori-Infected Patients with Gastric Premalignant Lesions. Cancers 2023, 15, 1662. https://doi.org/10.3390/cancers15061662
Della Bella C, D’Elios S, Coletta S, Benagiano M, Azzurri A, Cianchi F, de Bernard M, D’Elios MM. Increased IL-17A Serum Levels and Gastric Th17 Cells in Helicobacter pylori-Infected Patients with Gastric Premalignant Lesions. Cancers. 2023; 15(6):1662. https://doi.org/10.3390/cancers15061662
Chicago/Turabian StyleDella Bella, Chiara, Sofia D’Elios, Sara Coletta, Marisa Benagiano, Annalisa Azzurri, Fabio Cianchi, Marina de Bernard, and Mario Milco D’Elios. 2023. "Increased IL-17A Serum Levels and Gastric Th17 Cells in Helicobacter pylori-Infected Patients with Gastric Premalignant Lesions" Cancers 15, no. 6: 1662. https://doi.org/10.3390/cancers15061662
APA StyleDella Bella, C., D’Elios, S., Coletta, S., Benagiano, M., Azzurri, A., Cianchi, F., de Bernard, M., & D’Elios, M. M. (2023). Increased IL-17A Serum Levels and Gastric Th17 Cells in Helicobacter pylori-Infected Patients with Gastric Premalignant Lesions. Cancers, 15(6), 1662. https://doi.org/10.3390/cancers15061662