

Rapamycin-Induced Feedback Activation of eIF4E-EIF4A Dependent mRNA Translation in Pancreatic Cancer


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Ribosome Footprinting Identifies Translational Targets of mTOR and RPS6 (S6)
2.2. Rapamycin Inhibits the Translation of Genes Involved in the Cell Cycle and Cancer Cell Growth
2.3. Rapamycin Induces Translation of a Subset of mRNAs
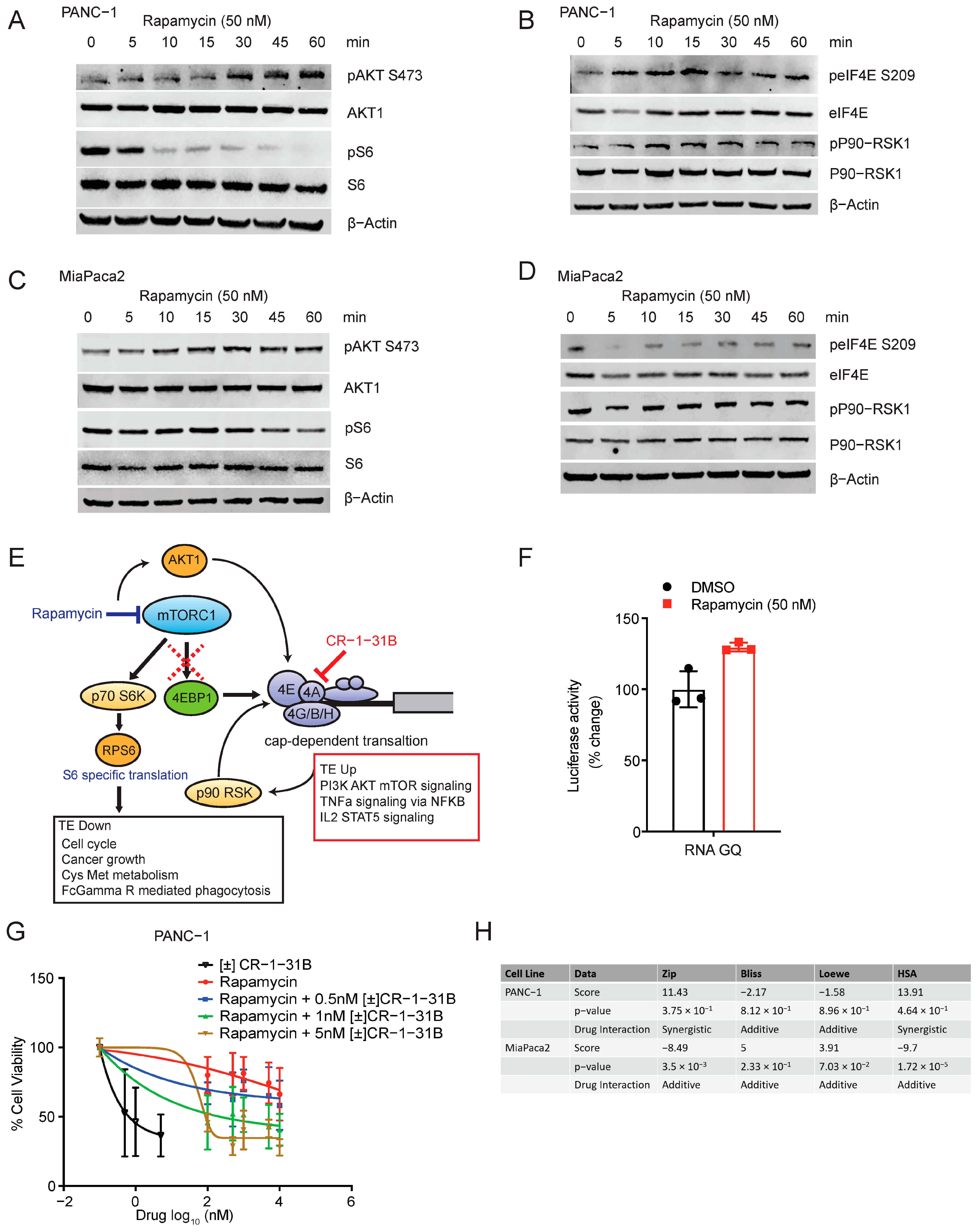
2.4. Rapamycin Activates Translation through Feedback Activation of AKT1 and eIF4E
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Ribosome Footprinting
4.3. Sequence Alignment
4.4. Footprint Profile Analysis
4.5. Metagene Analysis
4.6. Global mRNA Translation
4.7. Cell Viability Assay
4.8. Clonogenic Survival Assay
4.9. Immunoblotting
4.10. Luciferase Reporter Assay
4.11. Statistical Analysis
4.12. Online Content
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signaling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Kawasome, H.; Papst, P.; Webb, S.; Keller, G.M.; Johnson, G.L.; Gelfand, E.W.; Terada, N. Targeted disruption of p70s6k defines its role in protein synthesis and rapamycin sensitivity. Proc. Natl. Acad. Sci. USA 1998, 95, 5033–5038. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef]
- Mieulet, V.; Roceri, M.; Espeillac, C.; Sotiropoulos, A.; Ohanna, M.; Oorschot, V.; Klumperman, J.; Sandri, M.; Pende, M. S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am. J. Physiol. Cell Physiol. 2007, 293, C712–C722. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. [Google Scholar] [CrossRef]
- Rutkovsky, A.C.; Yeh, E.S.; Guest, S.T.; Findlay, V.J.; Muise-Helmericks, R.C.; Armeson, K.; Ethier, S.P. Eukaryotic initiation factor 4E-binding protein as an oncogene in breast cancer. BMC Cancer 2019, 19, 491. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef]
- Cui, J.; Guo, Y.; Wu, H.; Xiong, J.; Peng, T. Everolimus regulates the activity of gemcitabine-resistant pancreatic cancer cells by targeting the Warburg effect via PI3K/AKT/mTOR signaling. Mol. Med. 2021, 27, 38. [Google Scholar] [CrossRef]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Martineau, Y.; Azar, R.; Müller, D.; Lasfargues, C.; El Khawand, S.; Anesia, R.; Pelletier, J.; Bousquet, C.; Pyronnet, S. Pancreatic tumors escape from translational control through 4E-BP1 loss. Oncogene 2014, 33, 1367–1374. [Google Scholar] [CrossRef]
- Bohlen, J.; Roiuk, M.; Teleman, A.A. Phosphorylation of ribosomal protein S6 differentially affects mRNA translation based on ORF length. Nucleic Acids Res. 2021, 49, 13062–13074. [Google Scholar] [CrossRef]
- Marques-Ramos, A.; Candeias, M.M.; Menezes, J.; Lacerda, R.; Willcocks, M.; Teixeira, A.; Locker, N.; Romao, L. Cap-independent translation ensures mTOR expression and function upon protein synthesis inhibition. RNA 2017, 23, 1712–1728. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]
- Siddiqui, N.; Sonenberg, N. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015, 43, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signaling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef]
- Chen, X.G.; Liu, F.; Song, X.F.; Wang, Z.H.; Dong, Z.Q.; Hu, Z.Q.; Lan, R.Z.; Guan, W.; Zhou, T.G.; Xu, X.M.; et al. Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Mol. Carcinog. 2010, 49, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Topisirovic, I. Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell. Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef]
- Wolfe, A.L.; Singh, K.; Zhong, Y.; Drewe, P.; Rajasekhar, V.K.; Sanghvi, V.R.; Mavrakis, K.J.; Jiang, M.; Roderick, J.E.; Van der Meulen, J.; et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 2014, 513, 65–70. [Google Scholar] [CrossRef]
- Zhao, Y.; Schoeps, B.; Yao, D.; Zhang, Z.; Schuck, K.; Tissen, V.; Jager, C.; Schlitter, A.M.; van der Kammen, R.; Ludwig, C.; et al. mTORC1 and mTORC2 Converge on the Arp2/3 Complex to Promote Kras(G12D)-Induced Acinar-to-Ductal Metaplasia and Early Pancreatic Carcinogenesis. Gastroenterology 2021, 160, 1755–1770.e17. [Google Scholar] [CrossRef]
- Wendel, H.G.; Silva, R.L.; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007, 21, 3232–3237. [Google Scholar] [CrossRef]
- Mohan, P.; Pasion, J.; Ciriello, G.; Lailler, N.; de Stanchina, E.; Viale, A.; van den Berg, A.; Diepstra, A.; Wendel, H.G.; Sanghvi, V.R.; et al. Frequent 4EBP1 Amplification Induces Synthetic Dependence on FGFR Signaling in Cancer. Cancers 2022, 14, 2397. [Google Scholar] [CrossRef]
- Romagnoli, A.; Maracci, C.; D’Agostino, M.; Teana, A.; Marino, D.D. Targeting mTOR and eIF4E: A feasible scenario in ovarian cancer therapy. Cancer Drug Resist. 2021, 4, 596–606. [Google Scholar] [CrossRef]
- Pourdehnad, M.; Truitt, M.L.; Siddiqi, I.N.; Ducker, G.S.; Shokat, K.M.; Ruggero, D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 2013, 110, 11988–11993. [Google Scholar] [CrossRef]
- Schmitz, J.C.; Liu, J.; Lin, X.; Chen, T.M.; Yan, W.; Tai, N.; Gollerkeri, A.; Chu, E. Translational regulation as a novel mechanism for the development of cellular drug resistance. Cancer Metastasis Rev. 2001, 20, 33–41. [Google Scholar] [CrossRef]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef]
- Singh, K.; Lin, J.; Lecomte, N.; Mohan, P.; Gokce, A.; Sanghvi, V.R.; Jiang, M.; Grbovic-Huezo, O.; Burcul, A.; Stark, S.G.; et al. Targeting eIF4A-Dependent Translation of KRAS Signaling Molecules. Cancer Res. 2021, 81, 2002–2014. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef]
- Zhong, Y.; Karaletsos, T.; Drewe, P.; Sreedharan, V.T.; Kuo, D.; Singh, K.; Wendel, H.G.; Ratsch, G. RiboDiff: Detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics 2017, 33, 139–141. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Zheng, S.; Aldahdooh, J.; Shadbahr, T.; Wang, Y.; Aldahdooh, D.; Bao, J.; Wang, W.; Tang, J. DrugComb update: A more comprehensive drug sensitivity data repository and analysis portal. Nucleic Acids Res. 2021, 49, W174–W184. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.U.; Hector, H.; Pederson, E.N.; Lin, J.; Ouyang, Z.; Wendel, H.-G.; Singh, K. Rapamycin-Induced Feedback Activation of eIF4E-EIF4A Dependent mRNA Translation in Pancreatic Cancer. Cancers 2023, 15, 1444. https://doi.org/10.3390/cancers15051444
Nguyen TU, Hector H, Pederson EN, Lin J, Ouyang Z, Wendel H-G, Singh K. Rapamycin-Induced Feedback Activation of eIF4E-EIF4A Dependent mRNA Translation in Pancreatic Cancer. Cancers. 2023; 15(5):1444. https://doi.org/10.3390/cancers15051444
Chicago/Turabian StyleNguyen, Trang Uyen, Harrison Hector, Eric Nels Pederson, Jianan Lin, Zhengqing Ouyang, Hans-Guido Wendel, and Kamini Singh. 2023. "Rapamycin-Induced Feedback Activation of eIF4E-EIF4A Dependent mRNA Translation in Pancreatic Cancer" Cancers 15, no. 5: 1444. https://doi.org/10.3390/cancers15051444
APA StyleNguyen, T. U., Hector, H., Pederson, E. N., Lin, J., Ouyang, Z., Wendel, H.-G., & Singh, K. (2023). Rapamycin-Induced Feedback Activation of eIF4E-EIF4A Dependent mRNA Translation in Pancreatic Cancer. Cancers, 15(5), 1444. https://doi.org/10.3390/cancers15051444