

Dominant Gene Expression Profiles Define Adenoid Cystic Carcinoma (ACC) from Different Tissues: Validation of a Gene Signature Classifier for Poor Survival in Salivary Gland ACC

 , , , ,
, , , , 
 ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Salivary Gland ACC Samples
2.2. RNA Isolation and Sequencing
2.3. Data Analysis
2.4. Unsupervised Hierarchical Clustering
2.5. Statistical Analysis
2.6. Development and Evaluation of Prognostic Classifiers
3. Results
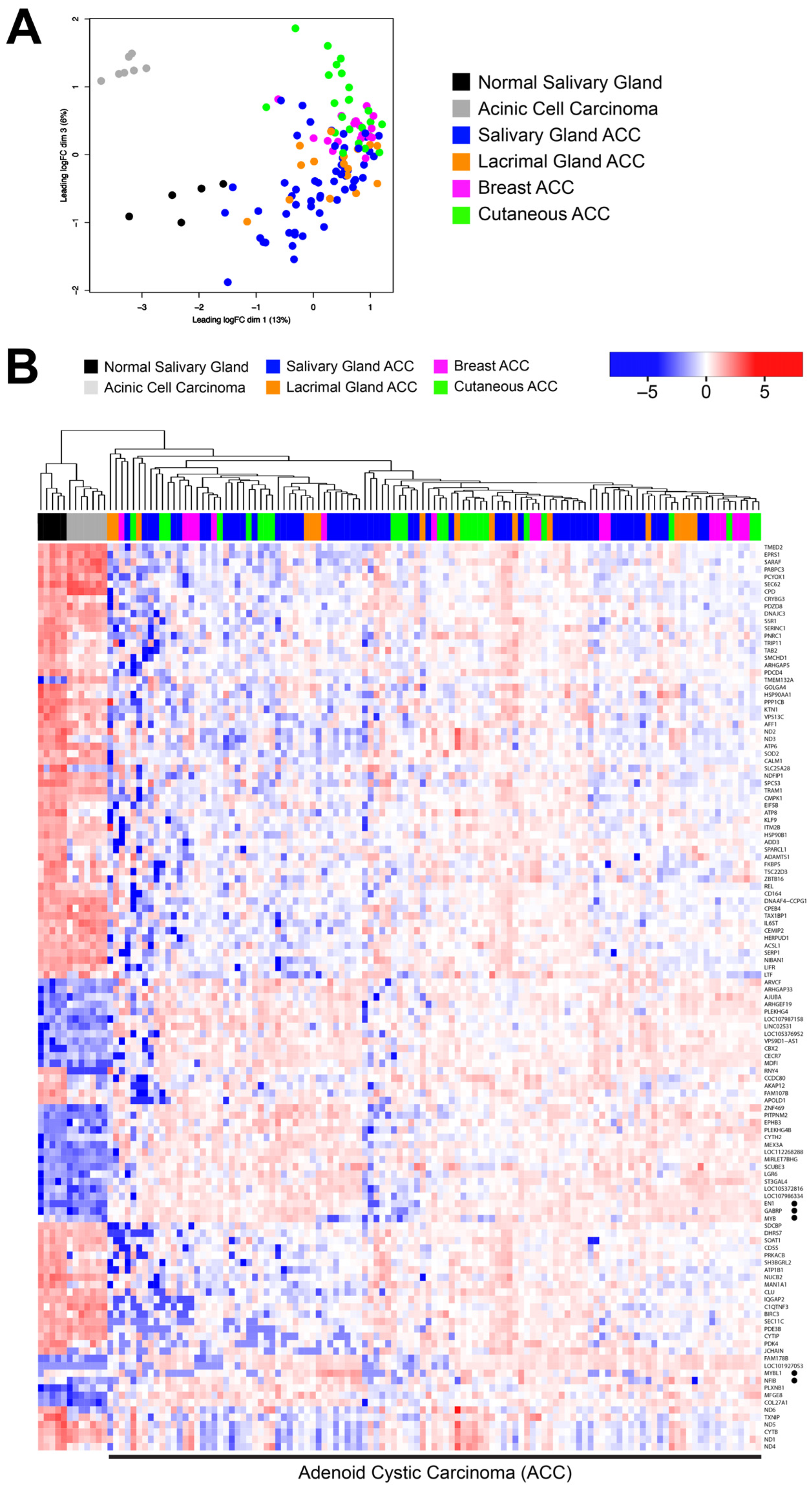
3.1. ACC Tumors from Different Organ Sites Share a Common Transcriptional Profile
3.2. Many ACC Tumors Fail to Express Tissue-Specific Gene Expression Markers
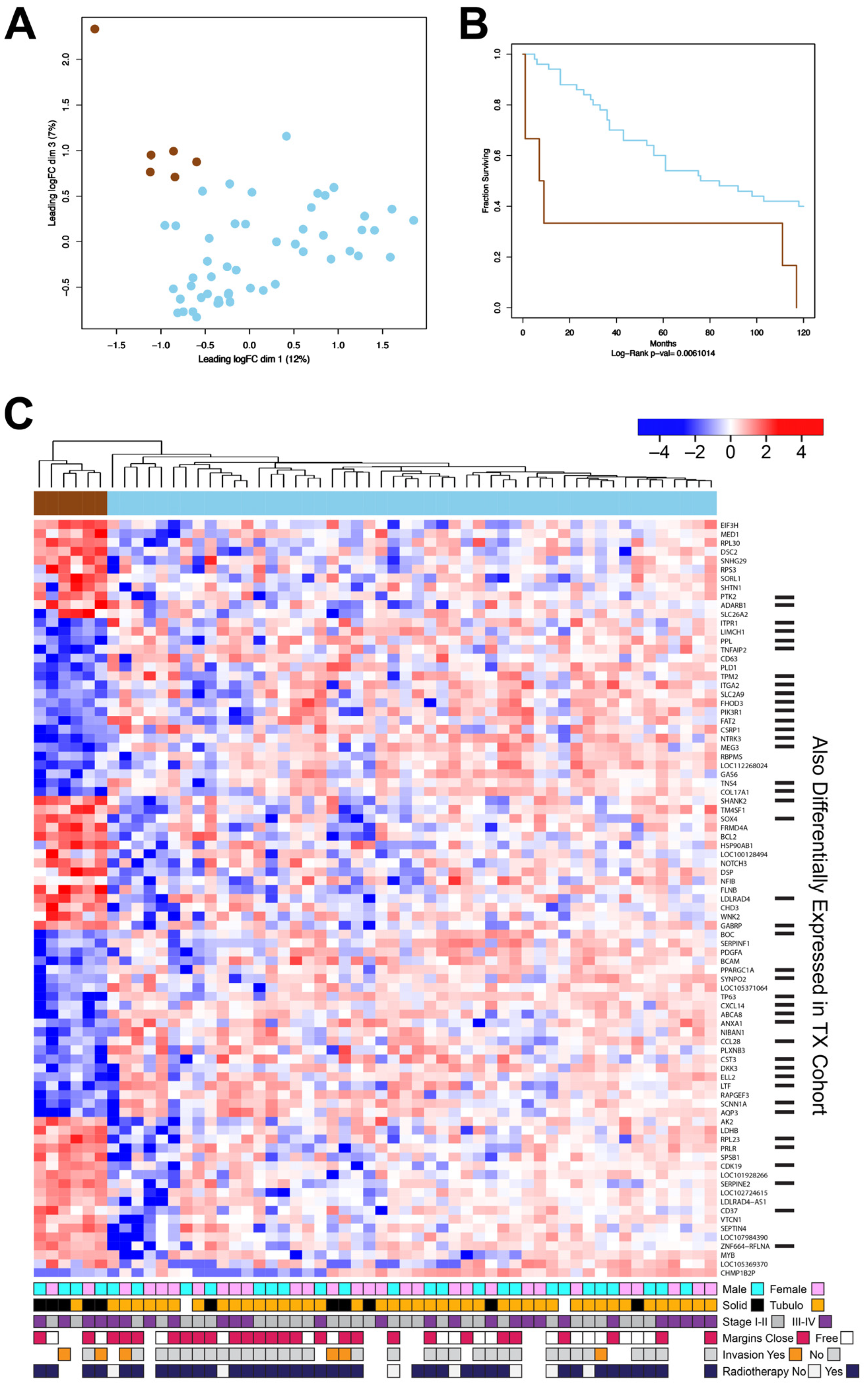
3.3. The New DK Cohort of ACC Tumors Also Contains a Poor-Survival Subgroup of Patients
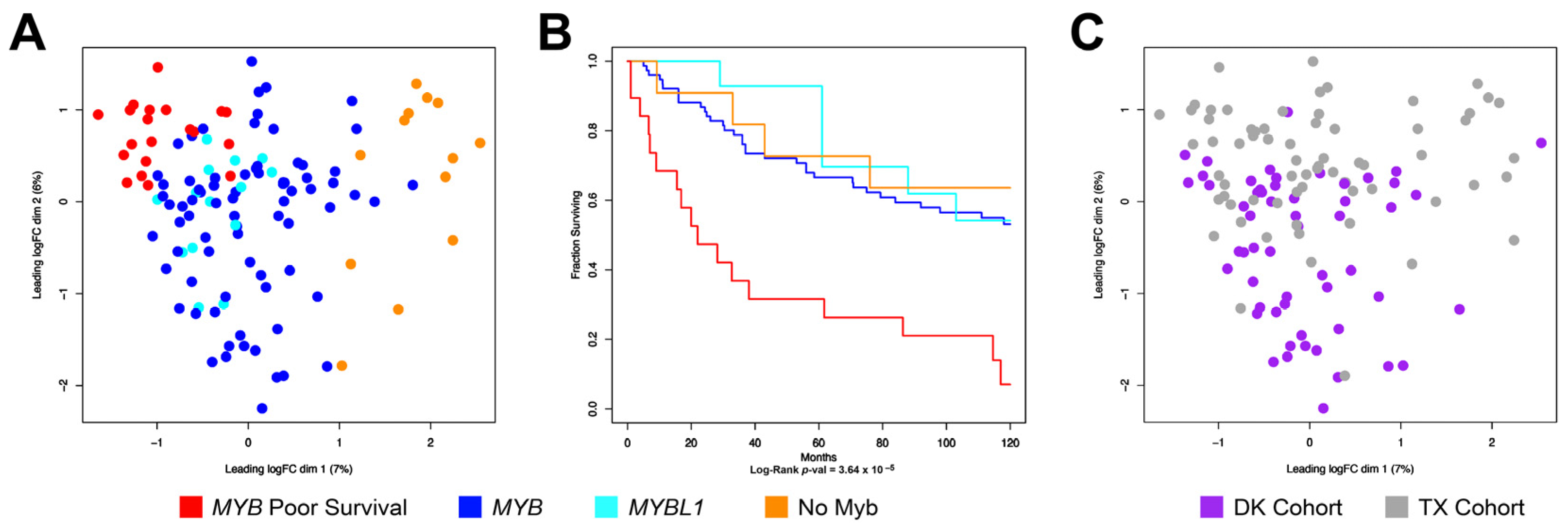
3.4. Combining the TX and DK Cohorts Provides Additional Details about Subgroups of ACC Tumors
3.5. ACC Tumors That Do Not Express MYB or MYBL1 Have a Unique Transcription Profile
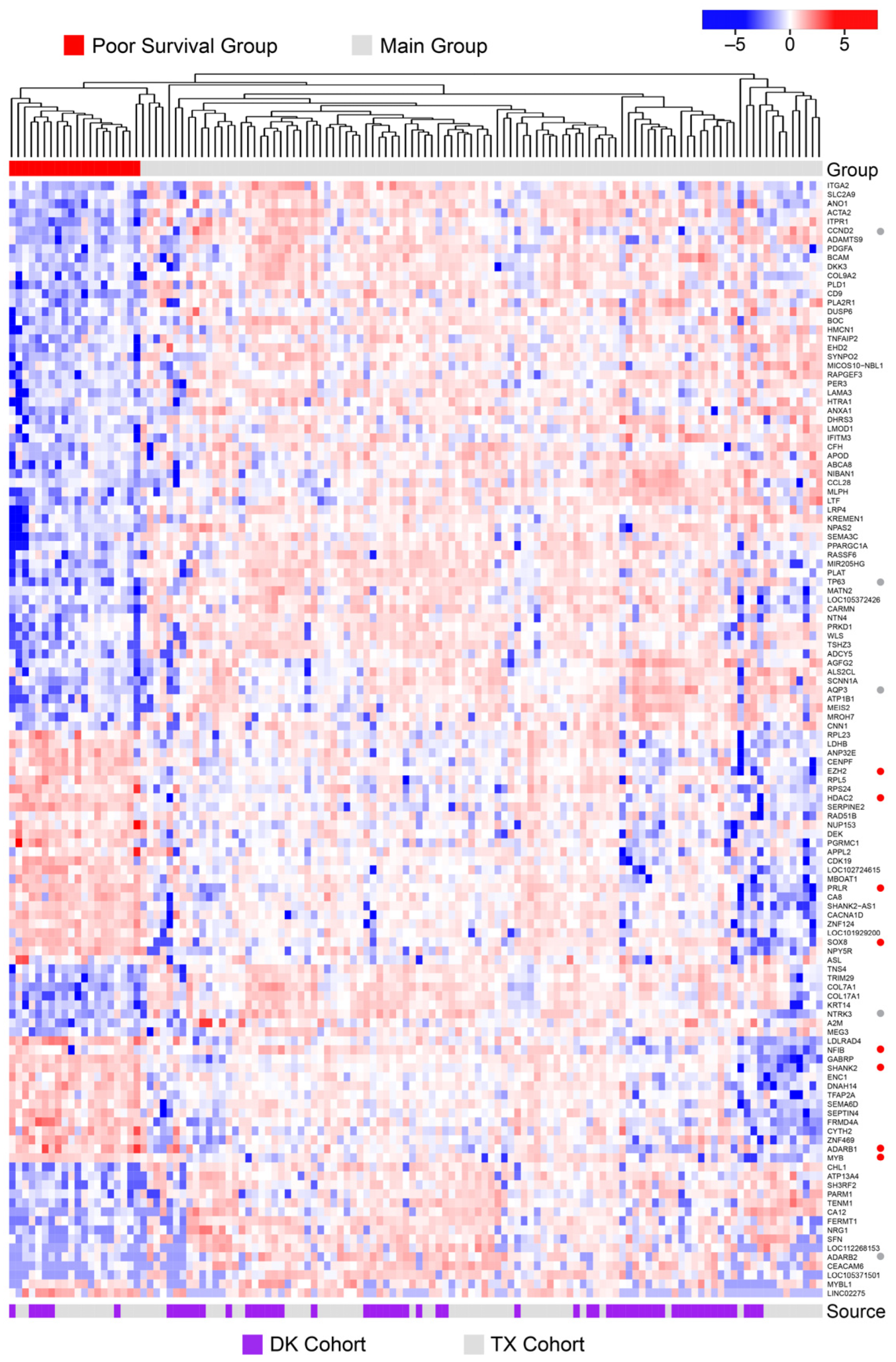
3.6. Poor Survival ACC Samples Have a Unique Transcription Profile
3.7. A Multi-Gene Classifier to Identify Poor Survival Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Li, J.; Rao, P.H.; Zhao, Y.J.; Bell, D.; Lippman, S.M.; Weber, R.S.; Caulin, C.; El-Naggar, A.K. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin. Cancer Res. 2010, 16, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Roberts, D.; Karpowicz, M.; Hanna, E.Y.; Weber, R.S.; El-Naggar, A.K. Clinical significance of Myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer Biol. Ther. 2011, 12, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Brill, L.B., II; Kanner, W.A.; Fehr, A.; Andren, Y.; Moskaluk, C.A.; Loning, T.; Stenman, G.; Frierson, H.F., Jr. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod. Pathol. 2011, 24, 1169–1176. [Google Scholar] [CrossRef]
- Brayer, K.J.; Frerich, C.A.; Kang, H.; Ness, S.A. Recurrent Fusions in MYB and MYBL1 Define a Common, Transcription Factor-Driven Oncogenic Pathway in Salivary Gland Adenoid Cystic Carcinoma. Cancer Discov. 2016, 6, 176–187. [Google Scholar] [CrossRef]
- Frerich, C.A.; Brayer, K.J.; Painter, B.M.; Kang, H.; Mitani, Y.; El-Naggar, A.; Ness, S.A. Transcriptomes define distinct subgroups of salivary gland adenoid cystic carcinoma with different driver mutations and outcomes. Oncotarget 2018, 9, 7341–7358. [Google Scholar] [CrossRef]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef]
- Ness, S.A. The myb oncoprotein: Regulating a regulator. Biochim. Biophys. Acta 1996, 1288, F123–F139. [Google Scholar] [CrossRef]
- Zhou, Y.; Ness, S.A. Myb proteins: Angels and demons in normal and transformed cells. Front. Biosci. 2011, 16, 1109–1131. [Google Scholar] [CrossRef]
- Frerich, C.A.; Sedam, H.N.; Kang, H.; Mitani, Y.; El-Naggar, A.K.; Ness, S.A. N-Terminal Truncated Myb with New Transcriptional Activity Produced Through Use of an Alternative MYB Promoter in Salivary Gland Adenoid Cystic Carcinoma. Cancers 2019, 12, 45. [Google Scholar] [CrossRef]
- Lei, W.; Rushton, J.J.; Davis, L.M.; Liu, F.; Ness, S.A. Positive and negative determinants of target gene specificity in Myb transcription factors. J. Biol. Chem. 2004, 279, 29519–29527. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lei, W.; O’Rourke, J.P.; Ness, S.A. Oncogenic mutations cause dramatic, qualitative changes in the transcriptional activity of c-Myb. Oncogene 2006, 25, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Rushton, J.J.; Davis, L.M.; Lei, W.; Mo, X.; Leutz, A.; Ness, S.A. Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene 2003, 22, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Fuglerud, B.M.; Ledsaak, M.; Rogne, M.; Eskeland, R.; Gabrielsen, O.S. The pioneer factor activity of c-Myb involves recruitment of p300 and induction of histone acetylation followed by acetylation-induced chromatin dissociation. Epigenet. Chromatin 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Abraham, B.J.; Berezovskaya, A.; Farah, N.; Liu, Y.; Leon, T.; Fielding, A.; Tan, S.H.; Sanda, T.; Weintraub, A.S.; et al. APOBEC signature mutation generates an oncogenic enhancer that drives LMO1 expression in T-ALL. Leukemia 2017, 31, 2057–2064. [Google Scholar] [CrossRef]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef]
- Frierson, H.F., Jr.; Moskaluk, C.A. Mutation signature of adenoid cystic carcinoma: Evidence for transcriptional and epigenetic reprogramming. J. Clin. Investig. 2013, 123, 2783–2785. [Google Scholar] [CrossRef]
- Introna, M.; Golay, J.; Frampton, J.; Nakano, T.; Ness, S.; Graf, T. Mutations in v-myb alter the differentiation of myelomonocytic cells transformed by the oncogene. Cell 1990, 63, 1289–1297. [Google Scholar] [CrossRef]
- Ness, S.A.; Beug, H.; Graf, T. v-myb dominance over v-myc in doubly transformed chick myelomonocytic cells. Cell 1987, 51, 41–50. [Google Scholar] [CrossRef]
- Ness, S.A.; Marknell, Å.; Graf, T. The v-myb oncogene product binds to and activates the promyelocyte-specific mim-1 gene. Cell 1989, 59, 1115–1125. [Google Scholar] [CrossRef]
- McNagny, K.M.; Graf, T. E26 leukemia virus converts primitive erythroid cells into cycling multilineage progenitors. Blood 2003, 101, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Duan, M.; Guo, F.; Tang, J.; Oybamiji, O.; Yu, H.; Ness, S.; Zhao, Y.Y.; Mao, P.; Guo, Y. SMDB: Pivotal somatic sequence alterations reprogramming regulatory cascades. NAR Cancer 2020, 2, zcaa030. [Google Scholar] [CrossRef] [PubMed]
- Kulessa, H.; Frampton, J.; Graf, T. GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes Dev. 1995, 9, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Lemma, R.B.; Ledsaak, M.; Fuglerud, B.M.; Sandve, G.K.; Eskeland, R.; Gabrielsen, O.S. Chromatin occupancy and target genes of the haematopoietic master transcription factor MYB. Sci. Rep. 2021, 11, 9008. [Google Scholar] [CrossRef]
- Coca-Pelaz, A.; Rodrigo, J.P.; Bradley, P.J.; Vander Poorten, V.; Triantafyllou, A.; Hunt, J.L.; Strojan, P.; Rinaldo, A.; Haigentz, M., Jr.; Takes, R.P.; et al. Adenoid cystic carcinoma of the head and neck--An update. Oral Oncol. 2015, 51, 652–661. [Google Scholar] [CrossRef]
- Brown, R.B.; Madrid, N.J.; Suzuki, H.; Ness, S.A. Optimized approach for Ion Proton RNA sequencing reveals details of RNA splicing and editing features of the transcriptome. PLoS ONE 2017, 12, e0176675. [Google Scholar] [CrossRef]
- Feeney, L.; Hapuarachi, B.; Adderley, H.; Rack, S.; Morgan, D.; Walker, R.; Rauch, R.; Herz, E.; Kaye, J.; Harrington, K.; et al. Clinical disease course and survival outcomes following disease recurrence in adenoid cystic carcinoma with and without NOTCH signaling pathway activation. Oral Oncol. 2022, 133, 106028. [Google Scholar] [CrossRef]
- Lee, D.Y.; Brayer, K.J.; Mitani, Y.; Burns, E.A.; Rao, A.; Bell, D.; Williams, M.D.; Ferrarotto, R.; Pytynia, K.B.; El-Naggar, A.K.; et al. Oncogenic Orphan Nuclear Receptor NR4A3 Interacts and Cooperates with MYB in Acinic Cell Carcinoma. Cancers 2020, 12, E2433. [Google Scholar] [CrossRef]
- Akbaba, S.; Bostel, T.; Lang, K.; Bahadir, S.; Lipman, D.; Schmidberger, H.; Matthias, C.; Rotter, N.; Knopf, A.; Freudlsperger, C.; et al. Large German Multicenter Experience on the Treatment Outcome of 207 Patients With Adenoid Cystic Carcinoma of the Major Salivary Glands. Front. Oncol. 2020, 10, 593379. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Mitani, Y.; Diao, L.; Guijarro, I.; Wang, J.; Zweidler-McKay, P.; Bell, D.; William, W.N., Jr.; Glisson, B.S.; Wick, M.J.; et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J. Clin. Oncol. 2017, 35, 352–360. [Google Scholar] [CrossRef]
- Davis, M.P.; van Dongen, S.; Abreu-Goodger, C.; Bartonicek, N.; Enright, A.J. Kraken: A set of tools for quality control and analysis of high-throughput sequence data. Methods 2013, 63, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Andreasen, S.; Tan, Q.; Agander, T.K.; Steiner, P.; Bjorndal, K.; Hogdall, E.; Larsen, S.R.; Erentaite, D.; Olsen, C.H.; Ulhoi, B.P.; et al. Adenoid cystic carcinomas of the salivary gland, lacrimal gland, and breast are morphologically and genetically similar but have distinct microRNA expression profiles. Mod. Pathol. 2018, 31, 1211–1225. [Google Scholar] [CrossRef]
- Morita, N.; Murase, T.; Ueda, K.; Nagao, T.; Kusafuka, K.; Nakaguro, M.; Urano, M.; Taguchi, K.I.; Yamamoto, H.; Kano, S.; et al. Pathological evaluation of tumor grade for salivary adenoid cystic carcinoma: A proposal of an objective grading system. Cancer Sci. 2021, 112, 1184–1195. [Google Scholar] [CrossRef]
- Xuan, L.; Yuan, J.; Zhang, H.; Zhang, Y.; Liu, H. Dominant cell type analysis predicts head and neck adenoid cystic carcinoma outcomes. Ann. Diagn. Pathol. 2022, 56, 151867. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Cheung, R.; Xu, H.; Jin, X.; Tian, W.; Pinney, K.; Bu, L.; Stone, S.; Woodward, R.N.; Agrawal, N.; Dholakia, S.; et al. Validation of a gene expression signature to measure immune quiescence in kidney transplant recipients in the CLIA setting. Biomark. Med. 2022, 16, 647–661. [Google Scholar] [CrossRef]
- Morris, J.S.; Luthra, R.; Liu, Y.; Duose, D.Y.; Lee, W.; Reddy, N.G.; Windham, J.; Chen, H.; Tong, Z.; Zhang, B.; et al. Development and Validation of a Gene Signature Classifier for Consensus Molecular Subtyping of Colorectal Carcinoma in a CLIA-Certified Setting. Clin. Cancer Res. 2021, 27, 120–130. [Google Scholar] [CrossRef]
- Rusch, M.; Nakitandwe, J.; Shurtleff, S.; Newman, S.; Zhang, Z.; Edmonson, M.N.; Parker, M.; Jiao, Y.; Ma, X.; Liu, Y.; et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun. 2018, 9, 3962. [Google Scholar] [CrossRef] [PubMed]
- Rettig, E.M.; Tan, M.; Ling, S.; Yonescu, R.; Bishop, J.A.; Fakhry, C.; Ha, P.K. MYB rearrangement and clinicopathologic characteristics in head and neck adenoid cystic carcinoma. Laryngoscope 2015, 125, E292–E299. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Number | Form | Gender | Stage | Age at Surgery | Source | Oncogene * |
|---|---|---|---|---|---|---|---|
| New Cohort (DK) | |||||||
| Breast | 17 | NA | NA | NA | NA | DK | MYB: 16 MYBL1: 1 |
| Cutaneous | 24 | NA | NA | NA | NA | DK | MYB: 17 MYBL1: 3 No_MYB: 4 |
| Lacrimal | 16 | NA | NA | NA | NA | DK: 10 FL: 6 | MYB: 11 MYBL1: 2 No_MYB: 3 |
| Salivary | 56 | Solid: 11 Tubulocribiform: 43 NA: 2 | F: 29 M: 27 | Stage I-II: 30 Stage III-IV: 26 | Age: 32–87 NA: 17 | DK | MYB: 47 MYBL1: 7 No_MYB: 2 |
| Cohort Total | 113 | MYB: 91 MYBL1: 13 No_MYB: 9 | |||||
| Previous Cohort (TX, Frerich et al. 2018 [6]) | |||||||
| Salivary | 68 | NA | F: 30 M: 38 | NA | NA | TX | MYB: 52 MYBL1: 7 No_MYB: 9 |
| Combined | 181 | MYB: 143 MYBL1: 20 No_MYB: 18 | |||||
| DE Genes | Top 10 Up-Regulated | FC | Adj p-Value | Top 10 Down-Regulated | FC | Adj p-Value |
|---|---|---|---|---|---|---|
| 881 | TG | 136.26 | 1.41 × 10−32 | LINC01833 | 0.02 | 1.39 × 10−5 |
| HMGA2 | 42.11 | 1.66 × 10−14 | MUC7 | 0.03 | 1.25 × 10−2 | |
| IGFN1 | 14.91 | 7.41 × 10−8 | LOC643201 | 0.03 | 2.04 × 10−3 | |
| LYZ | 12.38 | 4.19 × 10−6 | MUC19 | 0.03 | 1.93 × 10−3 | |
| FLG2 | 12.08 | 5.87 × 10−5 | CTNND2 | 0.04 | 6.12 × 10−5 | |
| COL1A1 | 9.62 | 9.72 × 10−20 | FIRRE | 0.04 | 4.73 × 10−10 | |
| FLG | 8.80 | 5.38 × 10−6 | LOC105378521 | 0.04 | 8.12 × 10−6 | |
| LTF | 8.05 | 1.37 × 10−6 | ART3 | 0.04 | 7.41 × 10−8 | |
| HSPB8 | 7.97 | 5.59 × 10−6 | LOC107984390 | 0.04 | 1.42 × 10−11 | |
| TNNT1 | 7.74 | 2.29 × 10−3 | SIX3 | 0.04 | 5.12 × 10−4 |
| GO.ID | Term | Annotated | Signif | Expected | Adj p-Value |
|---|---|---|---|---|---|
| GO:0043062 | extracellular structure organization | 218 | 87 | 31.6 | 2.40 × 10−19 |
| GO:0045229 | external encapsulating structure organization | 219 | 87 | 31.74 | 3.50 × 10−19 |
| GO:0045765 | regulation of angiogenesis | 128 | 47 | 18.55 | 2.40 × 10−10 |
| GO:0022610 | biological adhesion | 658 | 166 | 95.37 | 3.10 × 10−10 |
| GO:0008284 | positive regulation of cell population prolif. | 354 | 93 | 51.31 | 7.80 × 10−8 |
| GO:0009617 | response to bacterium | 178 | 54 | 25.8 | 2.10 × 10−7 |
| DE Genes | Top 10 Up-Regulated | FC | Adj p-Value | Top 10 Down-Regulated | FC | Adj p-Value |
|---|---|---|---|---|---|---|
| 729 | ANKRD1 | 10.28 | 3.99 × 10−5 | CST4 | 2.2 × 10−04 | 3.46 × 10−3 |
| LINC02275 | 8.57 | 2.77 × 10−5 | CST5 | 3.0 × 10−03 | 3.82 × 10−3 | |
| LINC02515 | 5.66 | 4.90 × 10−5 | CST2 | 3.0 × 10−03 | 8.83 × 10−3 | |
| NCAN | 5.31 | 5.79 × 10−3 | CST1 | 3.1 × 10−03 | 2.66 × 10−3 | |
| HPSE2 | 4.54 | 4.08 × 10−3 | SPRR3 | 3.3 × 10−03 | 6.00 × 10−3 | |
| NPY5R | 4.51 | 9.50 × 10−7 | KRT13 | 4.2 × 10−03 | 3.94 × 10−4 | |
| LINC01833 | 4.22 | 2.95 × 10−3 | SPRR1A | 5.3 × 10−03 | 2.05 × 10−3 | |
| SOX8 | 4.04 | 8.42 × 10−7 | KRT6C | 5.5 × 10−03 | 6.69 × 10−4 | |
| ASL | 3.88 | 3.82 × 10−5 | SMR3B | 5.9 × 10−03 | 2.82 × 10−3 | |
| PRLR | 3.81 | 1.99 × 10−8 | BPIFA2 | 7.1 × 10−03 | 1.67 × 10−2 |
| Symbol | Value | Symbol | Value | Symbol | Value |
|---|---|---|---|---|---|
| A2M | −0.11 | HMCN1 | −0.065 | PLA2R1 | −0.0434 |
| ABCA8 | −0.0129 | IPO9 | 0.376 | PLAT | −0.0448 |
| ACTA2 | −0.1264 | ITPR1 | −0.1121 | PLD1 | −0.0356 |
| ADAMTS9 | −0.0986 | KRT14 | −0.0359 | PPARGC1A | −0.0378 |
| ALDOA | −0.018 | LDLRAD4 | 0.0354 | PRUNE2 | −0.0026 |
| ANO1 | −0.099 | LGR4 | −0.0391 | RASSF6 | −0.1461 |
| APOL6 | −0.0977 | LIMA1 | −0.0793 | SEMA3C | −0.0582 |
| CARMN | −0.0091 | LIMCH1 | −0.145 | SH3D19 | −0.0562 |
| CD9 | −0.0886 | LOC107987158 | 0.0363 | SLPI | −0.1399 |
| CFH | −0.0226 | LTF | −0.0048 | SVIL | −0.168 |
| COL17A1 | −0.0151 | MAMLD1 | 0.0339 | SYNPO2 | −0.0159 |
| COL7A1 | −0.0095 | MIR205HG | −0.0511 | TAGLN | −0.0321 |
| COL9A2 | −0.0072 | MLPH | −0.013 | TNFRSF19 | −0.0054 |
| DMD | −0.3178 | MTUS1 | −0.0917 | TP63 | −0.1442 |
| EFS | 0.0074 | PARP14 | −0.0746 | TPM2 | −0.0175 |
| EGFR | −0.0288 | PCYOX1 | −0.1327 | ||
| FRMD4B | −0.0279 | PIK3R1 | −0.0097 |
| Clinical Covariates & Gene Classifier | Values | Hazard Ratio | 95% Confidence Interval | p-Value |
|---|---|---|---|---|
| Vascular Invasion | No | 1 | 0.097 | |
| Yes | 2.703 | 0.83–8.76 | ||
| Stage | I–II | 1 | 0.271 | |
| III–IV | 1.601 | 0.69–3.70 | ||
| Gene Classifier | Group 1 | 1 | 0.010 | |
| Group 2 | 26.01 | 2.19–309.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brayer, K.J.; Kang, H.; El-Naggar, A.K.; Andreasen, S.; Homøe, P.; Kiss, K.; Mikkelsen, L.; Heegaard, S.; Pelaez, D.; Moeyersoms, A.; et al. Dominant Gene Expression Profiles Define Adenoid Cystic Carcinoma (ACC) from Different Tissues: Validation of a Gene Signature Classifier for Poor Survival in Salivary Gland ACC. Cancers 2023, 15, 1390. https://doi.org/10.3390/cancers15051390
Brayer KJ, Kang H, El-Naggar AK, Andreasen S, Homøe P, Kiss K, Mikkelsen L, Heegaard S, Pelaez D, Moeyersoms A, et al. Dominant Gene Expression Profiles Define Adenoid Cystic Carcinoma (ACC) from Different Tissues: Validation of a Gene Signature Classifier for Poor Survival in Salivary Gland ACC. Cancers. 2023; 15(5):1390. https://doi.org/10.3390/cancers15051390
Chicago/Turabian StyleBrayer, Kathryn J., Huining Kang, Adel K. El-Naggar, Simon Andreasen, Preben Homøe, Katalin Kiss, Lauge Mikkelsen, Steffen Heegaard, Daniel Pelaez, Acadia Moeyersoms, and et al. 2023. "Dominant Gene Expression Profiles Define Adenoid Cystic Carcinoma (ACC) from Different Tissues: Validation of a Gene Signature Classifier for Poor Survival in Salivary Gland ACC" Cancers 15, no. 5: 1390. https://doi.org/10.3390/cancers15051390
APA StyleBrayer, K. J., Kang, H., El-Naggar, A. K., Andreasen, S., Homøe, P., Kiss, K., Mikkelsen, L., Heegaard, S., Pelaez, D., Moeyersoms, A., Tse, D. T., Guo, Y., Lee, D. Y., & Ness, S. A. (2023). Dominant Gene Expression Profiles Define Adenoid Cystic Carcinoma (ACC) from Different Tissues: Validation of a Gene Signature Classifier for Poor Survival in Salivary Gland ACC. Cancers, 15(5), 1390. https://doi.org/10.3390/cancers15051390