
Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.2. OGM: DNA Extraction, Labeling, and Chip Run
2.3. OGM Data Analysis
2.4. Determining Lowest Limits of Detection by In Silico Dilution Series
3. Results
3.1. Technical Metrics and Overall Number of SVs and CNVs
3.2. OGM Results Compared to FISH
3.3. Lowest Limits of Detection with OGM
3.4. Genome-Wide Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.D. Predicting clinical outcome in CLL: How and why. Hematology 2009, 2009, 421–429. [Google Scholar] [CrossRef]
- Mato, A.; Nabhan, C.; Kay, N.E.; Lamanna, N.; Kipps, T.J.; Grinblatt, D.L.; Flowers, C.R.; Farber, C.M.; Davids, M.S.; Kiselev, P.; et al. Prognostic Testing Patterns and Outcomes of Chronic Lymphocytic Leukemia Patients Stratified by Fluorescence In Situ Hybridization/Cytogenetics: A Real-World Clinical Experience in the Connect CLL Registry. Clin. Lymphoma Myeloma Leuk. 2018, 18, 114–124.e2. [Google Scholar] [CrossRef] [PubMed]
- Rack, K.A.; van den Berg, E.; Haferlach, C.; Beverloo, H.B.; Costa, D.; Espinet, B.; Foot, N.; Jeffries, S.; Martin, K.; O’Connor, S.; et al. European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia 2019, 33, 1851–1867. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. IwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic Aberrations and Survival in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, M.; Cavazzini, F.; Bardi, A.; Volta, E.; Melandri, A.; Tammiso, E.; Saccenti, E.; Lista, E.; Quaglia, F.M.; Urso, A.; et al. Biological significance and prognostic/predictive impact of complex karyotype in chronic lymphocytic leukemia. Oncotarget 2018, 9, 34398–34412. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, Z.; Rigolin, G.M.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood 2019, 133, 1205–1216. [Google Scholar] [CrossRef]
- Baliakas, P.; Espinet, B.; Mellink, C.; Jarosova, M.; Athanasiadou, A.; Ghia, P.; Kater, A.P.; Oscier, D.; Haferlach, C.; Stamatopoulos, K. Cytogenetics in Chronic Lymphocytic Leukemia: ERIC Perspectives and Recommendations. Hemasphere 2022, 6, e707. [Google Scholar] [CrossRef]
- Chatzikonstantinou, T.; Demosthenous, C.; Baliakas, P. Biology and Treatment of High-Risk CLL: Significance of Complex Karyotype. Front. Oncol. 2021, 11, 788761. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.M.N.; Chartrand, S.; LaBarge, B.; Claxton, D.F.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical Genome Mapping in Acute Myeloid Leukemia: A Multicenter Evaluation. Blood Adv. 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.R.; Chaubey, A.; Savage, N.M.; et al. Clinical Validation and Diagnostic Utility of Optical Genome Mapping for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML / MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosom. Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef]
- Balducci, E.; Kaltenbach, S.; Villarese, P.; Duroyon, E.; Zalmai, L.; Friedrich, C.; Suarez, F.; Marcais, A.; Bouscary, D.; Decroocq, J.; et al. Optical genome mapping refines cytogenetic diagnostics, prognostic stratification and provides new molecular insights in adult MDS/AML patients. Blood Cancer J. 2022, 12, 1–4. [Google Scholar] [CrossRef]
- Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers 2022, 14, 3376. [Google Scholar] [CrossRef]
- Ramos-Campoy, S.; Puiggros, A.; Kamaso, J.; Beà, S.; Bougeon, S.; Larráyoz, M.J.; Costa, D.; Parker, H.; Rigolin, G.M.; Blanco, M.L.; et al. TP53 Abnormalities Are Underlying the Poor Outcome Associated with Chromothripsis in Chronic Lymphocytic Leukemia Patients with Complex Karyotype. Cancers 2022, 14, 3715. [Google Scholar] [CrossRef]
- O’Keefe, C.; McDevitt, M.A.; Maciejewski, J.P. Copy neutral loss of heterozygosity: A novel chromosomal lesion in myeloid malignancies. Blood 2010, 115, 2731–2739. [Google Scholar] [CrossRef]
- Blanco, G.; Puiggros, A.; Baliakas, P.; Athanasiadou, A.; García-Malo, M.; Collado, R.; Xochelli, A.; Rodríguez-Rivera, M.; Ortega, M.; Calasanz, M.J.; et al. Karyotypic complexity rather than chromosome 8 abnormalities aggravates the outcome of chronic lymphocytic leukemia patients with TP53 aberrations. Oncotarget 2016, 7, 80916–80924. [Google Scholar] [CrossRef] [PubMed]
- Fabris, S.; Mosca, L.; Cutrona, G.; Lionetti, M.; Agnelli, L.; Ciceri, G.; Barbieri, M.; Maura, F.; Matis, S.; Colombo, M.; et al. Chromosome 2p gain in monoclonal B-cell lymphocytosis and in early stage chronic lymphocytic leukemia. Am. J. Hematol. 2013, 88, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Chapiro, E.; Leporrier, N.; Radford-Weiss, I.; Bastard, C.; Mossafa, H.; Leroux, D.; Tigaud, I.; de Braekeleer, M.; Terré, C.; Brizard, F.; et al. Gain of the short arm of chromosome 2 (2p) is a frequent recurring chromosome aberration in untreated chronic lymphocytic leukemia (CLL) at advanced stages. Leuk. Res. 2010, 34, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Houldsworth, J.; Guttapalli, A.; Thodima, V.; Yan, X.J.; Mendiratta, G.; Zielonka, T.; Nanjangud, G.; Chen, W.; Patil, S.; Mato, A.; et al. Genomic imbalance defines three prognostic groups for risk stratification of patients with chronic lymphocytic leukemia. Leuk. Lymphoma 2014, 55, 920–928. [Google Scholar] [CrossRef]
- De Paoli, L.; Cerri, M.; Monti, S.; Rasi, S.; Spina, V.; Bruscaggin, A.; Greco, M.; Ciardullo, C.; Famà, R.; Cresta, S.; et al. MGA, a suppressor of MYC, is recurrently inactivated in high risk chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1087–1090. [Google Scholar] [CrossRef]
- Edelmann, J.; Holzmann, K.; Miller, F.; Winkler, D.; Bühler, A.; Zenz, T.; Bullinger, L.; Kühn, M.W.M.; Gerhardinger, A.; Bloehdorn, J.; et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 2012, 120, 4783–4794. [Google Scholar] [CrossRef]
- Nguyen-Khac, F.; Chapiro, E.; Lesty, C.; Grelier, A.; Luquet, I.; Radford-Weiss, I.; Lefebvre, C.; Fert-Ferrer, S.; Callet-Bauchu, E.; Lippert, E.; et al. Specific chromosomal IG translocations have different prognoses in chronic lymphocytic leukemia. Am. J. blood Res. 2011, 1, 13–21. [Google Scholar]
- Fang, H.; Reichard, K.K.; Rabe, K.G.; Hanson, C.A.; Call, T.G.; Ding, W.; Kenderian, S.S.; Muchtar, E.; Schwager, S.M.; Leis, J.F.; et al. IGH translocations in chronic lymphocytic leukemia: Clinicopathologic features and clinical outcomes. Am. J. Hematol. 2019, 94, 338–345. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Filip, D.; Mraz, M. The role of MYC in the transformation and aggressiveness of ‘indolent’ B-cell malignancies. Leuk. Lymphoma 2019, 61, 510–524. [Google Scholar] [CrossRef]
- Puiggros, A.; Collado, R.; Calasanz, M.J.; Ortega, M.; Ruiz-Xivillé, N.; Rivas-Delgado, A.; Luño, E.; González, T.; Navarro, B.; García-Malo, M.; et al. Patients with chronic lymphocytic leukemia and complex karyotype show an adverse outcome even in absence of TP53/ATM FISH deletions. Oncotarget 2017, 8, 54297–54303. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.; Wenger, G.D.; Chaubey, A.; Dash, D.; Kanagal-Shamanna, R.; Kantarci, S.; Kolhe, R.; Van Dyke, D.L.; Wang, L.; Wolff, D.J.; et al. Assessing copy number aberrations and copy-neutral loss-of-heterozygosity across the genome as best practice: An evidence-based review from the Cancer Genomics Consortium (CGC) working group for chronic lymphocytic leukemia. Cancer Genet. 2018, 228, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Ouillette, P.; Collins, R.; Shakhan, S.; Li, J.; Li, C.; Shedden, K.; Malek, S.N. The Prognostic Significance of Various 13q14 Deletions in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2011, 17, 6778–6790. [Google Scholar] [CrossRef] [PubMed]
- Diop, F.; Moia, R.; Favini, C.; Spaccarotella, E.; De Paoli, L.; Bruscaggin, A.; Spina, V.; Terzi-Di-Bergamo, L.; Arruga, F.; Tarantelli, C.; et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 2020, 105, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Maguire, O.; Wallace, P.; Minderman, H. Fluorescent In Situ Hybridization in Suspension by Imaging Flow Cytometry. Methods Mol. Biol. 2016, 1389, 111–126. [Google Scholar] [CrossRef]
- Robbe, P.; Ridout, K.E.; Vavoulis, D.V.; Dréau, H.; Ben Kinnersley, B.; Denny, N.; Chubb, D.; Appleby, N.; Cutts, A.; Cornish, A.J.; et al. Whole-genome sequencing of chronic lymphocytic leukemia identifies subgroups with distinct biological and clinical features. Nat. Genet. 2022, 54, 1675–1689. [Google Scholar] [CrossRef]
- Visentin, A.; Bonaldi, L.; Rigolin, G.M.; Mauro, F.R.; Martines, A.; Frezzato, F.; Imbergamo, S.; Scomazzon, E.; Pravato, S.; Bardi, M.A.; et al. The combination of complex karyotype subtypes and IGHV mutational status identifies new prognostic and predictive groups in chronic lymphocytic leukaemia. Br. J. Cancer 2019, 121, 150–156. [Google Scholar] [CrossRef]
- Pfeifer, D.; Pantic, M.; Skatulla, I.; Rawluk, J.; Kreutz, C.; Martens, U.M.; Fisch, P.; Timmer, J.; Veelken, H. Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays. Blood 2007, 109, 1202–1210. [Google Scholar] [CrossRef]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. Am. J. Hematol. 2022, 97, 975–982. [Google Scholar] [CrossRef]


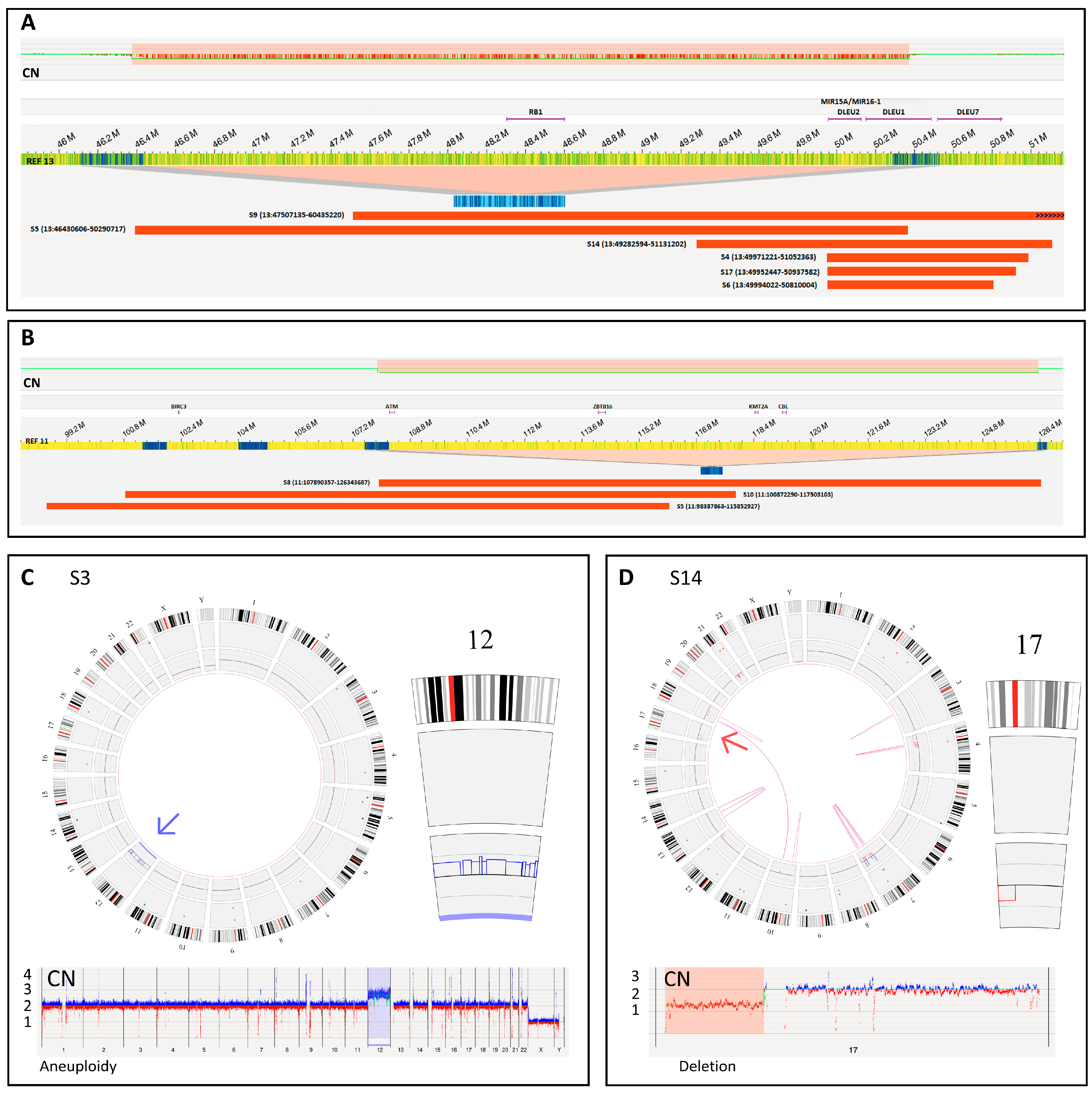{kind=link}
| FISH | OGM | ||||
|---|---|---|---|---|---|
| Sample ID | Material | Result (% of Positive Cells) | Material | Result (% Variant Allele Fraction) | Concordant with FISH |
| S1 | BM | Trisomy 12 (68%) | BM | (12) × 3 (49%) | Yes |
| S2 | Blood | Trisomy 12 (49%) | BM | (12) × 3 (45%) | Yes |
| S3 | Blood | Trisomy 12 (60%) | BM | (12) × 3 (35%) | Yes |
| S4 | Blood | Del(13q14.3) (87%) | BM | Del(13q14.2q14.3) (49,971,221–51,052,363) (89%) a | Yes |
| S5 | Blood | Del(11q22.3) (82%) Del(13q14.3) (89%) | BM | Del(11q22.1q23.3) (98,387,863–115,852,927) (43%) Del(13q14.13q14.3) (46,430,606–50,290,717) (57%) | Yes Yes |
| S6 | Blood | Del(13q14.3) (96%) | BM | Del(13q14.2q14.3) (49,994,022–50,810,004) (94%) a | Yes |
| S7 | BM | Monosomy 12 (60%) Del(17p13.1) (70%) | BM | Del(12p13.1q12) (27,123,509–45,992,905) (31%) b Del(17p13.3p11.2) (66,653–21,732,588) (47%) | Yes Yes |
| S8 | Blood | Del(11q22.3) (78%) | BM | Del(11q22.3q24.2) (107,890,357–126,343,687) (47%) | Yes |
| S9 | BM | Del(13q14.3) (35%) | BM | Del(13q14.2q21.2) (47,507,135–60,435,220) (42%) | Yes |
| S10 | Blood | Del(11q22.3) (18%) | BM | Del(11q22.1q23.3) (100,872,290–117,503,103) (11%) | Yes |
| S11 | Blood | Negative | BM | Negative | Yes |
| S12 | Blood | Negative | Blood | Negative | Yes |
| S13 | Blood | Negative | BM | Negative | Yes |
| S14 | Blood | Del(13q14.3) (13.5%) Del(17p13.1) (70%) | Blood | Del(13q14.q14.3) (49,282,594–51,131,202) (10%) Del(17p13.3p11.2) (66,653–22,079,438) (36%) | Yes Yes |
| S15 | Blood | Negative | BM | Negative | Yes |
| S16 | Blood | Negative | BM | Negative | Yes |
| S17 | Blood | Del(13q14.3) (77%) | BM | Del(13q14.2q14.3) (49,952,447–50,937,582) (48%) | Yes |
| S18 | Blood | Trisomy 12 (78%) | BM | (12) × 3 (47%) | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valkama, A.; Vorimo, S.; Kumpula, T.A.; Räsänen, H.; Savolainen, E.-R.; Pylkäs, K.; Mantere, T. Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia. Cancers 2023, 15, 1294. https://doi.org/10.3390/cancers15041294
Valkama A, Vorimo S, Kumpula TA, Räsänen H, Savolainen E-R, Pylkäs K, Mantere T. Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia. Cancers. 2023; 15(4):1294. https://doi.org/10.3390/cancers15041294
Chicago/Turabian StyleValkama, Andriana, Sandra Vorimo, Timo A. Kumpula, Hannele Räsänen, Eeva-Riitta Savolainen, Katri Pylkäs, and Tuomo Mantere. 2023. "Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia" Cancers 15, no. 4: 1294. https://doi.org/10.3390/cancers15041294
APA StyleValkama, A., Vorimo, S., Kumpula, T. A., Räsänen, H., Savolainen, E.-R., Pylkäs, K., & Mantere, T. (2023). Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia. Cancers, 15(4), 1294. https://doi.org/10.3390/cancers15041294