
Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL)

 , , , , , ,
, , , , , ,  ,
,  , add
Show full author list
, add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. DNA Extraction, Labelling and Data Collection for Optical Mapping
2.3. Data Assembly, Structural Variant Calling and Filtering
2.4. Chromosome Banding Analyses and Fluorescence In Situ Hybridization
2.5. Chromosomal Microarray Analyses
2.6. Comparison among Techniques
2.7. Statistical Analyses
3. Results
3.1. Global SV and CNV Detection by OGM
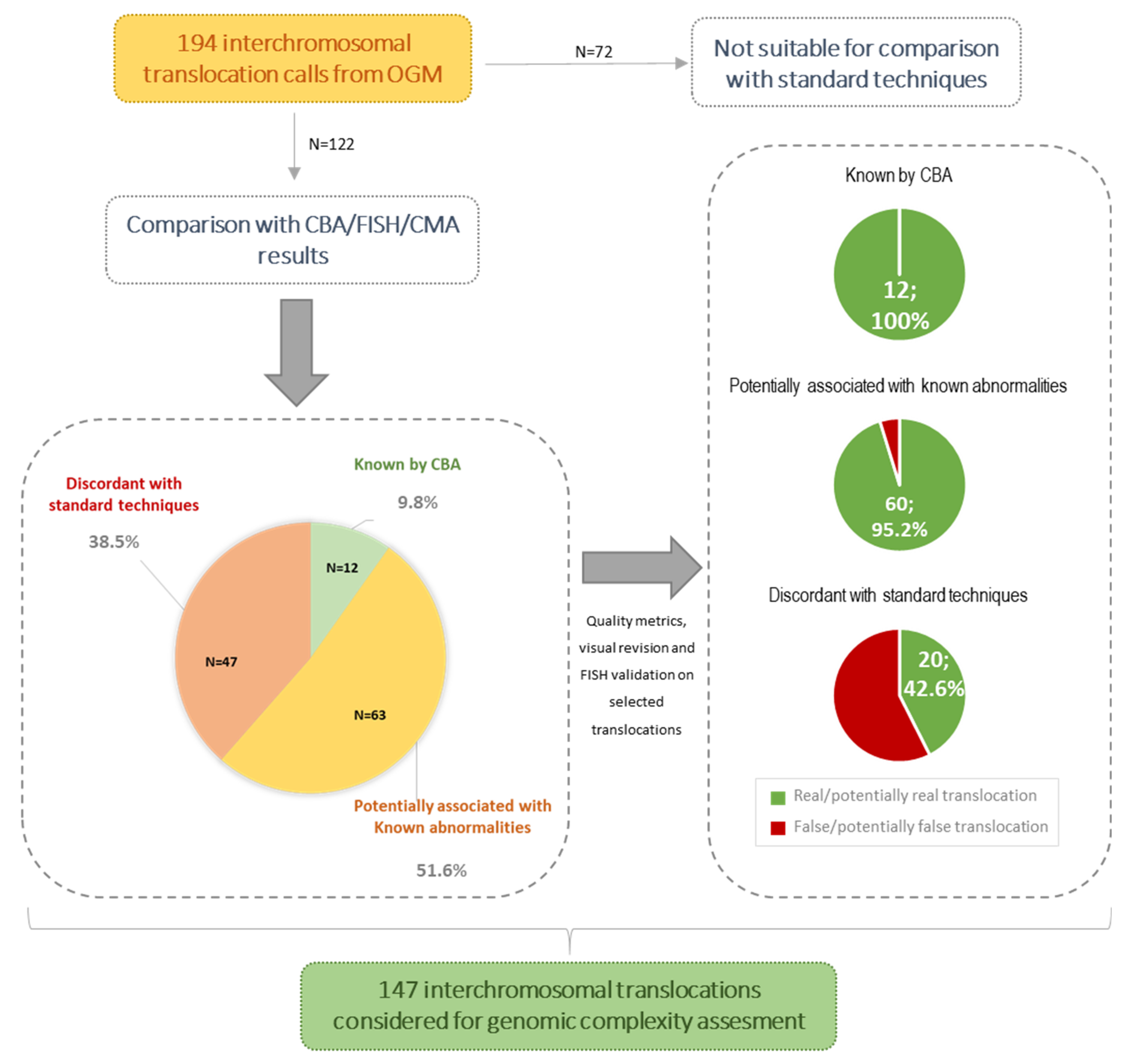
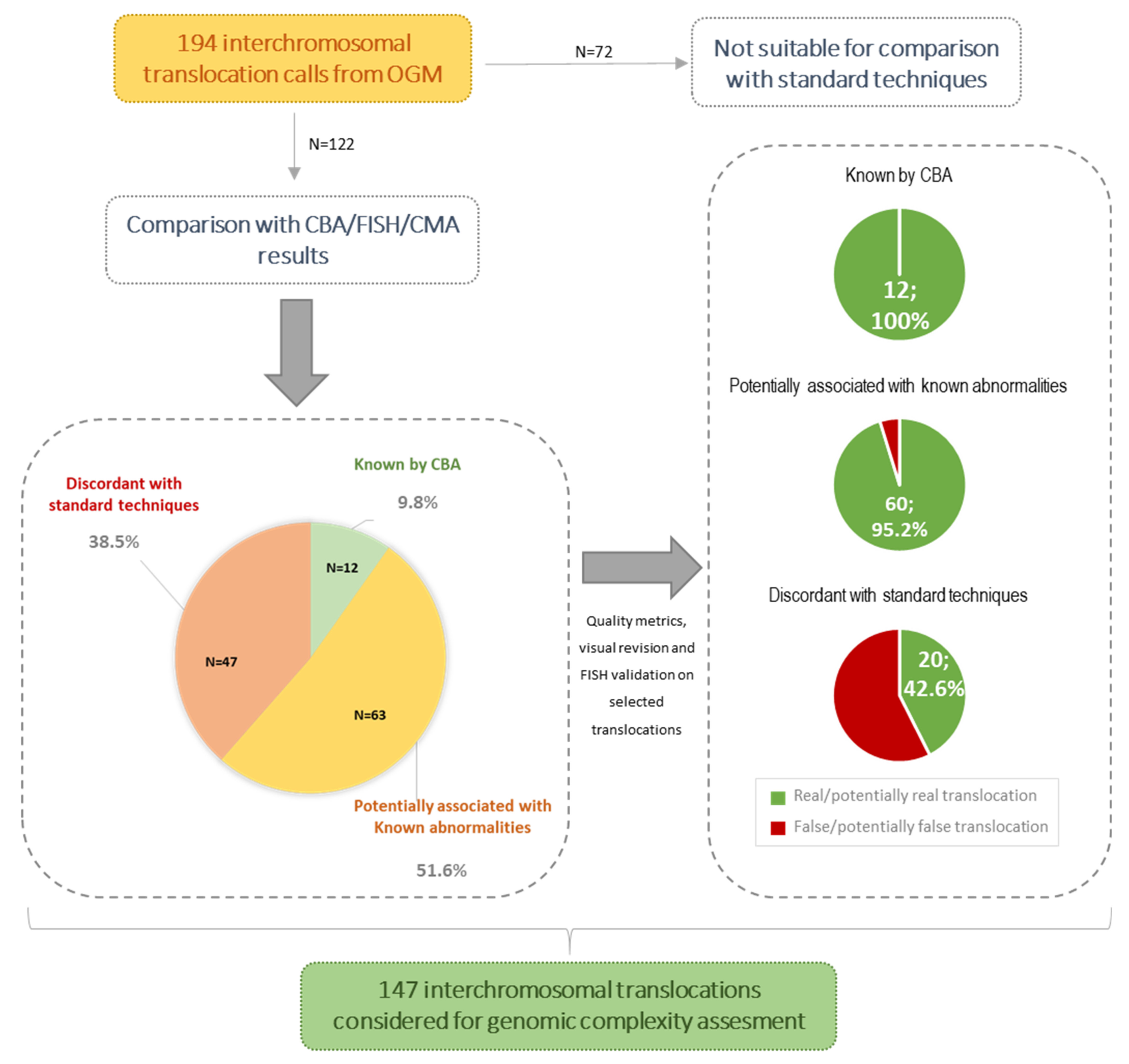
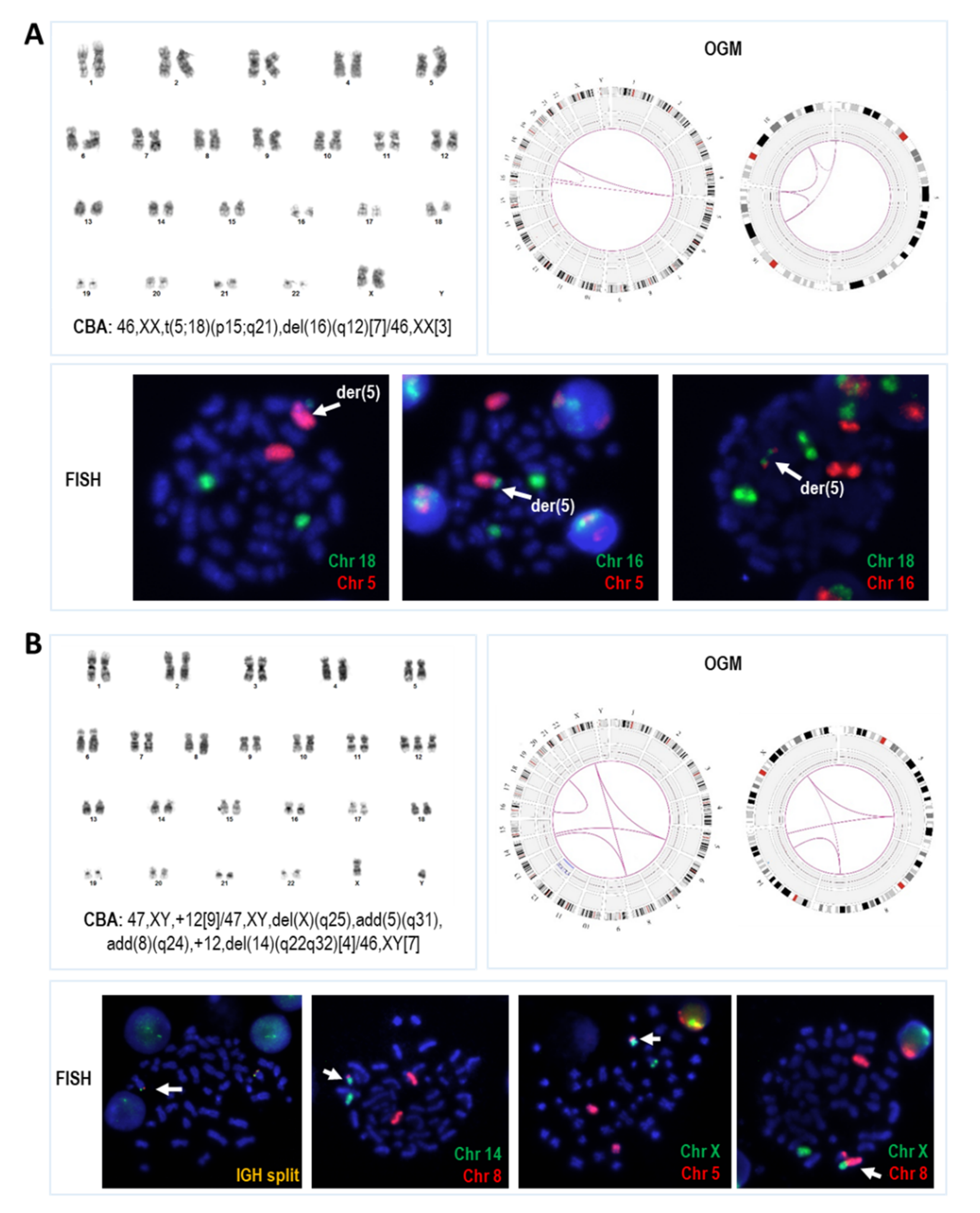
3.2. Concordance Rate of OGM Results with Standard Techniques
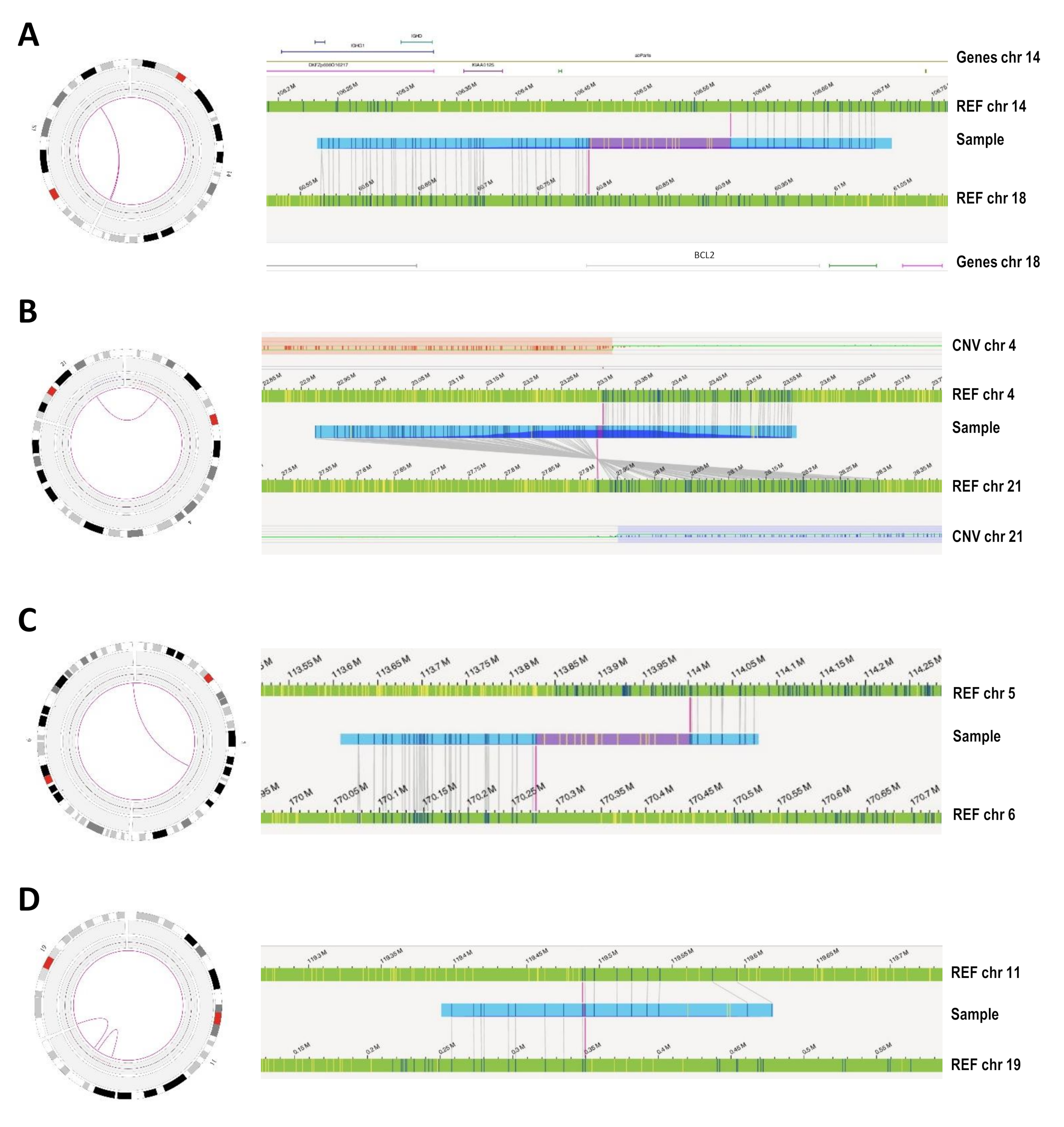
3.3. Novel Abnormalities Detected by OGM
3.4. Global Genomic Complexity Found by OGM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- González-Gascón, I.; Muñoz-Novas, C.; Rodríguez-Vicente, A.E.; Quijada-Álamo, M.; Hernández-Sánchez, M.; Pérez-Carretero, C.; Ramos-Ascanio, V.; Hernández-Rivas, J.A. From Biomarkers to Models in the Changing Landscape of Chronic Lymphocytic Leukemia: Evolve or Become Extinct. Cancers 2021, 13, 1782. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicker, F.; Schnittger, S.; Haferlach, T.; Kern, W.; Schoch, C. Immunostimulatory oligonucleotide-induced metaphase cytogenetics detect chromosomal aberrations in 80% of CLL patients: A study of 132 CLL cases with correlation to FISH, IgVH status, and CD38 expression. Blood 2006, 108, 3152–3160. [Google Scholar] [CrossRef] [Green Version]
- Rigolin, G.M.; Cibien, F.; Martinelli, S.; Formigaro, L.; Rizzotto, L.; Tammiso, E.; Saccenti, E.; Bardi, A.; Cavazzini, F.; Ciccone, M.; et al. Chromosome aberrations detected by conventional karyotyping using novel mitogens in chronic lymphocytic leukemia with ”normal” FISH: Correlations with clinicobiologic parameters. Blood 2012, 119, 2310–2313. [Google Scholar] [CrossRef] [PubMed]
- Herling, C.D.; Klaumünzer, M.; Rocha, C.K.; Altmüller, J.; Thiele, H.; Bahlo, J.; Kluth, S.; Crispatzu, G.; Herling, M.; Schiller, J.; et al. Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood 2016, 128, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Puiggros, A.; Collado, R.; Calasanz, M.J.; Ortega, M.; Ruiz-Xivillé, N.; Rivas-Delgado, A.; Luño, E.; González, T.; Navarro, B.; García-Malo, M.; et al. Patients with chronic lymphocytic leukemia and complex karyotype show an adverse outcome even in absence of TP53/ATM FISH deletions. Oncotarget 2017, 8, 54297–54303. [Google Scholar] [CrossRef] [Green Version]
- Rigolin, G.M.; Saccenti, E.; Guardalben, E.; Cavallari, M.; Formigaro, L.; Zagatti, B.; Visentin, A.; Mauro, F.R.; Lista, E.; Bassi, C.; et al. In chronic lymphocytic leukaemia with complex karyotype, major structural abnormalities identify a subset of patients with inferior outcome and distinct biological characteristics. Br. J. Haematol. 2018, 181, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, Z.; Rigolin, G.M.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood 2019, 133, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.A.; O’Brien, S.M.; Wierda, W.G.; Ferrajoli, A.; Stingo, F.; Smith, S.C.; Burger, J.A.; Estrov, Z.; Jain, N.; Kantarjian, H.M.; et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer 2015, 121, 3612–3621. [Google Scholar] [CrossRef] [Green Version]
- Chanan-Khan, A.; Cramer, P.; Demirkan, F.; Fraser, G.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): A randomised, double-blind, phase 3 study. Lancet Oncol. 2016, 17, 200–211. [Google Scholar] [CrossRef]
- Anderson, M.A.; Tam, C.; Lew, T.E.; Juneja, S.; Juneja, M.; Westerman, D.; Wall, M.; Lade, S.; Gorelik, A.; Huang, D.C.S.; et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood 2017, 129, 3362–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, S.; Furman, R.R.; Coutre, S.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Wierda, W.; Jones, J.; Zhao, W.; et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: A 5-year experience. Blood 2018, 131, 1910–1919. [Google Scholar] [CrossRef]
- Mato, A.R.; Thompson, M.; Allan, J.N.; Brander, D.M.; Pagel, J.M.; Ujjani, C.S.; Hill, B.T.; Lamanna, N.; Lansigan, F.; Jacobs, R.; et al. Real-world outcomes and management strategies for venetoclax-treated chronic lymphocytic leukemia patients in the United States. Haematologica 2018, 103, 1511–1517. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Heerema, N.A.; Zhao, W.; Booth, A.M.; Ding, W.; Bartlett, N.L.; Brander, D.M.; Barr, P.M.; Rogers, K.A.; et al. Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N. Engl. J. Med. 2018, 379, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Fraser, G.; Coutre, S.E.; Brown, J.R.; Barrientos, J.C.; Barr, P.M.; Byrd, J.C.; O’Brien, S.M.; Dilhuydy, M.S.; Hillmen, P.; et al. Long-Term Studies Assessing Outcomes of Ibrutinib Therapy in Patients With Del(11q) Chronic Lymphocytic Leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 715–722.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sawaf, O.; Lilienweiss, E.; Bahlo, J.; Robrecht, S.; Fink, A.M.; Patz, M.; Tandon, M.; Jiang, Y.; Schary, W.; Ritgen, M.; et al. High efficacy of venetoclax plus obinutuzumab in patients with complex karyotype and chronic lymphocytic leukemia. Blood 2020, 135, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Wierda, W.G.; Schuh, A.; Devereux, S.; Chaves, J.M.; Brown, J.R.; Hillmen, P.; Martin, P.; Awan, F.T.; Stephens, D.M.; et al. Acalabrutinib monotherapy in patients with relapsed/refractory chronic lymphocytic leukemia: Updated phase 2 results. Blood 2020, 135, 1204–1213. [Google Scholar] [CrossRef]
- Kater, A.P.; Wu, J.Q.; Kipps, T.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Robak, T.; de la Serna, J.; et al. Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations From the MURANO Phase III Study. J. Clin. Oncol. 2020, 38, 4042–4054. [Google Scholar] [CrossRef]
- Kreuzer, K.A.; Furman, R.R.; Stilgenbauer, S.; Dubowy, R.L.; Kim, Y.; Munugalavadla, V.; Lilienweiss, E.; Reinhardt, H.C.; Cramer, P.; Eichhorst, B.; et al. The impact of complex karyotype on the overall survival of patients with relapsed chronic lymphocytic leukemia treated with idelalisib plus rituximab. Leukemia 2020, 34, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Kittai, A.S.; Miller, C.; Goldstein, D.; Huang, Y.; Abruzzo, L.V.; Beckwith, K.; Bhat, S.A.; Bond, D.A.; Grever, M.R.; Heerema, N.A.; et al. The impact of increasing karyotypic complexity and evolution on survival in patients with CLL treated with ibrutinib. Blood 2021, 138, 2372–2382. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Schnittger, S.; Kern, W.; Haferlach, T. Comprehensive genetic characterization of CLL: A study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia 2007, 21, 2442–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badoux, X.C.; Keating, M.J.; Wang, X.; O’Brien, S.M.; Ferrajoli, A.; Faderl, S.; Burger, J.; Koller, C.; Lerner, S.; Kantarjian, H.; et al. Cyclophosphamide, fludarabine, alemtuzumab, and rituximab as salvage therapy for heavily pretreated patients with chronic lymphocytic leukemia. Blood 2011, 118, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Puiggros, A.; Xochelli, A.; Sutton, L.A.; Nguyen-Khac, F.; Gardiner, A.; Plevova, K.; Minga, E.; Hadzidimitriou, A.; Walewska, R.; et al. Additional trisomies amongst patients with chronic lymphocytic leukemia carrying trisomy 12: The accompanying chromosome makes a difference. Haematologica 2016, 101, e299–e302. [Google Scholar] [CrossRef] [Green Version]
- Visentin, A.; Bonaldi, L.; Rigolin, G.M.; Mauro, F.R.; Martines, A.; Frezzato, F.; Imbergamo, S.; Scomazzon, E.; Pravato, S.; Bardi, M.A.; et al. The combination of complex karyotype subtypes and IGHV mutational status identifies new prognostic and predictive groups in chronic lymphocytic leukaemia. Br. J. Cancer 2019, 121, 150–156. [Google Scholar] [CrossRef]
- Heerema, N.A.; Muthusamy, N.; Zhao, Q.; Ruppert, A.S.; Breidenbach, H.; Andritsos, L.A.; Grever, M.R.; Maddocks, K.J.; Woyach, J.; Awan, F.; et al. Prognostic significance of translocations in the presence of mutated IGHV and of cytogenetic complexity at diagnosis of chronic lymphocytic leukemia. Haematologica 2021, 106, 1608–1615. [Google Scholar] [CrossRef]
- Leeksma, A.C.; Baliakas, P.; Moysiadis, T.; Puiggros, A.; Plevova, K.; Van der Kevie-Kersemaekers, A.M.; Posthuma, H.; Rodriguez-Vicente, A.E.; Tran, A.N.; Barbany, G.; et al. Genomic arrays identify high-risk chronic lymphocytic leukemia with genomic complexity: A multi-center study. Haematologica 2021, 106, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Campoy, S.; Puiggros, A.; Beà, S.; Bougeon, S.; Larráyoz, M.J.; Costa, D.; Parker, H.; Rigolin, G.M.; Ortega, M.; Blanco, M.L.; et al. Chromosome banding analysis and genomic microarrays are both useful but not equivalent methods for genomic complexity risk stratification in chronic lymphocytic leukemia patients. Haematologica 2022, 107, 593–603. [Google Scholar] [CrossRef]
- Baliakas, P.; Espinet, B.; Mellink, C.; Jarosova, M.; Athanasiadou, A.; Ghia, P.; Kater, A.P.; Oscier, D.; Haferlach, C.; Stamatopoulos, K. Cytogenetics in Chronic Lymphocytic Leukemia: ERIC Perspectives and Recommendations. Hemasphere 2022, 25, e707. [Google Scholar] [CrossRef]
- Puiggros, A.; Puigdecanet, E.; Salido, M.; Ferrer, A.; Abella, E.; Gimeno, E.; Nonell, L.; Herranz, M.J.; Galván, A.B.; Rodríguez-Rivera, M.; et al. Genomic arrays in chronic lymphocytic leukemia routine clinical practice: Are we ready to substitute conventional cytogenetics and fluorescence in situ hybridization techniques? Leuk. Lymphoma 2013, 54, 986–995. [Google Scholar] [CrossRef]
- Urbankova, H.; Papajik, T.; Plachy, R.; Holzerova, M.; Balcarkova, J.; Divoka, M.; Prochazka, V.; Pikalova, Z.; Indrak, K.; Jarosova, M. Array-based karyotyping in chronic lymphocytic leukemia (CLL) detects new unbalanced abnormalities that escape conventional cytogenetics and CLL FISH panel. Biomed. Pap. Med. Fac. Palacky Univ. Olomouc Czech Repub. 2014, 158, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastie, A.R.; Lam, E.T.; Chun Pang, A.W.; Zhang, X.; Andrews, W.; Lee, J.; Liang, T.Y.; Wang, J.; Zhou, X.; Zhu, Z.; et al. Rapid Automated Large Structural Variation Detection in a Diploid Genome by NanoChannel Based Next-Generation Mapping. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Bionano Genomics. Bionano Solve Theory of Operation: Structural Variant Calling; Bionano Genomics: San Diego, CA, USA, 2021. [Google Scholar]
- Levy, B.; Baughn, L.B.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, A.; Akkari, Y.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. A National Multicenter Evaluation of the Clinical Utility of Optical Genome Mapping for Assessment of Genomic Aberrations in Acute Myeloid Leukemia. medRxiv 2020. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML/MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Rack, K.; De Bie, J.; Ameye, G.; Gielen, O.; Demeyer, S.; Cools, J.; De Keersmaecker, K.; Vermeesch, J.R.; Maertens, J.; Segers, H.; et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am. J. Hematol. 2022, 97, 548–561. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. ISCN 2020: An International System for Human Cytogenomic Nomenclature; Karger: Basel, Switzerland, 2020. [Google Scholar]
- Kriegova, E.; Fillerova, R.; Minarik, J.; Savara, J.; Manakova, J.; Petrackova, A.; Dihel, M.; Balcarkova, J.; Krhovska, P.; Pika, T.; et al. Whole-genome optical mapping of bone-marrow myeloma cells reveals association of extramedullary multiple myeloma with chromosome 1 abnormalities. Sci. Rep. 2021, 11, 14671. [Google Scholar] [CrossRef]
- Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Göhring, G.; Möricke, A.; Cario, G.; et al. The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 4388. [Google Scholar] [CrossRef]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosomes Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Rush, D.; Montalban-Bravo, G.; Mallampati, S.; Medeiros, L.J.; Levy, B.; Luthra, R.; Kanagal-Shamanna, R. Application of Optical Genome Mapping For Comprehensive Assessment of Chromosomal Structural Variants for Clinical Evaluation of Myelodysplastic Syndromes. medRxiv 2021. [Google Scholar] [CrossRef]
- Smith, A.C.; Neveling, K.; Kanagal-Shamanna, R. Optical genome mapping for structural variation analysis in hematologic malignancies. Am. J. Hematol. 2022, 97, 975–982. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-CK Group | CK Group | p-Value | |
|---|---|---|---|
| n = 24; n (%) | n = 18; n (%) | ||
| Gender | |||
| Male | 17 (70.8%) | 11 (61.1%) | 0.530 |
| Median age at diagnosis (range) | 66 (42–85) | 69 (37–88) | 0.297 |
| Stage at diagnosis | |||
| MBL | 0 (0.0%) | 3 (16.7%) | 0.071 |
| CLL | 24 (100%) | 15 (83.3%) | |
| Binet A | 22 (91.7%) | 10 (66.7%) | 0.085 |
| Binet B/C | 2 (8.4%) | 5 (33.3%) | |
| Common CLL genomic aberrations * | |||
| del(13)(q14) | 19 (79.2%) | 11 (61.1%) | 0.302 |
| Trisomy 12 | 3 (12.5%) | 5 (27.8%) | 0.256 |
| del(11)(q22q23) | 5 (20.8%) | 7 (38.9%) | 0.302 |
| Aberrations in TP53 | 0 (0.0%) | 8 (44.4%) | <0.001 |
| del(17)(p13) | 0 (0.0%) | 7 (38.9%) | 0.001 |
| TP53 mutation | 0 (0.0%) | 7 (38.9%) | 0.001 |
| Unmutated IGHV | 12/23 (52.2%) | 12/18 (66.7%) | 0.524 |
| Time from diagnosis to cytogenetic study (range) | 22.5 months (0–123) | 6 months (0–174) | 0.867 |
| Median follow-up (range) | 44.5 months (0–95) | 31.5 months (0–78) | 0.065 |
| Treatment | |||
| Treated patients | 13 (54.2%) | 13 (72.2%) | 0.338 |
| Median time to first treatment (95% CI) | 43 months (26–54) | 9 months (8–30) | 0.165 |
| Patient ID | Undetected Alteration | Size (Kb) | Potential Cause of Discrepancy | Comments |
|---|---|---|---|---|
| 12 | Translocation t(13;?)(p11;?) | - | Involvement of (peri-)centromeric regions | |
| CK4 | Translocation t(17;?)(p11;?) | - | Involvement of (peri-)centromeric regions | |
| CK8 | Translocation t(17;18)(q10;q10) | - | Involvement of (peri-)centromeric regions | |
| CK9 | Translocation t(13;21)(q11;p11) | - | Involvement of (peri-)centromeric regions | |
| CK10 | Translocation t(14;?)(p11;?) | - | Involvement of (peri-)centromeric regions | |
| CK12 | Translocation t(11;?)(p11;?) | - | Involvement of (peri-)centromeric regions | |
| CK13 | Translocation t(14;17)(q11;p11) | - | Involvement of (peri-)centromeric regions | |
| CK16 | Translocation t(14;18)(p11;q11) | - | Involvement of (peri-)centromeric regions | |
| CK16 | Translocation t(15;22)(p11;q15) | - | Involvement of (peri-)centromeric regions | |
| CK17 | Translocation t(15;?)(p11;?) | - | Involvement of (peri-)centromeric regions | |
| CK4 | Translocation t(6;19)(q12;p13) | - | Involvement of telomeric region | |
| 7 | Gain Yq11.223q11.23(24637115_28799654) | 4163 | Masked region (partial CNV on chr. Y) | Detected at 50% by CMA, and visually suggested in the whole genome CNV view |
| CK2 | Deletion 17p13.3p13.3(525_2489182) | 2489 | Masked region (telomere) | |
| CK6 | Deletion 1p36.33p36.32(1997349_3740109) | 1743 | Masked region (telomere) | |
| 20 | Deletion 6q21 | NA | Sensitivity | Detected in 12% of nuclei by FISH (CMA not available) |
| 21 | Deletion 13q14 (D13S319) | NA | Sensitivity | Detected in 17% of nuclei by FISH (not detected by CMA) |
| CK3 | Gain 6pterq16 | NA | Sensitivity | Detected in 3% of nuclei by FISH, confirmed in metaphases (not detected by CMA) |
| CK8 | Deletion 17p13.3p11.2(526_21347924) | 21347 | Sensitivity | Detected in 15% of nuclei by FISH (also visually found by CMA) |
| CK12 | Gain 11q14.2q14.2(86177075_86856206) | 679 | Sensitivity | Detected at 25% by CMA, and visually suggested in the whole genome CNV view |
| CK12 | Gain 11q22.3q22.3(106600681_106991146) | 390 | Sensitivity | Detected at 25% by CMA, and visually suggested in the whole genome CNV view |
| CK12 | Gain 11q24.3q24.3(129345165_130249509) | 904 | Sensitivity | Detected at 25% by CMA, and visually suggested in the whole genome CNV view |
| CK12 | Gain 19q13.2q13.42(41644540_54499334) | 12855 | Sensitivity | Detected at 20% by CMA |
| CK13 | Gain 1p22pter | NA | Sensitivity | Detected in 5% of nuclei by FISH (not detected by CMA) |
| 7 | Translocation t(X;?)(p22;?) | Sensitivity | Percentage not available, could be a minor clone expanded during CBA culture | |
| CK4 | Translocation t(12;?)(q24;?) | Sensitivity | Percentage not available, could be a minor clone (“add(12)(q24)” detected as clonal evolution in only two out of eight abnormal metaphases) | |
| CK6 | Translocation t(9;?)(q34;?) | Sensitivity | Percentage not available, could be a minor clone expanded during CBA culture (CK defined as a composite karyotype) | |
| CK8 | Translocation t(6;?)(p25;?) | Sensitivity | Percentage not available, could be a minor clone (“add(6)(p25)” detected as clonal evolution from abnormal cells with monosomy of chr. 17, as the TP53 deletion was detected at 15% the abnormality was probably below that percentage) | |
| 8 | Deletion 13q32.1(95520821_95658848) | 138 | Small deletion within chromothripsis | Small deletion, part of a complex CMA profile on chr. 13 properly detected |
| CK16 | Translocation t(11;?)(q23;?) | Unknown cause of discrepancy | The “add(11)(q23)” was present in the main clone. Although not called as SV, some imaged molecules showed fusions between different regions of chr 11; WCP for chr. 11 confirmed hybridization in the whole abnormal chromosome | |
| CK17 | Translocation t(9;?)(q34;?) | Unknown cause of discrepancy | Percentage not available; although the “add(9)(q34)” could be a minor clone (detected as clonal evolution in only two out of 13 abnormal metaphases), other abnormalities from the same clone were properly detected. WCP revealed that the additional material was from chr. 17 and, as seen by CBA, the chr. 17 telomeric region could be involved in the fusion (telomeric regions are masked by the SV pipeline) |
| Characteristics | NC-OGM (n = 30) | C-OGM (n = 12) | p-Value |
|---|---|---|---|
| Age at diagnosis | 68 (37–85) | 67 (55–88) | 0.944 |
| Males | 20 (66.7%) | 8 (66.7%) | 1.000 |
| Advanced Binet stage (B or C) | 3 (10.0%) | 4 (33.3%) | 0.088 |
| Time from diagnosis to OGM study (months) | 8 (0–174) | 25 (0–125) | 0.854 |
| Number of abnormalities by OGM (filtered data) | 4 (1–11) | 32 (16–70) | <0.001 |
| Copy number variants (CNV gains and losses) | 1 (0–4) | 12 (2–25) | <0.001 |
| Translocations (intra and interchromosomal) | 2 (0–8) | 22 (9–42) | <0.001 |
| Genomic complexity by conventional methods | |||
| Low/intermediate-CK by CBA (3–4 abn.) | 6 (21.4%) | 2 (7.1%) | 1.000 |
| High-CK by CBA (≥5 abn.) | 2 (6.7%) | 8 (66.7%) | <0.001 |
| Intermediate-GC by CMA (3–4 abn. *) (n = 40) | 6/28 (21.4%) | 3/12 (25.0%) | 1.000 |
| High-GC by CMA (≥5 abn. *) (n = 40) | 2/28 (7.1%) | 8/12 (66.7%) | 0.001 |
| High complexity by CBA and/or CMA (≥5 abn.) | 2 (6.7%) | 11 (91.7%) | <0.001 |
| Chromothripsis | 0 (0.0%) | 9 (75.0%) | <0.001 |
| FISH abnormalities | |||
| del(13q) | 22 (73.3%) | 8 (66.7%) | 0.715 |
| Trisomy 12 | 7 (23.3%) | 1 (8.3%) | 0.402 |
| del(11q) [ATM] | 8 (26.7%) | 4 (33.3%) | 0.715 |
| del(17p) [TP53] | 1 (3.3%) | 6 (50.0%) | 0.001 |
| TP53 abnormalities (del/mut) | 1 (3.3%) | 7 (58.3%) | <0.001 |
| Unmutated IGHV (n = 41) | 15/29 (51.7%) | 9/12 (75.0%) | 0.296 |
| Last follow-up (n = 40) ¥ | |||
| Treated patients | 16/30 (53.3%) | 8/10 (80.0%) | 0.090 |
| Time to first treatment (months, 95% CI) | 43 (28.0–52.6) | 2 (1.9–23.0) | 0.014 |
| Follow-up (months) | 42 (0–95) | 26 (0–82) | 0.070 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers 2022, 14, 3376. https://doi.org/10.3390/cancers14143376
Puiggros A, Ramos-Campoy S, Kamaso J, de la Rosa M, Salido M, Melero C, Rodríguez-Rivera M, Bougeon S, Collado R, Gimeno E, et al. Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers. 2022; 14(14):3376. https://doi.org/10.3390/cancers14143376
Chicago/Turabian StylePuiggros, Anna, Silvia Ramos-Campoy, Joanna Kamaso, Mireia de la Rosa, Marta Salido, Carme Melero, María Rodríguez-Rivera, Sandrine Bougeon, Rosa Collado, Eva Gimeno, and et al. 2022. "Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL)" Cancers 14, no. 14: 3376. https://doi.org/10.3390/cancers14143376
APA StylePuiggros, A., Ramos-Campoy, S., Kamaso, J., de la Rosa, M., Salido, M., Melero, C., Rodríguez-Rivera, M., Bougeon, S., Collado, R., Gimeno, E., García-Serra, R., Alonso, S., Moro-García, M. A., García-Malo, M. D., Calvo, X., Arenillas, L., Ferrer, A., Mantere, T., Hoischen, A., ... Espinet, B. (2022). Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers, 14(14), 3376. https://doi.org/10.3390/cancers14143376