

Unraveling the Peculiar Features of Mitochondrial Metabolism and Dynamics in Prostate Cancer



Abstract
Simple Summary
Abstract
1. Introduction
1.1. Mitochondrial Metabolism in Cancer Cells
1.2. Mitochondrial Dynamics in Cancer Cells
2. Mitochondrial Metabolism and Dynamics in PCa
2.1. Mitochondrial Metabolism
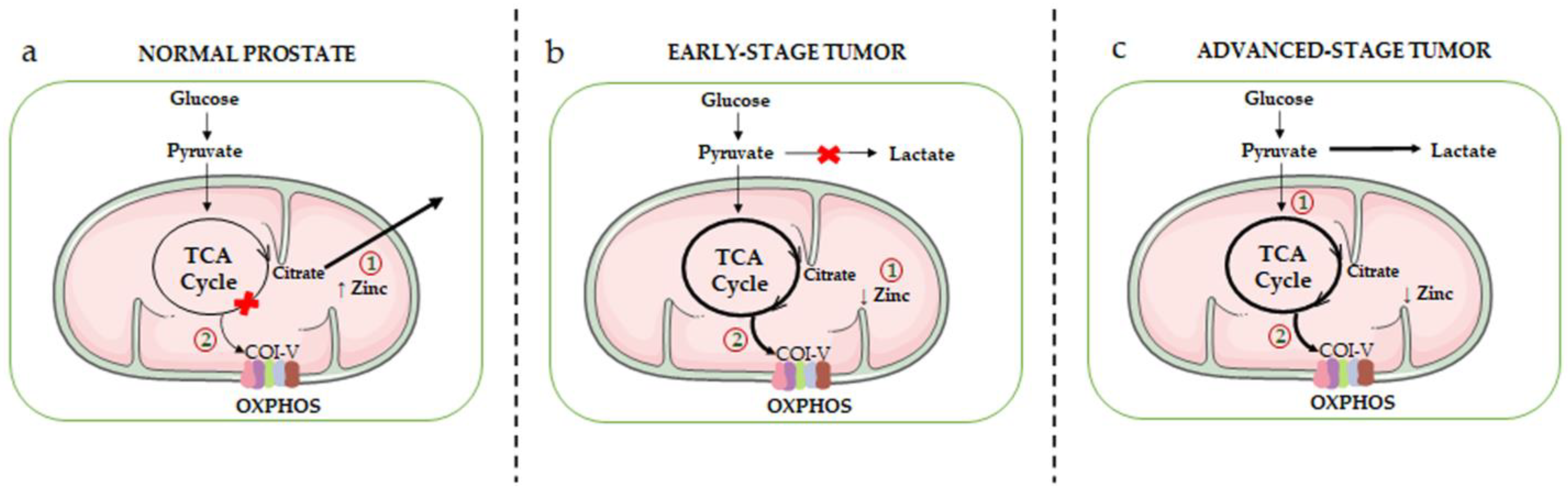
2.1.1. Metabolic Rewiring
2.1.2. The AR-Mitochondria Axis
2.1.3. mtDNA Mutations
2.2. Mitochondrial Dynamics
3. Targeting Mitochondrial Reprogramming and Dynamics in PCa

{kind=link}
| Drug | Therapy Class | Effects | References |
|---|---|---|---|
| Docetaxel + ADT | Taxane drugs + GnRH analogs | Inhibition of TCA cycle; decrease of the GSH/GSSG ratio; downregulation of glutamine | [221] |
| Metformin, phenformin canagliflozin | Antidiabetic (type 2 diabetes) drugs | Inhibition of ETC complex I; decreased O2 consumption; AMPK activation; suppression of the OXPHOS pathway and ATP production; upregulation of ROS levels | [31,52,167,177,229,236,242,243,244,245,246,247,248,249,250,251] |
| Amiodarone | Class III antiarrythmic drugs | Inhibition of ETC complex I and II; suppression of ATP production | [260] |
| IACS-010759, CB-839 | Small molecule inhibitors | Inhibition of ETC complex I; inhibition of mitochondrial glutaminase and glutamate supply to the TCA cycle; decreased OXPHOS pathway | [179] |
| Pd(II)COS@GbA | Pd(II) anticancer complexes | Inhibition of ETC complex V (ATP synthase); suppression of ATP production; mitochondrial fission | [262] |
| Doxycycline, azithromycin, tigecycline, pyrvinium pamoate, chloramphenicol | Antibiotics | Inhibition of the OXPHOS pathway and mitochondrial respiration; dysregulations of ETC complexes; suppression of ATP production; impairment of mitochondrial biogenesis | [176,264,265,266,267] |
| Buforin IIb | Antimicrobial peptides | Inhibition of glycolysis and ATP production | [268] |
| Vitamin E-derived tocotrienols, curcumin, resveratrol, triterpenoids, sulforaphane, phenethyl isothiocyanate (PEITC), silibinin, atpenin A5 analogs, jasmonates, alternol | Natural antioxidants | Reduced O2 consumption, ETC protein (complexes COI-V) levels and activity; suppression of the OXPHOS pathway and ATP production; decreased glycolytic pathway; AMPK activation; altered Ca2+/ROS homeostasis; mitochondrial fission, mitophagy | [78,180,261,289,290,291,292,293,294,295,296,297,298,299,300,301,302,303,304,305,306] |
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef]
- Granchi, C.; Fancelli, D.; Minutolo, F. An update on therapeutic opportunities offered by cancer glycolytic metabolism. Bioorg. Med. Chem. Lett. 2014, 24, 4915–4925. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019, 38, 157–164. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2020, 599, 1745–1757. [Google Scholar] [CrossRef]
- Fontana, F.; Limonta, P. The multifaceted roles of mitochondria at the crossroads of cell life and death in cancer. Free. Radic. Biol. Med. 2021, 176, 203–221. [Google Scholar] [CrossRef]
- Niu, D.; Wu, Y.; Lei, Z.; Zhang, M.; Xie, Z.; Tang, S. Lactic acid, a driver of tumor-stroma interactions. Int. Immunopharmacol. 2022, 106, 108597. [Google Scholar] [CrossRef]
- Wang, X.; Liu, H.; Ni, Y.; Shen, P.; Han, X. Lactate shuttle: From substance exchange to regulatory mechanism. Hum. Cell 2022, 35, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van De Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef]
- Mosier, J.A.; Schwager, S.C.; Boyajian, D.A.; Reinhart-King, C.A. Cancer cell metabolic plasticity in migration and metastasis. Clin. Exp. Metastasis 2021, 38, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.Q.; Zhang, X.; Zhang, S.; Zhu, T.; Garg, M.; Lobie, P.E.; Pandey, V. Mitochondria: The metabolic switch of cellular oncogenic transformation. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2021, 1876, 188534. [Google Scholar] [CrossRef]
- Zhang, X.; Su, Q.; Zhou, J.; Yang, Z.; Liu, Z.; Ji, L.; Gao, H.; Jiang, G. To betray or to fight? The dual identity of the mitochondria in cancer. Futur. Oncol. 2021, 17, 723–743. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Grasso, C.; Berridge, M.V. Metabolic reprogramming of mitochondrial respiration in metastatic cancer. Cancer Metastasis Rev. 2018, 37, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Bacci, M.; Lorito, N.; Ippolito, L.; Ramazzotti, M.; Luti, S.; Romagnoli, S.; Parri, M.; Bianchini, F.; Cappellesso, F.; Virga, F.; et al. Reprogramming of Amino Acid Transporters to Support Aspartate and Glutamate Dependency Sustains Endocrine Resistance in Breast Cancer. Cell Rep. 2019, 28, 104–118 e108. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Lau, P.; Kwan, Y.; Kong, S. Mitochondrial Fuel Dependence on Glutamine Drives Chemo-Resistance in the Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3315. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef]
- Mortezaee, K. Redox tolerance and metabolic reprogramming in solid tumors. Cell Biol. Int. 2021, 45, 273–286. [Google Scholar] [CrossRef]
- Oliveira, G.L.; Coelho, A.R.; Marques, R.; Oliveira, P.J. Cancer cell metabolism: Rewiring the mitochondrial hub. Biochim. et Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1867, 166016. [Google Scholar] [CrossRef]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cao, M.; Liu, J.; Yang, Q.; Miao, X.; Go, V.L.W.; Lee, P.W.N.; Xiao, G.G. Metabolic Regulation in Mitochondria and Drug Resistance. Adv. Exp. Med. Biol. 2017, 1038, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, S.; Mishra, S.V.; Ghorai, A.; Hole, A.; Chandrani, P.; Dutt, A.; Chilakapati, M.; Dutt, S. Metabolic rewiring in drug resistant cells exhibit higher OXPHOS and fatty acids as preferred major source to cellular energetics. Biochim. Biophys. Acta (BBA)—Bioenerg. 2020, 1861, 148300. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- Fiorillo, M.; Sotgia, F.; Lisanti, M.P. “Energetic” Cancer Stem Cells (e-CSCs): A New Hyper-Metabolic and Proliferative Tumor Cell Phenotype, Driven by Mitochondrial Energy. Front. Oncol. 2018, 8, 677. [Google Scholar] [CrossRef] [PubMed]
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature 2020, 588, 712–716. [Google Scholar] [CrossRef]
- Marzagalli, M.; Raimondi, M.; Fontana, F.; Marelli, M.M.; Moretti, R.M.; Limonta, P. Cellular and molecular biology of cancer stem cells in melanoma: Possible therapeutic implications. Semin. Cancer Biol. 2019, 59, 221–235. [Google Scholar] [CrossRef]
- Sotgia, F.; Fiorillo, M.; Lisanti, M.P. Hallmarks of the cancer cell of origin: Comparisons with “energetic” cancer stem cells (e-CSCs). Aging 2019, 11, 1065–1068. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Luo, Q.; Li, H.; Liu, Q.; Ju, Y.; Song, G. Increased Oxidative Phosphorylation is Required for Stemness Maintenance in Liver Cancer Stem Cells from Hepatocellular Carcinoma Cell Line HCCLM3 Cells. Int. J. Mol. Sci. 2020, 21, 5276. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Bhattacharyya, S. Cancer Stem Cells: Metabolic Characterization for Targeted Cancer Therapy. Front. Oncol. 2021, 11, 756888. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Fontana, F.; Raimondi, M.; Limonta, P. Cancer Stem Cells—Key Players in Tumor Relapse. Cancers 2021, 13, 376. [Google Scholar] [CrossRef] [PubMed]
- Zanotelli, M.R.; Goldblatt, Z.E.; Miller, J.P.; Bordeleau, F.; Li, J.; VanderBurgh, J.A.; Lampi, M.C.; King, M.R.; Reinhart-King, C.A. Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol. Biol. Cell 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016, 380, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef] [PubMed]
- Bacci, M.; Ippolito, L.; Magnelli, L.; Giannoni, E.; Chiarugi, P. Stromal-induced mitochondrial re-education: Impact on epithelial-to-mesenchymal transition and cancer aggressiveness. Semin. Cell Dev. Biol. 2020, 98, 71–79. [Google Scholar] [CrossRef]
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Weissig, V. Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. Int. J. Mol. Sci. 2015, 16, 17394–17421. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. Natural Compounds in Prostate Cancer Prevention and Treatment: Mechanisms of Action and Molecular Targets. Cells 2020, 9, 460. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Lee, H.; Jang, H.; Woo, S.M.; Park, J.B.; Lee, S.-H.; Kang, J.H.; Kim, H.Y.; Song, J.; Kim, S.-Y. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells 2020, 9, 2013. [Google Scholar] [CrossRef]
- Iessi, E.; Vona, R.; Cittadini, C.; Matarrese, P. Targeting the Interplay between Cancer Metabolic Reprogramming and Cell Death Pathways as a Viable Therapeutic Path. Biomedicines 2021, 9, 1942. [Google Scholar] [CrossRef]
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca(2+) overload- and ROS-associated mitochondrial dysfunction contributes to delta-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292. [Google Scholar] [CrossRef]
- Wu, Z.; Ho, W.S.; Lu, R. Targeting Mitochondrial Oxidative Phosphorylation in Glioblastoma Therapy. NeuroMol. Med. 2022, 24, 18–22. [Google Scholar] [CrossRef]
- Yu, H.-J.; Xiao, G.-L.; Zhao, Y.-Y.; Wang, X.-X.; Lan, R. Targeting Mitochondrial Metabolism and RNA Polymerase POLRMT to Overcome Multidrug Resistance in Cancer. Front. Chem. 2021, 9, 775226. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332. [Google Scholar] [CrossRef]
- Choudhury, F.K. Mitochondrial Redox Metabolism: The Epicenter of Metabolism during Cancer Progression. Antioxidants 2021, 10, 1838. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Parascandolo, A.; Laukkanen, M.O. Carcinogenesis and Reactive Oxygen Species Signaling: Interaction of the NADPH Oxidase NOX1-5 and Superoxide Dismutase 1-3 Signal Transduction Pathways. Antioxid. Redox Signal. 2019, 30, 443–486. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Kohan, R.; Collin, A.; Guizzardi, S.; Tolosa De Talamoni, N.; Picotto, G. Reactive oxygen species in cancer: A paradox between pro- and anti-tumour activities. Cancer Chemother. Pharmacol. 2020, 86, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and Downsides of Reactive Oxygen Species for Cancer: The Roles of Reactive Oxygen Species in Tumorigenesis, Prevention, and Therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.-C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxid. Med. Cell. Longev. 2021, 2021, 9965916. [Google Scholar] [CrossRef]
- Bekhet, O.H.; Eid, M.E. The interplay between reactive oxygen species and antioxidants in cancer progression and therapy: A narrative review. Transl. Cancer Res. 2021, 10, 4196–4206. [Google Scholar] [CrossRef]
- Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, 432. [Google Scholar] [CrossRef] [PubMed]
- Macasoi, I.; Mioc, A.; Mioc, M.; Racoviceanu, R.; Soica, I.; Chevereșan, A.; Dehelean, C.; Dumitrașcu, V. Targeting Mitochondria through the Use of Mitocans as Emerging Anticancer Agents. Curr. Med. Chem. 2019, 27, 5730–5757. [Google Scholar] [CrossRef] [PubMed]
- Sterea, A.M.; El Hiani, Y. The Role of Mitochondrial Calcium Signaling in the Pathophysiology of Cancer Cells. Adv. Exp. Med. Biol. 2019, 1131, 747–770. [Google Scholar] [CrossRef]
- Genovese, I.; Carinci, M.; Modesti, L.; Aguiari, G.; Pinton, P.; Giorgi, C. Mitochondria: Insights into Crucial Features to Overcome Cancer Chemoresistance. Int. J. Mol. Sci. 2021, 22, 4770. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Mitochondrial functional and structural impairment is involved in the antitumor activity of delta-tocotrienol in prostate cancer cells. Free. Radic. Biol. Med. 2020, 160, 376–390. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- Lee, Y.G.; Park, D.H.; Chae, Y.C. Role of Mitochondrial Stress Response in Cancer Progression. Cells 2022, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zang, C.; Yuan, F.; Ju, C.; Shang, M.; Ning, J.; Yang, Y.; Ma, J.; Li, G.; Bao, X.; et al. The role of FUNDC1 in mitophagy, mitochondrial dynamics and human diseases. Biochem. Pharmacol. 2022, 197, 114891. [Google Scholar] [CrossRef]
- Wang, S.; Tan, J.; Miao, Y.; Zhang, Q. Mitochondrial Dynamics, Mitophagy, and Mitochondria–Endoplasmic Reticulum Contact Sites Crosstalk Under Hypoxia. Front. Cell Dev. Biol. 2022, 10, 848214. [Google Scholar] [CrossRef]
- Grasso, D.; Medeiros, H.; Zampieri, L.X.; Bol, V.; Danhier, P.; Van Gisbergen, M.W.; Bouzin, C.; Brusa, D.; Grégoire, V.; Smeets, H.; et al. Fitter Mitochondria Are Associated with Radioresistance in Human Head and Neck SQD9 Cancer Cells. Front. Pharmacol. 2020, 11, 263. [Google Scholar] [CrossRef]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar]
- Zhang, Q.; Chen, W.; Xie, C.; Dai, X.; Ma, J.; Lu, J. The Role of PGC-1α in Digestive System Malignant Tumours. Anti-Cancer Agents Med. Chem. 2020, 20, 276–285. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef]
- Raggi, C.; Taddei, M.L.; Sacco, E.; Navari, N.; Correnti, M.; Piombanti, B.; Pastore, M.; Campani, C.; Pranzini, E.; Iorio, J.; et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J. Hepatol. 2021, 74, 1373–1385. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Panigrahi, D.P.; Bhol, C.S.; Patra, S.; Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Singh, A.; Patil, S.; Bhutia, S.K. Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: A potential target for anti-CSC cancer therapy. Cancer Lett. 2021, 498, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef]
- Martens, S.; McMahon, H.T. Mechanisms of membrane fusion: Disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008, 9, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, Y. Mitochondrial fission and fusion. Biochem. Soc. Trans. 2016, 44, 1725–1735. [Google Scholar] [CrossRef]
- Steffen, J.; Koehler, C.M. ER–mitochondria contacts: Actin dynamics at the ER control mitochondrial fission via calcium release. J. Cell Biol. 2017, 217, 15–17. [Google Scholar] [CrossRef]
- Banerjee, R.; Mukherjee, A.; Nagotu, S. Mitochondrial dynamics and its impact on human health and diseases: Inside the DRP1 blackbox. J. Mol. Med. 2021, 100, 1–21. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Kulikov, A.V.; Luchkina, E.A.; Gogvadze, V.; Zhivotovsky, B. Mitophagy: Link to cancer development and therapy. Biochem. Biophys. Res. Commun. 2017, 482, 432–439. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Gogvadze, V.; Zhivotovsky, B. Mitophagy in carcinogenesis and cancer treatment. Discov. Oncol. 2021, 12, 58. [Google Scholar] [CrossRef]
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2021, 11, 30. [Google Scholar] [CrossRef]
- Poole, L.P.; Macleod, K.F. Mitophagy in tumorigenesis and metastasis. Cell. Mol. Life Sci. 2021, 78, 3817–3851. [Google Scholar] [CrossRef]
- Qiu, Y.-H.; Zhang, T.-S.; Wang, X.-W.; Wang, M.-Y.; Zhao, W.-X.; Zhou, H.-M.; Zhang, C.-H.; Cai, M.-L.; Chen, X.-F.; Zhao, W.-L.; et al. Mitochondria autophagy: A potential target for cancer therapy. J. Drug Target. 2021, 29, 576–591. [Google Scholar] [CrossRef]
- Ray, S.K.; Mukherjee, S. Mitophagy in Carcinogenesis and Tumour Progression- A New Paradigm with Emerging Importance. Anti-Cancer Agents Med. Chem. 2021, 21, 2130–2141. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Labrie, F.; Candas, B.; Gomez, J.-L.; Cusan, L. Can combined androgen blockade provide long-term control or possible cure of localized prostate cancer? Urology 2002, 60, 115–119. [Google Scholar] [CrossRef]
- James, N.D.; De Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Onozawa, M.; Akaza, H.; Hinotsu, S.; Oya, M.; Ogawa, O.; Kitamura, T.; Suzuki, K.; Naito, S.; Namiki, M.; Nishimura, K.; et al. Combined androgen blockade achieved better oncological outcome in androgen deprivation therapy for prostate cancer: Analysis of community-based multi-institutional database across Japan using propensity score matching. Cancer Med. 2018, 7, 4893–4902. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juarez Soto, A.; Merseburger, A.S.; Ozguroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Kim, T.; Lee, Y.; Koo, K. Current Status and Future Perspectives of Androgen Receptor Inhibition Therapy for Prostate Cancer: A Comprehensive Review. Biomolecules 2021, 11, 492. [Google Scholar] [CrossRef]
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015, 6, 35542–35555. [Google Scholar] [CrossRef]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef]
- Marelli, M.M.; Moretti, R.M.; Januszkiewicz-Caulier, J.; Motta, M.; Limonta, P. Gonadotropin-Releasing Hormone (GnRH) Receptors in Tumors: A New Rationale for the Therapeutical Application of GnRH Analogs in Cancer Patients? Curr. Cancer Drug Targets 2006, 6, 257–269. [Google Scholar] [CrossRef]
- Limonta, P.; Marelli, M.M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH Receptors in Cancer: From Cell Biology to Novel Targeted Therapeutic Strategies. Endocr. Rev. 2012, 33, 784–811. [Google Scholar] [CrossRef]
- Limonta, P.; Manea, M. Gonadotropin-releasing hormone receptors as molecular therapeutic targets in prostate cancer: Current options and emerging strategies. Cancer Treat. Rev. 2013, 39, 647–663. [Google Scholar] [CrossRef]
- Manea, M.; Marelli, M.M.; Moretti, R.; Maggi, R.; Marzagalli, M.; Limonta, P. Targeting Hormonal Signaling Pathways in Castration Resistant Prostate Cancer. Recent Patents Anti-Cancer Drug Discov. 2014, 9, 267–285. [Google Scholar] [CrossRef]
- Jang, H.S.; Koo, K.C.; Cho, K.S.; Chung, B.H. Survival Outcomes of Concurrent Treatment with Docetaxel and Androgen Deprivation Therapy in Metastatic Castration-Resistant Prostate Cancer. Yonsei Med. J. 2016, 57, 1070–1078. [Google Scholar] [CrossRef]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef]
- Saad, F.; Shore, N.; Zhang, T.; Sharma, S.; Cho, H.K.; Jacobs, I.A. Emerging therapeutic targets for patients with advanced prostate cancer. Cancer Treat. Rev. 2019, 76, 1–9. [Google Scholar] [CrossRef]
- Fontana, F.; Marzagalli, M.; Marelli, M.M.; Raimondi, M.; Moretti, R.; Limonta, P. Gonadotropin-Releasing Hormone Receptors in Prostate Cancer: Molecular Aspects and Biological Functions. Int. J. Mol. Sci. 2020, 21, 9511. [Google Scholar] [CrossRef]
- Fontana, F.; Limonta, P. Dissecting the Hormonal Signaling Landscape in Castration-Resistant Prostate Cancer. Cells 2021, 10, 1133. [Google Scholar] [CrossRef]
- Hou, Z.; Huang, S.; Li, Z. Androgens in prostate cancer: A tale that never ends. Cancer Lett. 2021, 516, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Anselmi, M.; Limonta, P. Molecular mechanisms and genetic alterations in prostate cancer: From diagnosis to targeted therapy. Cancer Lett. 2022, 534, 215619. [Google Scholar] [CrossRef] [PubMed]
- Grigor, E.J.; Fergusson, D.; Kekre, N.; Montroy, J.; Atkins, H.; Seftel, M.D.; Daugaard, M.; Presseau, J.; Thavorn, K.; Hutton, B.; et al. Risks and Benefits of Chimeric Antigen Receptor T-Cell (CAR-T) Therapy in Cancer: A Systematic Review and Meta-Analysis. Transfus. Med. Rev. 2019, 33, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; Shenderov, E.; Eisenberger, M.A.; Kachhap, S.; Pardoll, D.M.; Denmeade, S.R.; Antonarakis, E.S. Extreme responses to immune checkpoint blockade following bipolar androgen therapy and enzalutamide in patients with metastatic castration resistant prostate cancer. Prostate 2020, 80, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.L. Immunotherapy in treatment of metastatic prostate cancer: An approach to circumvent immunosuppressive tumor microenvironment. Prostate 2021, 81, 1125–1134. [Google Scholar] [CrossRef]
- Siewe, N.; Friedman, A. Combination therapy for mCRPC with immune checkpoint inhibitors, ADT and vaccine: A mathematical model. PLoS ONE 2022, 17, e0262453. [Google Scholar] [CrossRef]
- Grupp, K.; Jedrzejewska, K.; Tsourlakis, M.C.; Koop, C.; Wilczak, W.; Adam, M.; Quaas, A.; Sauter, G.; Simon, R.; Izbicki, J.R.; et al. High mitochondria content is associated with prostate cancer disease progression. Mol. Cancer 2013, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Vayalil, P.K.; Landar, A. Mitochondrial oncobioenergetic index: A potential biomarker to predict progression from indolent to aggressive prostate cancer. Oncotarget 2015, 6, 43065–43080. [Google Scholar] [CrossRef]
- Kelly, R.S.; Sinnott, J.A.; Rider, J.R.; Ebot, E.M.; Gerke, T.; Bowden, M.; Pettersson, A.; Loda, M.; Sesso, H.D.; Kantoff, P.W.; et al. The role of tumor metabolism as a driver of prostate cancer progression and lethal disease: Results from a nested case-control study. Cancer Metab. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef]
- Costello, L.C.; Feng, P.; Milon, B.; Tan, M.; Franklin, R.B. Role of zinc in the pathogenesis and treatment of prostate cancer: Critical issues to resolve. Prostate Cancer Prostatic Dis. 2004, 7, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: Connecting the dots. Mol. Cancer 2006, 5, 17. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B.; Zou, J.; Feng, P.; Bok, R.; Swanson, M.G.; Kurhanewicz, J. Human prostate cancer ZIP1/zinc/citrate genetic/metabolic relationship in the TRAMP prostate cancer animal model. Cancer Biol. Ther. 2011, 12, 1078–1084. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef]
- Hacioglu, C.; Kacar, S.; Kar, F.; Kanbak, G.; Sahinturk, V. Concentration-Dependent Effects of Zinc Sulfate on DU-145 Human Prostate Cancer Cell Line: Oxidative, Apoptotic, Inflammatory, and Morphological Analyzes. Biol. Trace Element Res. 2020, 195, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L.; Afyounian, E.; Jylhä, A.; Nättinen, J.; Aapola, U.; Annala, M.; Kivinummi, K.K.; Tammela, T.T.L.; Beuerman, R.W.; Uusitalo, H.; et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 2018, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Ye, G.; Ren, S.; Piao, H.-L.; Zhao, X.; Lu, X.; Wang, F.; Ma, W.; Li, J.; Yin, P.; et al. Metabolomics and transcriptomics profiles reveal the dysregulation of the tricarboxylic acid cycle and related mechanisms in prostate cancer. Int. J. Cancer 2018, 143, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef]
- Kolenko, V.; Teper, E.; Kutikov, A.; Uzzo, R. Zinc and zinc transporters in prostate carcinogenesis. Nat. Rev. Urol. 2013, 10, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.-C.; Anderle, P.; Bürzle, M.; Suzuki, Y.; Freeman, M.; Hediger, M.; Kovacs, G. Zinc transporters in prostate cancer. Mol. Asp. Med. 2013, 34, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.B.; Golovine, K.V.; Kutikov, A.; Canter, D.J.; Rybko, V.A.; Roshchin, D.A.; Matveev, V.B.; Uzzo, R.G.; Kolenko, V.M. Reversal of epigenetic silencing of AP-2alpha results in increased zinc uptake in DU-145 and LNCaP prostate cancer cells. Carcinogenesis 2011, 32, 1773–1781. [Google Scholar] [CrossRef]
- Xue, Y.N.; Yu, B.B.; Liu, Y.N.; Guo, R.; Li, L.J.; Zhang, L.C.; Su, J.; Sun, L.K.; Li, Y. Zinc promotes prostate cancer cell chemosensitivity to paclitaxel by inhibiting epithelial-mesenchymal transition and inducing apoptosis. Prostate 2019, 79, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Sauer, A.K.; Vela, H.; Vela, G.; Stark, P.; Barrera-Juarez, E.; Grabrucker, A.M. Zinc Deficiency in Men Over 50 and Its Implications in Prostate Disorders. Front. Oncol. 2020, 10, 1293. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef]
- Bazylianska, V.; Kalpage, H.A.; Wan, J.; Vaishnav, A.; Mahapatra, G.; Turner, A.A.; Chowdhury, D.D.; Kim, K.; Morse, P.T.; Lee, I.; et al. Lysine 53 Acetylation of Cytochrome c in Prostate Cancer: Warburg Metabolism and Evasion of Apoptosis. Cells 2021, 10, 802. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Shen, Z.; Huang, S.; Zhao, L.; Chen, S.; Mak, T.W.; Wang, X. The ER UDPase ENTPD5 Promotes Protein N-Glycosylation, the Warburg Effect, and Proliferation in the PTEN Pathway. Cell 2010, 143, 711–724. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.-J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-Mediated Warburg Effect Is Required for PTEN- and p53-Deficiency-Driven Prostate Cancer Growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Mills, I.G. The Interplay Between Prostate Cancer Genomics, Metabolism, and the Epigenome: Perspectives and Future Prospects. Front. Oncol. 2021, 11, 704353. [Google Scholar] [CrossRef]
- Wang, C.; Tao, W.; Ni, S.; Chen, Q. SENP1 Interacts with HIF1α to Regulate Glycolysis of Prostatic Carcinoma Cells. Int. J. Biol. Sci. 2019, 15, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Huang, Q.; Peng, F.; Wang, J.; Zhao, W.; Guo, G. Expression and Clinical Significance of HKII and HIF-1α in Grade Groups of Prostate Cancer. Front. Genet. 2021, 12, 680928. [Google Scholar] [CrossRef] [PubMed]
- Nassar, Z.D.; Aref, A.T.; Miladinovic, D.; Mah, C.Y.; Raj, G.V.; Hoy, A.J.; Butler, L.M. Peri-prostatic adipose tissue: The metabolic microenvironment of prostate cancer. BJU Int. 2018, 121 (Suppl. S3), 9–21. [Google Scholar] [CrossRef] [PubMed]
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the Adipose Microenvironment and the Obesity–Cancer Link—A Systematic Review. Cancer Prev. Res. 2017, 10, 494–506. [Google Scholar] [CrossRef]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumorsviaHIF-1α activation. Oncotarget 2016, 7, 64854–64877. [Google Scholar] [CrossRef] [PubMed]
- Cutruzzolà, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Anselmi, M.; Carollo, E.; Sartori, P.; Procacci, P.; Carter, D.; Limonta, P. Adipocyte-Derived Extracellular Vesicles Promote Prostate Cancer Cell Aggressiveness by Enabling Multiple Phenotypic and Metabolic Changes. Cells 2022, 11, 2388. [Google Scholar] [CrossRef] [PubMed]
- Dakubo, G.D.; Parr, R.L.; Costello, L.C.; Franklin, R.B.; Thayer, R.E. Altered metabolism and mitochondrial genome in prostate cancer. J. Clin. Pathol. 2006, 59, 10–16. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef]
- Oberhuber, M.; Pecoraro, M.; Rusz, M.; Oberhuber, G.; Wieselberg, M.; Haslinger, P.; Gurnhofer, E.; Schlederer, M.; Limberger, T.; Lagger, S.; et al. STAT 3 -dependent analysis reveals PDK 4 as independent predictor of recurrence in prostate cancer. Mol. Syst. Biol. 2020, 16, e9247. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Lin, C.-Y.; Kung, H.-J. Targeting Mitochondrial OXPHOS and Their Regulatory Signals in Prostate Cancers. Int. J. Mol. Sci. 2021, 22, 13435. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, L.C.A.; Mui, E.; Nixon, C.; Hedley, A.; Strachan, D.; MacKay, G.; Sumpton, D.; Sansom, O.J.; Leung, H.Y.; Ahmad, I. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene 2021, 40, 2355–2366. [Google Scholar] [CrossRef]
- Giannoni, E.; Taddei, M.L.; Morandi, A.; Comito, G.; Calvani, M.; Bianchini, F.; Richichi, B.; Raugei, G.; Wong, N.; Tang, D.; et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs OXPHOS and prostate cancer metastatic spread. Oncotarget 2015, 6, 24061–24074. [Google Scholar] [CrossRef]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM 2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef]
- Hsu, M.-C.; Hung, W.-C. Pyruvate kinase M2 fuels multiple aspects of cancer cells: From cellular metabolism, transcriptional regulation to extracellular signaling. Mol. Cancer 2018, 17, 35. [Google Scholar] [CrossRef]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2018, 1, 70–85. [Google Scholar] [CrossRef]
- Lee, Y.G.; Nam, Y.; Shin, K.J.; Yoon, S.; Park, W.S.; Joung, J.Y.; Seo, J.K.; Jang, J.; Lee, S.; Nam, D.; et al. Androgen-induced expression of DRP1 regulates mitochondrial metabolic reprogramming in prostate cancer. Cancer Lett. 2020, 471, 72–87. [Google Scholar] [CrossRef]
- Kumar, R.P.; Ray, S.; Home, P.; Saha, B.; Bhattacharya, B.; Wilkins, H.M.; Chavan, H.; Ganguly, A.; Milano-Foster, J.; Paul, A.; et al. Regulation of energy metabolism during early mammalian development: TEAD4 controls mitochondrial transcription. Development 2018, 145, dev162644. [Google Scholar] [CrossRef]
- Chen, C.-L.; Hsu, S.-C.; Chung, T.-Y.; Chu, C.-Y.; Wang, H.-J.; Hsiao, P.-W.; Yeh, S.-D.; Ann, D.K.; Yen, Y.; Kung, H.-J. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nat. Commun. 2021, 12, 2398. [Google Scholar] [CrossRef]
- Skvortsov, S.; Skvortsova, I.-I.; Tang, D.G.; Dubrovska, A. Concise Review: Prostate Cancer Stem Cells: Current Understanding. Stem Cells 2018, 36, 1457–1474. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Lisanti, M.P. Cancer Metabolism: New Validated Targets for Drug Discovery. Oncotarget 2013, 4, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Ozsvari, B.; Fiorillo, M.; De Francesco, E.M.; Bonuccelli, G.; Lisanti, M.P. A mitochondrial based oncology platform for targeting cancer stem cells (CSCs): MITO-ONC-RX. Cell Cycle 2018, 17, 2091–2100. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.J.; Klotz, L.H.; Venkateswaran, V. Metformin and prostate cancer stem cells: A novel therapeutic target. Prostate Cancer Prostatic Dis. 2015, 18, 303–309. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, X.; Yu, D.; Li, X.; Li, Y.; Long, Y.; Yuan, Y.; Ji, Z.; Zhang, M.; Wen, J.-G.; et al. Application of mitochondrial pyruvate carrier blocker UK5099 creates metabolic reprogram and greater stem-like properties in LnCap prostate cancer cells in vitro. Oncotarget 2015, 6, 37758–37769. [Google Scholar] [CrossRef]
- Basu, H.S.; Wilganowski, N.; Robertson, S.; Reuben, J.M.; Cohen, E.N.; Zurita, A.; Ramachandran, S.; Xiao, L.; Titus, M.; Wilding, G. Prostate cancer cells survive anti-androgen and mitochondrial metabolic inhibitors by modulating glycolysis and mitochondrial metabolic activities. Prostate 2021, 81, 799–811. [Google Scholar] [CrossRef]
- Mamouni, K.; Kallifatidis, G.; Lokeshwar, B. Targeting Mitochondrial Metabolism in Prostate Cancer with Triterpenoids. Int. J. Mol. Sci. 2021, 22, 2466. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 2013, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, É.; Vernier, M.; Yee, T.; E Laflamme, C.; Li, S.; Chen, Y.; Giguère, V. SREBF1 Activity Is Regulated by an AR/mTOR Nuclear Axis in Prostate Cancer. Mol. Cancer Res. 2018, 16, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Gonthier, K.; Poluri, R.T.K.; Audet-Walsh, E. Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. J. Steroid Biochem. Mol. Biol. 2019, 191, 105367. [Google Scholar] [CrossRef] [PubMed]
- Stone, L. Mitochondrial metabolism: A target in AR-driven disease. Nat. Rev. Urol. 2019, 16, 1. [Google Scholar] [CrossRef]
- Xu, H.; Liu, Z.; Gao, D.; Li, P.; Shen, Y.; Sun, Y.; Xu, L.; Song, N.; Wang, Y.; Zhan, M.; et al. Reprogramming hormone-sensitive prostate cancer to a lethal neuroendocrine cancer lineage by mitochondrial pyruvate carrier (MPC). Mol. Metab. 2022, 59, 101466. [Google Scholar] [CrossRef]
- Wu, M.-J.; Chen, C.-J.; Lin, T.-Y.; Liu, Y.-Y.; Tseng, L.-L.; Cheng, M.-L.; Chuu, C.-P.; Tsai, H.-K.; Kuo, W.-L.; Kung, H.-J.; et al. Targeting KDM4B that coactivates c-Myc-regulated metabolism to suppress tumor growth in castration-resistant prostate cancer. Theranostics 2021, 11, 7779–7796. [Google Scholar] [CrossRef]
- Bajpai, P.; Koc, E.; Sonpavde, G.; Singh, R.; Singh, K.K. Mitochondrial localization, import, and mitochondrial function of the androgen receptor. J. Biol. Chem. 2019, 294, 6621–6634. [Google Scholar] [CrossRef]
- Valcarcel-Jimenez, L.; Gaude, E.; Torrano, V.; Frezza, C.; Carracedo, A. Mitochondrial Metabolism: Yin and Yang for Tumor Progression. Trends Endocrinol. Metab. 2017, 28, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free. Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef]
- Chen, J.Z.; Gokden, N.; Greene, G.F.; Mukunyadzi, P.; Kadlubar, F.F. Extensive somatic mitochondrial mutations in primary prostate cancer using laser capture microdissection. Cancer Res. 2002, 62, 6470–6474. [Google Scholar] [PubMed]
- Chen, J.Z.; Kadlubar, F.F.; Chen, J.Z. Mitochondrial Mutagenesis and Oxidative Stress in Human Prostate Cancer. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2004, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Mills, I.G.; Klevebring, D.; Liu, W.; Neiman, M.; Xu, J.; Wikström, P.; Wiklund, P.; Wiklund, F.; Egevad, L.; et al. The Mitochondrial and Autosomal Mutation Landscapes of Prostate Cancer. Eur. Urol. 2013, 63, 702–708. [Google Scholar] [CrossRef]
- McCrow, J.P.; Petersen, D.C.; Louw, M.; Chan, E.K.F.; Harmeyer, K.; Vecchiarelli, S.; Lyons, R.J.; Bornman, M.S.R.; Hayes, V.M. Spectrum of mitochondrial genomic variation and associated clinical presentation of prostate cancer in South African men. Prostate 2016, 76, 349–358. [Google Scholar] [CrossRef]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef]
- Gómez-Zaera, M.; Abril, J.; González, L.; Aguiló, F.; Condom, E.; Nadal, M.; Nunes, V. Identification of somatic and germline mitochondrial DNA sequence variants in prostate cancer patients. Mutat. Res. 2006, 595, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Kloss-Brandstätter, A.; Schäfer, G.; Erhart, G.; Hüttenhofer, A.; Coassin, S.; Seifarth, C.; Summerer, M.; Bektic, J.; Klocker, H.; Kronenberg, F. Somatic Mutations throughout the Entire Mitochondrial Genome Are Associated with Elevated PSA Levels in Prostate Cancer Patients. Am. J. Hum. Genet. 2010, 87, 802–812. [Google Scholar] [CrossRef]
- Niedzwiecka, K.; Tisi, R.; Penna, S.; Lichocka, M.; Plochocka, D.; Kucharczyk, R. Two mutations in mitochondrial ATP6 gene of ATP synthase, related to human cancer, affect ROS, calcium homeostasis and mitochondrial permeability transition in yeast. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 117–131. [Google Scholar] [CrossRef]
- Schöpf, B.; Weissensteiner, H.; Schäfer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1487. [Google Scholar] [CrossRef]
- Sun, Q.; Arnold, R.S.; Sun, C.Q.; Petros, J.A. A mitochondrial DNA mutation influences the apoptotic effect of statins on prostate cancer. Prostate 2015, 75, 1916–1925. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Audano, M.; Pedretti, S.; Ligorio, S.; Crestani, M.; Caruso, D.; De Fabiani, E.; Mitro, N. “The Loss of Golden Touch”: Mitochondria-Organelle Interactions, Metabolism, and Cancer. Cells 2020, 9, 2519. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, A.; Venero, J.L.; Iwasawa, R.; Hankir, M.-K.; Rahman, S.; Boobis, A.; Hajji, N. PGC-1α controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging 2015, 7, 629–647. [Google Scholar] [CrossRef]
- Piyarathna, D.W.B.; Balasubramanian, A.; Arnold, J.M.; Lloyd, S.M.; Karanam, B.; Castro, P.; Ittmann, M.M.; Putluri, N.; Navone, N.; Jones, J.A.; et al. ERR1- and PGC1α-associated mitochondrial alterations correlate with pan-cancer disparity in African Americans. J. Clin. Investig. 2019, 129, 2351–2356. [Google Scholar] [CrossRef]
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Fujimoto, N.; Seki, N.; Naito, S. Peroxisome Proliferator-Activated Receptor γ Coactivator-1α Interacts with the Androgen Receptor (AR) and Promotes Prostate Cancer Cell Growth by Activating the AR. Mol. Endocrinol. 2010, 24, 114–127. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Chen, L.; Yu, B.; Xue, Y.; Guo, R.; Su, J.; Liu, Y.; Sun, L. p53/PGC-1α-mediated mitochondrial dysfunction promotes PC3 prostate cancer cell apoptosis. Mol. Med. Rep. 2020, 22, 155–164. [Google Scholar] [CrossRef]
- Burch, T.C.; Rhim, J.S.; Nyalwidhe, J.O. Mitochondria Biogenesis and Bioenergetics Gene Profiles in Isogenic Prostate Cells with Different Malignant Phenotypes. BioMed Res. Int. 2016, 2016, 1785201. [Google Scholar] [CrossRef]
- Kaddour-Djebbar, I.; Choudhary, V.; Brooks, C.; Ghazaly, T.; Lakshmikanthan, V.; Dong, Z.; Kumar, M.V. Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells. Int. J. Oncol. 2010, 36, 1437–1444. [Google Scholar] [CrossRef]
- Philley, J.V.; Kannan, A.; Qin, W.; Sauter, E.R.; Ikebe, M.; Hertweck, K.L.; Troyer, D.A.; Semmes, O.J.; Dasgupta, S. Complex-I Alteration and Enhanced Mitochondrial Fusion Are Associated With Prostate Cancer Progression. J. Cell. Physiol. 2016, 231, 1364–1374. [Google Scholar] [CrossRef]
- Civenni, G.; Bosotti, R.; Timpanaro, A.; Vàzquez, R.; Merulla, J.; Pandit, S.; Rossi, S.; Albino, D.; Allegrini, S.; Mitra, A.; et al. Epigenetic Control of Mitochondrial Fission Enables Self-Renewal of Stem-like Tumor Cells in Human Prostate Cancer. Cell Metab. 2019, 30, 303–318 e306. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Carbone, G.M.; Catapano, C.V. Mitochondrial fission promotes self-renewal and tumorigenic potential in prostate cancer. Mol. Cell. Oncol. 2019, 6, e1644598. [Google Scholar] [CrossRef]
- Yan, C.; Li, T.-S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer. Res. 2018, 38, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pan, W.; Xu, G.; Chen, L. Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clin. Chim. Acta 2020, 502, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin. Cancer Biol. 2020, 66, 45–58. [Google Scholar] [CrossRef]
- Dai, K.; Radin, D.P.; Leonardi, D. Deciphering the dual role and prognostic potential of PINK1 across cancer types. Neural Regen. Res. 2021, 16, 659–665. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Zhang, W.; Zhang, D.; Li, Y.; Zhang, J.; Zhang, Y.; Diao, T.; Cui, L.; Li, W.; et al. Abiraterone and MDV3100 inhibits the proliferation and promotes the apoptosis of prostate cancer cells through mitophagy. Cancer Cell Int. 2019, 19, 332. [Google Scholar] [CrossRef]
- Katreddy, R.R.; Bollu, L.R.; Su, F.; Xian, N.; Srivastava, S.; Thomas, R.; Dai, Y.; Wu, B.; Xu, Y.; Rea, M.A.; et al. Targeted reduction of the EGFR protein, but not inhibition of its kinase activity, induces mitophagy and death of cancer cells through activation of mTORC2 and Akt. Oncogenesis 2018, 7, 5. [Google Scholar] [CrossRef]
- Zou, J.; Yue, F.; Li, W.; Song, K.; Jiang, X.; Yi, J.; Liu, L. Autophagy Inhibitor LRPPRC Suppresses Mitophagy through Interaction with Mitophagy Initiator Parkin. PLoS ONE 2014, 9, e94903. [Google Scholar] [CrossRef]
- Balvan, J.; Gumulec, J.; Raudenska, M.; Krizova, A.; Štěpka, P.; Babula, P.; Kizek, R.; Adam, V.; Masarik, M. Oxidative Stress Resistance in Metastatic Prostate Cancer: Renewal by Self-Eating. PLoS ONE 2015, 10, e0145016. [Google Scholar] [CrossRef]
- Qu, F.; Gu, Y.; Xue, M.; He, M.; Zhou, F.; Wang, G.; Peng, Y. Impact of therapy on cancer metabolism in high-risk localized prostate cancer treated with neoadjuvant docetaxel and androgen deprivation therapy. Prostate 2021, 81, 560–571. [Google Scholar] [CrossRef]
- Hatoum, D.; McGowan, E.M. Recent Advances in the Use of Metformin: Can Treating Diabetes Prevent Breast Cancer? BioMed Res. Int. 2015, 2015, 548436. [Google Scholar] [CrossRef]
- Maruthur, N.M.; Tseng, E.; Hutfless, S.; Wilson, L.M.; Suarez-Cuervo, C.; Berger, Z.; Chu, Y.; Iyoha, E.; Segal, J.B.; Bolen, S. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: A Systematic Review and Meta-analysis. Ann. Intern. Med. 2016, 164, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. 2017, 219, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Del Barco, S.; Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martín-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.N. Investigating Metformin for Cancer Prevention and Treatment: The End of the Beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Zaidi, S.; Gandhi, J.; Joshi, G.; Smith, N.L.; Khan, S.A. The anticancer potential of metformin on prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 351–361. [Google Scholar] [CrossRef]
- DeCensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and Cancer Risk in Diabetic Patients: A Systematic Review and Meta-analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer Risk in Diabetic Patients Treated with Metformin: A Systematic Review and Meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef]
- León-González, A.J.; Jiménez-Vacas, J.M.; Fuentes-Fayos, A.C.; Sarmento-Cabral, A.; Herrera-Martínez, A.D.; Gahete, M.D.; Luque, R.M. Role of metformin and other metabolic drugs in the prevention and therapy of endocrine-related cancers. Curr. Opin. Pharmacol. 2021, 60, 17–26. [Google Scholar] [CrossRef]
- Li, H.-X.; Gao, J.-M.; Liang, J.-Q.; Xi, J.-M.; Fu, M.; Wu, Y.-J. Vitamin D3potentiates the growth inhibitory effects of metformin in DU145 human prostate cancer cells mediated by AMPK/mTOR signalling pathway. Clin. Exp. Pharmacol. Physiol. 2015, 42, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, C.; He, T.; Mao, J.; Li, C.; Lyu, J.; Meng, Q.H. Metformin inhibits prostate cancer cell proliferation, migration, and tumor growth through upregulation of PEDF expression. Cancer Biol. Ther. 2016, 17, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Li, C.; Gao, X.; Liu, Z.; Chen, C.; Luo, D. Metformin inhibits tumor growth and affects intestinal flora in diabetic tumor-bearing mice. Eur. J. Pharmacol. 2021, 912, 174605. [Google Scholar] [CrossRef]
- Morale, M.G.; Tamura, R.E.; Rubio, I.G.S. Metformin and Cancer Hallmarks: Molecular Mechanisms in Thyroid, Prostate and Head and Neck Cancer Models. Biomolecules 2022, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Demir, Ü.; Köhler, A.; Schneider, R.; Schweiger, S.; Klocker, H. Metformin anti-tumor effect via disruption of the MID1 translational regulator complex and AR downregulation in prostate cancer cells. BMC Cancer 2014, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sekine, Y.; Furuya, Y.; Miyazawa, Y.; Koike, H.; Suzuki, K. Metformin inhibits the proliferation of human prostate cancer PC-3 cells via the downregulation of insulin-like growth factor 1 receptor. Biochem. Biophys. Res. Commun. 2015, 461, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, G.; Tong, D.; Parmar, H.; Hasenmayer, D.; Yuan, W.; Zhang, D.; Jiang, J. Metformin represses androgen-dependent and androgen-independent prostate cancers by targeting androgen receptor. Prostate 2015, 75, 1187–1196. [Google Scholar] [CrossRef]
- Sun, S.; Gong, F.; Liu, P.; Miao, Q. Metformin combined with quercetin synergistically repressed prostate cancer cells via inhibition of VEGF/PI3K/Akt signaling pathway. Gene 2018, 664, 50–57. [Google Scholar] [CrossRef]
- Yang, B.; Damodaran, S.; Khemees, T.A.; Filon, M.J.; Schultz, A.; Gawdzik, J.; Etheridge, T.; Malin, D.; Richards, K.A.; Cryns, V.L.; et al. Synthetic Lethal Metabolic Targeting of Androgen-Deprived Prostate Cancer Cells with Metformin. Mol. Cancer Ther. 2020, 19, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Ofori, S.; Mertens, R.T.; Parkin, S.; Awuah, S.G. Water-Soluble Gold(III)–Metformin Complex Alters Mitochondrial Bioenergetics in Breast Cancer Cells. Chemmedchem 2021, 16, 3222–3230. [Google Scholar] [CrossRef]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming Metabolism with Metformin Improves Tumor Oxygenation and Radiotherapy Response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Lee, Y.; Koo, K. Current Status and Application of Metformin for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 8540. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Vacas, J.M.; Herrero-Aguayo, V.; Montero-Hidalgo, A.J.; Sáez-Martínez, P.; Gómez-Gómez, E.; León-González, A.J.; Fuentes-Fayos, A.C.; Yubero-Serrano, E.M.; Requena-Tapia, M.J.; López, M.; et al. Clinical, Cellular, and Molecular Evidence of the Additive Antitumor Effects of Biguanides and Statins in Prostate Cancer. J. Clin. Endocrinol. Metab. 2021, 106, e696–e710. [Google Scholar] [CrossRef] [PubMed]
- Loubière, C.; Goiran, T.; Laurent, K.; Djabari, Z.; Tanti, J.-F.; Bost, F. Metformin-induced energy deficiency leads to the inhibition of lipogenesis in prostate cancer cells. Oncotarget 2015, 6, 15652–15661. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, C.; Wang, L.; Ma, Q.; Xia, P.; Qi, M.; Yang, M.; Han, B. Metformin inhibits epithelial–mesenchymal transition in prostate cancer cells: Involvement of the tumor suppressor miR30a and its target gene SOX4. Biochem. Biophys. Res. Commun. 2014, 452, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.; Beretov, J.; Ni, J.; Bai, X.; Bucci, J.; Graham, P.; Li, Y. Cancer stem cells in prostate cancer radioresistance. Cancer Lett. 2019, 465, 94–104. [Google Scholar] [CrossRef]
- Villani, L.A.; Smith, B.K.; Marcinko, K.; Ford, R.J.; Broadfield, L.A.; Green, A.E.; Houde, V.P.; Muti, P.; Tsakiridis, T.; Steinberg, G.R. The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol. Metab. 2016, 5, 1048–1056. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting Cancer Cell Metabolism: The Combination of Metformin and 2-Deoxyglucose Induces p53-Dependent Apoptosis in Prostate Cancer Cells. Cancer Res 2010, 70, 2465–2475. [Google Scholar] [CrossRef]
- Tsakiridis, E.E.; Broadfield, L.; Marcinko, K.; Biziotis, O.-D.; Ali, A.; Mekhaeil, B.; Ahmadi, E.; Singh, K.; Mesci, A.; Zacharidis, P.G.; et al. Combined metformin-salicylate treatment provides improved anti-tumor activity and enhanced radiotherapy response in prostate cancer; drug synergy at clinically relevant doses. Transl. Oncol. 2021, 14, 101209. [Google Scholar] [CrossRef] [PubMed]
- Joentausta, R.M.; Rannikko, A.; Murtola, T.J. Prostate Cancer–specific Survival After Radical Prostatectomy Is Improved Among Metformin Users but Not Among Other Antidiabetic Drug Users. Eur. Urol. Open Sci. 2021, 34, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Chang, S.-H.; Hicks, V.; Wang, M.; Grubb, R.L.; Drake, B.F., 3rd. Improved survival with post-diagnostic metformin and statin use in a racially diverse cohort of US Veterans with advanced prostate cancer. Prostate Cancer Prostatic Dis. 2021, 25, 707–712. [Google Scholar] [CrossRef]
- Yao, X.; Liu, H.; Xu, H. The Impact of Metformin Use with Survival Outcomes in Urologic Cancers: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2021, 2021, 5311828. [Google Scholar] [CrossRef]
- Aboelnaga, E.M.; Aboelnaga, M.M.; Elkalla, H.M. Metformin addition to androgen deprivation therapy effect on cancer prostate patients with type 2 diabetes. Diabetes Metab. Syndr. 2021, 15, 102251. [Google Scholar] [CrossRef]
- Alghandour, R.; Ebrahim, M.A.; Elshal, A.M.; Ghobrial, F.; Elzaafarany, M.; Elbaiomy, M.A. Repurposing metformin as anticancer drug: Randomized controlled trial in advanced prostate cancer (MANSMED). Urol. Oncol. 2021, 39, 831.e1–831.e10. [Google Scholar] [CrossRef]
- Cui, H.; Wang, Y.; Yang, S.; He, G.; Jiang, Z.; Gang, X.; Wang, G. Antidiabetic medications and the risk of prostate cancer in patients with diabetes mellitus: A systematic review and meta-analysis. Pharmacol. Res. 2022, 177, 106094. [Google Scholar] [CrossRef]
- Bilusic, M.; Toney, N.J.; Donahue, R.N.; Wroblewski, S.; Zibelman, M.; Ghatalia, P.; Ross, E.A.; Karzai, F.; Madan, R.A.; Dahut, W.L.; et al. A randomized phase 2 study of bicalutamide with or without metformin for biochemical recurrence in overweight or obese prostate cancer patients (BIMET-1). Prostate Cancer Prostatic Dis. 2022, 25, 735–740. [Google Scholar] [CrossRef]
- Martin, M.P.; Borchiellini, D.; Thamphya, B.; Guillot, A.; Paoli, J.-B.; Besson, D.; Hilgers, W.; Priou, F.; El Kouri, C.; Hoch, B.; et al. TAXOMET: A French Prospective Multicentric Randomized Phase II Study of Docetaxel Plus Metformin Versus Docetaxel Plus Placebo in Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2021, 19, 501–509. [Google Scholar] [CrossRef]
- Mitrakas, A.G.; Kalamida, D.; Koukourakis, M.I. Effect of mitochondrial metabolism-interfering agents on cancer cell mitochondrial function and radio/chemosensitivity. Anti-Cancer Drugs 2014, 25, 1182–1191. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.-L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther.—Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Elbehairi, S.E.I.; Alfaifi, M.Y.; Shati, A.A.; AlShehri, M.A.; Elshaarawy, R.F.; Hafez, H.S. Role of Pd(II)–chitooligosaccharides–Gboxin analog in oxidative phosphorylation inhibition and energy depletion: Targeting mitochondrial dynamics. Chem. Biol. Drug Des. 2020, 96, 1148–1161. [Google Scholar] [CrossRef]
- Gao, Y.; Shang, Q.; Li, W.; Guo, W.; Stojadinovic, A.; Mannion, C.; Man, Y.-G.; Chen, T. Antibiotics for cancer treatment: A double-edged sword. J. Cancer 2020, 11, 5135–5149. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed]
- Ozsvari, B.; Fiorillo, M.; Bonuccelli, G.; Cappello, A.R.; Frattaruolo, L.; Sotgia, F.; Trowbridge, R.; Foster, R.; Lisanti, M.P. Mitoriboscins: Mitochondrial-based therapeutics targeting cancer stem cells (CSCs), bacteria and pathogenic yeast. Oncotarget 2017, 8, 67457–67472. [Google Scholar] [CrossRef]
- Ozsvari, B.; Bonuccelli, G.; Sanchez-Alvarez, R.; Foster, R.; Sotgia, F.; Lisanti, M.P. Targeting flavin-containing enzymes eliminates cancer stem cells (CSCs), by inhibiting mitochondrial respiration: Vitamin B2 (Riboflavin) in cancer therapy. Aging 2017, 9, 2610–2628. [Google Scholar] [CrossRef]
- Fiorillo, M.; Tóth, F.; Sotgia, F.; Lisanti, M.P. Doxycycline, Azithromycin and Vitamin C (DAV): A potent combination therapy for targeting mitochondria and eradicating cancer stem cells (CSCs). Aging 2019, 11, 2202–2216. [Google Scholar] [CrossRef]
- Wanyan, Y.; Xu, X.; Liu, K.; Zhang, H.; Zhen, J.; Zhang, R.; Wen, J.; Liu, P.; Chen, Y. 2-Deoxy-d-glucose Promotes Buforin IIb-Induced Cytotoxicity in Prostate Cancer DU145 Cells and Xenograft Tumors. Molecules 2020, 25, 5778. [Google Scholar] [CrossRef]
- Shahidi, F.; De Camargo, A.C. Tocopherols and Tocotrienols in Common and Emerging Dietary Sources: Occurrence, Applications, and Health Benefits. Int. J. Mol. Sci. 2016, 17, 1745. [Google Scholar] [CrossRef]
- Beretta, G.; Gelmini, F.; Fontana, F.; Moretti, R.M.; Marelli, M.M.; Limonta, P. Semi-preparative HPLC purification of δ-tocotrienol (δ-T3) from Elaeis guineensis Jacq. and Bixa orellana L. and evaluation of its in vitro anticancer activity in human A375 melanoma cells. Nat. Prod. Res. 2018, 32, 1130–1135. [Google Scholar] [CrossRef]
- Chin, K.-Y.; Pang, K.-L.; Soelaiman, I.-N. Tocotrienol and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 97–130. [Google Scholar] [CrossRef]
- Comitato, R.; Ambra, R.; Virgili, F. Tocotrienols: A Family of Molecules with Specific Biological Activities. Antioxidants 2017, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.-Y.; Tay, S.S. A Review on the Relationship between Tocotrienol and Alzheimer Disease. Nutrients 2018, 10, 881. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, N.; Tan, E.; Loh, L.J.; Soh, B.S.; Yap, W.N. Tocotrienol is a cardioprotective agent against ageing-associated cardiovascular disease and its associated morbidities. Nutr. Metab. 2018, 15, 6. [Google Scholar] [CrossRef]
- Casati, L.; Pagani, F.; Limonta, P.; Vanetti, C.; Stancari, G.; Sibilia, V. Beneficial effects of δ-tocotrienol against oxidative stress in osteoblastic cells: Studies on the mechanisms of action. Eur. J. Nutr. 2019, 59, 1975–1987. [Google Scholar] [CrossRef]
- Wong, S.K.; Kamisah, Y.; Mohamed, N.; Muhammad, N.; Masbah, N.; Fahami, N.A.M.; Mohamed, I.N.; Shuid, A.N.; Saad, Q.M.; Abdullah, A.; et al. Potential Role of Tocotrienols on Non-Communicable Diseases: A Review of Current Evidence. Nutrients 2020, 12, 259. [Google Scholar] [CrossRef] [PubMed]
- Naomi, R.; Shafie, N.H.; Kaniappan, P.; Bahari, H. An Interactive Review on the Role of Tocotrienols in the Neurodegenerative Disorders. Front. Nutr. 2021, 8, 754086. [Google Scholar] [CrossRef]
- Montagnani Marelli, M.; Marzagalli, M.; Moretti, R.M.; Beretta, G.; Casati, L.; Comitato, R.; Gravina, G.L.; Festuccia, C.; Limonta, P. Vitamin E delta-tocotrienol triggers endoplasmic reticulum stress-mediated apoptosis in human melanoma cells. Sci. Rep. 2016, 6, 30502. [Google Scholar] [CrossRef]
- Marzagalli, M.; Moretti, R.M.; Messi, E.; Marelli, M.M.; Fontana, F.; Anastasia, A.; Bani, M.R.; Beretta, G.; Limonta, P. Targeting melanoma stem cells with the Vitamin E derivative δ-tocotrienol. Sci. Rep. 2018, 8, 587. [Google Scholar] [CrossRef] [PubMed]
- Sailo, B.L.; Banik, K.; Padmavathi, G.; Javadi, M.; Bordoloi, D.; Kunnumakkara, A.B. Tocotrienols: The promising analogues of vitamin E for cancer therapeutics. Pharmacol. Res. 2018, 130, 259–272. [Google Scholar] [CrossRef]
- Aggarwal, V.; Kashyap, D.; Sak, K.; Tuli, H.S.; Jain, A.; Chaudhary, A.; Garg, V.K.; Sethi, G.; Yerer, M.B. Molecular Mechanisms of Action of Tocotrienols in Cancer: Recent Trends and Advancements. Int. J. Mol. Sci. 2019, 20, 656. [Google Scholar] [CrossRef]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Moretti, R.M.; Marelli, M.M.; Limonta, P. Tocotrienols and Cancer: From the State of the Art to Promising Novel Patents. Recent Patents Anti-Cancer Drug Discov. 2019, 14, 5–18. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Marelli, M.M. Role of Endoplasmic Reticulum Stress in the Anticancer Activity of Natural Compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef] [PubMed]
- Montagnani Marelli, M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Moretti, R.M.; Limonta, P. Anticancer properties of tocotrienols: A review of cellular mechanisms and molecular targets. J. Cell. Physiol. 2019, 234, 1147–1164. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Marzagalli, M.; Raimondi, M.; Zuco, V.; Zaffaroni, N.; Limonta, P. δ-Tocotrienol sensitizes and re-sensitizes ovarian cancer cells to cisplatin via induction of G1 phase cell cycle arrest and ROS/MAPK-mediated apoptosis. Cell Prolif. 2021, 54, e13111. [Google Scholar] [CrossRef]
- Theodosis-Nobelos, P.; Papagiouvannis, G.; Rekka, E.A. A Review on Vitamin E Natural Analogues and on the Design of Synthetic Vitamin E Derivatives as Cytoprotective Agents. Mini-Reviews Med. Chem. 2021, 21, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Kharbanda, A.; Corley, C.; Simmons, P.; Allen, A.R. Tocotrienols as an Anti-Breast Cancer Agent. Antioxidants 2021, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Moretti, R.M.; Raimondi, M.; Marzagalli, M.; Beretta, G.; Procacci, P.; Sartori, P.; Marelli, M.M.; Limonta, P. δ-Tocotrienol induces apoptosis, involving endoplasmic reticulum stress and autophagy, and paraptosis in prostate cancer cells. Cell Prolif. 2019, 52, e12576. [Google Scholar] [CrossRef]
- Fontana, F.; Anselmi, M.; Limonta, P. Exploiting the Metabolic Consequences of PTEN Loss and Akt/Hexokinase 2 Hyperactivation in Prostate Cancer: A New Role for δ-Tocotrienol. Int. J. Mol. Sci. 2022, 23, 5269. [Google Scholar] [CrossRef]
- Ossikbayeva, S.; Khanin, M.; Sharoni, Y.; Trachtenberg, A.; Tuleukhanov, S.; Sensenig, R.; Rom, S.; Danilenko, M.; Orynbayeva, Z. Curcumin and Carnosic Acid Cooperate to Inhibit Proliferation and Alter Mitochondrial Function of Metastatic Prostate Cancer Cells. Antioxidants 2021, 10, 1591. [Google Scholar] [CrossRef]
- Chen, Q.-H. Curcumin-based anti-prostate cancer agents. Anti-Cancer Agents Med. Chem. 2015, 15, 138–156. [Google Scholar] [CrossRef] [PubMed]
- García-Zepeda, S.P.; García-Villa, E.; Díaz-Chávez, J.; Hernández-Pando, R.; Gariglio, P. Resveratrol induces cell death in cervical cancer cells through apoptosis and autophagy. Eur. J. Cancer Prev. 2013, 22, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Enriquez, S.; Pacheco-Velazquez, S.C.; Marin-Hernandez, A.; Gallardo-Perez, J.C.; Robledo-Cadena, D.X.; Hernandez-Resendiz, I.; Garcia-Garcia, J.D.; Belmont-Diaz, J.; Lopez-Marure, R.; Hernandez-Esquivel, L.; et al. Resveratrol inhibits cancer cell proliferation by impairing oxidative phosphorylation and inducing oxidative stress. Toxicol. Appl. Pharmacol. 2019, 370, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, N.; Ma, L.; Liu, M.; Liu, G.; Zhang, Y.; Lin, X. Oleanolic Acid Suppresses Aerobic Glycolysis in Cancer Cells by Switching Pyruvate Kinase Type M Isoforms. PLoS ONE 2014, 9, e91606. [Google Scholar] [CrossRef]
- Lodi, A.; Saha, A.; Lu, X.; Wang, B.; Sentandreu, E.; Collins, M.; Kolonin, M.G.; DiGiovanni, J.; Tiziani, S. Erratum: Combinatorial treatment with natural compounds in prostate cancer inhibits prostate tumor growth and leads to key modulations of cancer cell metabolism. NPJ Precis. Oncol. 2017, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, L.; Wu, N.; Ma, L.; Zhong, J.; Liu, G.; Lin, X. Oleanolic Acid Induces Metabolic Adaptation in Cancer Cells by Activating the AMP-Activated Protein Kinase Pathway. J. Agric. Food Chem. 2014, 62, 5528–5537. [Google Scholar] [CrossRef]
- Akhtar, N.; Syed, D.N.; Khan, M.I.; Adhami, V.M.; Mirza, B.; Mukhtar, H. The pentacyclic triterpenoid, plectranthoic acid, a novel activator of AMPK induces apoptotic death in prostate cancer cells. Oncotarget 2015, 7, 3819–3831. [Google Scholar] [CrossRef]
- Younis, T.; Khan, M.I.; Khan, M.R.; Rasul, A.; Majid, M.; Adhami, V.M.; Mukhtar, H. Nummularic acid, a triterpenoid, from the medicinal plant Fraxinus xanthoxyloides, induces energy crisis to suppress growth of prostate cancer cells. Mol. Carcinog. 2018, 57, 1267–1277. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Pelageev, D.N.; Hauschild, J.; Borisova, K.L.; Kaune, M.; Krisp, C.; Venz, S.; Sabutskii, Y.E.; Khmelevskaya, E.A.; Busenbender, T.; et al. Successful Targeting of the Warburg Effect in Prostate Cancer by Glucose-Conjugated 1,4-Naphthoquinones. Cancers 2019, 11, 1690. [Google Scholar] [CrossRef]
- Negrette-Guzmán, M.; Huerta-Yepez, S.; Vega, M.I.; León-Contreras, J.C.; Hernández-Pando, R.; Medina-Campos, O.N.; Rodríguez, E.; Tapia, E.; Pedraza-Chaverri, J. Sulforaphane induces differential modulation of mitochondrial biogenesis and dynamics in normal cells and tumor cells. Food Chem. Toxicol. 2017, 100, 90–102. [Google Scholar] [CrossRef]
- Xiao, D.; Powolny, A.A.; Moura, M.B.; Kelley, E.E.; Bommareddy, A.; Kim, S.-H.; Hahm, E.-R.; Normolle, D.; Van Houten, B.; Singh, S.V. Phenethyl Isothiocyanate Inhibits Oxidative Phosphorylation to Trigger Reactive Oxygen Species-mediated Death of Human Prostate Cancer Cells. J. Biol. Chem. 2010, 285, 26558–26569. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Kim, K.-Y.; Yu, S.-N.; Seo, Y.-K.; Chun, S.-S.; Yu, H.-S.; Ahn, S.-C. Silibinin induces mitochondrial NOX4-mediated endoplasmic reticulum stress response and its subsequent apoptosis. BMC Cancer 2016, 16, 452. [Google Scholar] [CrossRef]
- Wang, H.; Huwaimel, B.; Verma, K.; Miller, J.; Germain, T.M.; Kinarivala, N.; Pappas, D.; Brookes, P.S.; Trippier, P.C. Synthesis and Antineoplastic Evaluation of Mitochondrial Complex II (Succinate Dehydrogenase) Inhibitors Derived from Atpenin A5. Chemmedchem 2017, 12, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Goldin, N.; Heyfets, A.; Reischer, D.; Flescher, E. Mitochondria-mediated ATP depletion by anti-cancer agents of the jasmonate family. J. Bioenerg. Biomembr. 2007, 39, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; He, C.; Xu, Y.; Xu, H.; Tang, Y.; Chavan, H.; Duan, S.; Artigues, A.; Forrest, M.L.; Krishnamurthy, P.; et al. Alternol eliminates excessive ATP production by disturbing Krebs cycle in prostate cancer. Prostate 2019, 79, 628–639. [Google Scholar] [CrossRef]
- Naguib, A.; Mathew, G.; Reczek, C.R.; Watrud, K.; Ambrico, A.; Herzka, T.; Salas, I.C.; Lee, M.F.; El-Amine, N.; Zheng, W.; et al. Mitochondrial Complex I Inhibitors Expose a Vulnerability for Selective Killing of Pten-Null Cells. Cell Rep. 2018, 23, 58–67. [Google Scholar] [CrossRef]
- Khan, M.A.; Gahlot, S.; Majumdar, S. Oxidative Stress Induced by Curcumin Promotes the Death of Cutaneous T-cell Lymphoma (HuT-78) by Disrupting the Function of Several Molecular Targets. Mol. Cancer Ther. 2012, 11, 1873–1883. [Google Scholar] [CrossRef]
- Larasati, Y.A.; Yoneda-Kato, N.; Nakamae, I.; Yokoyama, T.; Meiyanto, E.; Kato, J.-Y. Curcumin targets multiple enzymes involved in the ROS metabolic pathway to suppress tumor cell growth. Sci. Rep. 2018, 8, 2039. [Google Scholar] [CrossRef]
- Mortezaee, K.; Salehi, E.; Mirtavoos-Mahyari, H.; Motevaseli, E.; Najafi, M.; Farhood, B.; Rosengren, R.J.; Sahebkar, A. Mechanisms of apoptosis modulation by curcumin: Implications for cancer therapy. J. Cell. Physiol. 2019, 234, 12537–12550. [Google Scholar] [CrossRef]
- Jiang, Z.; Chen, K.; Cheng, L.; Yan, B.; Qian, W.; Cao, J.; Li, J.; Wu, E.; Ma, Q.; Yang, W. Resveratrol and cancer treatment: Updates. Ann. N. Y. Acad. Sci. 2017, 1403, 59–69. [Google Scholar] [CrossRef]
- Gal, R.; Deres, L.; Toth, K.; Halmosi, R.; Habon, T. The Effect of Resveratrol on the Cardiovascular System from Molecular Mechanisms to Clinical Results. Int. J. Mol. Sci. 2021, 22, 10152. [Google Scholar] [CrossRef]
- Zhang, L.-X.; Li, C.-X.; Kakar, M.U.; Khan, M.S.; Wu, P.-F.; Amir, R.M.; Dai, D.-F.; Naveed, M.; Li, Q.-Y.; Saeed, M.; et al. Resveratrol (RV): A pharmacological review and call for further research. Biomed. Pharmacother. 2021, 143, 112164. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Bin Dukhyil, A.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Deb, P.K.; Devi, R. Dietary Polyphenols and Their Role in Oxidative Stress-Induced Human Diseases: Insights Into Protective Effects, Antioxidant Potentials and Mechanism(s) of Action. Front. Pharmacol. 2022, 13, 806470. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.; Moradi, F.; Maddalena, L.A.; Ferreira-Tollstadius, B.; Selim, S.; Stuart, J.A. Resveratrol integrates metabolic and growth effects in PC3 prostate cancer cells-involvement of prolyl hydroxylase and hypoxia inducible factor-1. Oncol. Lett. 2019, 17, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.; Yore, M.M.; Sporn, M.B. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer 2007, 7, 357–369. [Google Scholar] [CrossRef]
- Banno, N.; Akihisa, T.; Yasukawa, K.; Tokuda, H.; Tabata, K.; Nakamura, Y.; Nishimura, R.; Kimura, Y.; Suzuki, T. Anti-inflammatory activities of the triterpene acids from the resin of Boswellia carteri. J. Ethnopharmacol. 2006, 107, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M. In Silico Prediction of Steroids and Triterpenoids as Potential Regulators of Lipid Metabolism. Mar. Drugs 2021, 19, 650. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Ahmed, S.; Brankov, N.; Perloff, M. Triterpenoids as potential agents for the chemoprevention and therapy of breast cancer. Front. Biosci. 2011, 16, 980–996. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Nguyen, A.H.; Kumar, A.P.; Tan, B.K.; Sethi, G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: Potential role in prevention and therapy of cancer. Cancer Lett. 2012, 320, 158–170. [Google Scholar] [CrossRef]
- Ghante, M.H.; Jamkhande, P.G. Role of Pentacyclic Triterpenoids in Chemoprevention and Anticancer Treatment: An Overview on Targets and Underling Mechanisms. J. Pharmacopunct. 2019, 22, 55–67. [Google Scholar] [CrossRef]
- Li, S.; Kuo, H.-C.D.; Yin, R.; Wu, R.; Liu, X.; Wang, L.; Hudlikar, R.; Peter, R.M.; Kong, A.-N. Epigenetics/epigenomics of triterpenoids in cancer prevention and in health. Biochem. Pharmacol. 2020, 175, 113890. [Google Scholar] [CrossRef]
- Şoica, C.; Voicu, M.; Ghiulai, R.; Dehelean, C.; Racoviceanu, R.; Trandafirescu, C.; Roșca, O.-J.; Nistor, G.; Mioc, M.; Mioc, A. Natural Compounds in Sex Hormone-Dependent Cancers: The Role of Triterpenes as Therapeutic Agents. Front. Endocrinol. 2020, 11, 612396. [Google Scholar] [CrossRef] [PubMed]
- El-Baba, C.; Baassiri, A.; Kiriako, G.; Dia, B.; Fadlallah, S.; Moodad, S.; Darwiche, N. Terpenoids’ anti-cancer effects: Focus on autophagy. Apoptosis 2021, 26, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.K.; Sharma, P.; Dubey, A.K.; Gundamaraju, R.; Kumar, D.; Kumar, S.; Madaan, R.; Shri, R.; Tsagkaris, C.; Parisi, S.; et al. Natural Product-Based Studies for the Management of Castration-Resistant Prostate Cancer: Computational to Clinical Studies. Front. Pharmacol. 2021, 12, 732266. [Google Scholar] [CrossRef] [PubMed]
- Pandita, A.; Kumar, B.; Manvati, S.; Vaishnavi, S.; Singh, S.K.; Bamezai, R.N.K. Synergistic Combination of Gemcitabine and Dietary Molecule Induces Apoptosis in Pancreatic Cancer Cells and Down Regulates PKM2 Expression. PLoS ONE 2014, 9, e107154. [Google Scholar] [CrossRef] [PubMed]
- Amara, S.; Zheng, M.; Tiriveedhi, V. Oleanolic Acid Inhibits High Salt-Induced Exaggeration of Warburg-like Metabolism in Breast Cancer Cells. Cell Biochem. Biophys. 2016, 74, 427–434. [Google Scholar] [CrossRef]
- Guerra, A.R.; Duarte, M.F.; Duarte, I.F. Targeting Tumor Metabolism with Plant-Derived Natural Products: Emerging Trends in Cancer Therapy. J. Agric. Food Chem. 2018, 66, 10663–10685. [Google Scholar] [CrossRef]
- Lu, J.J.; Bao, J.L.; Wu, G.S.; Xu, W.S.; Huang, M.Q.; Chen, X.P.; Wang, Y.T. Quinones derived from plant secondary metabolites as anti-cancer agents. Anticancer. Agents Med. Chem. 2013, 13, 456–463. [Google Scholar]
- Negrette-Guzmán, M.; Huerta-Yepez, S.; Tapia, E.; Pedraza-Chaverri, J. Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: A seemingly contradictory dual role and an integrative hypothesis. Free. Radic. Biol. Med. 2013, 65, 1078–1089. [Google Scholar] [CrossRef]
- Delmas, D.; Xiao, J.; Vejux, A.; Aires, V. Silymarin and Cancer: A Dual Strategy in Both in Chemoprevention and Chemosensitivity. Molecules 2020, 25, 2009. [Google Scholar] [CrossRef]
- Koltai, T.; Fliegel, L. Role of Silymarin in Cancer Treatment: Facts, Hypotheses, and Questions. J. Evid.-Based Integr. Med. 2022, 27, 2515690X211068826. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, F.; Anselmi, M.; Limonta, P. Unraveling the Peculiar Features of Mitochondrial Metabolism and Dynamics in Prostate Cancer. Cancers 2023, 15, 1192. https://doi.org/10.3390/cancers15041192
Fontana F, Anselmi M, Limonta P. Unraveling the Peculiar Features of Mitochondrial Metabolism and Dynamics in Prostate Cancer. Cancers. 2023; 15(4):1192. https://doi.org/10.3390/cancers15041192
Chicago/Turabian StyleFontana, Fabrizio, Martina Anselmi, and Patrizia Limonta. 2023. "Unraveling the Peculiar Features of Mitochondrial Metabolism and Dynamics in Prostate Cancer" Cancers 15, no. 4: 1192. https://doi.org/10.3390/cancers15041192
APA StyleFontana, F., Anselmi, M., & Limonta, P. (2023). Unraveling the Peculiar Features of Mitochondrial Metabolism and Dynamics in Prostate Cancer. Cancers, 15(4), 1192. https://doi.org/10.3390/cancers15041192