
Sensitive Detection of Cell-Free Tumour DNA Using Optimised Targeted Sequencing Can Predict Prognosis in Gastro-Oesophageal Cancer

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
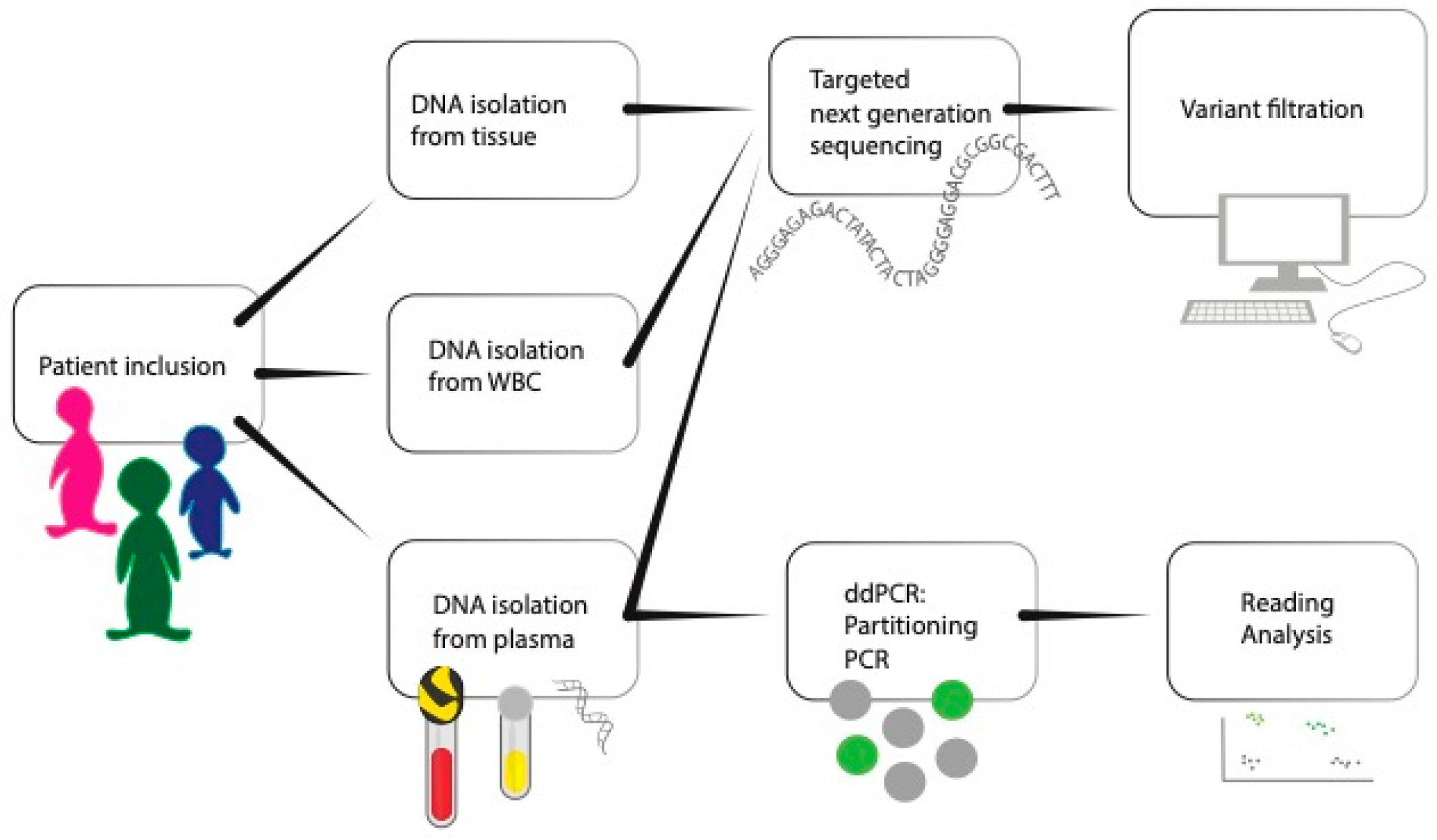
2. Materials and Methods
2.1. Study Design
2.2. Inclusion
2.3. Tissue DNA Isolation
2.4. Plasma DNA Isolation
2.5. White Blood Cell (WBC) DNA Isolation
2.6. Design of the GI cfDNA Panel
2.7. Validation of the GI cfDNA Panel
2.8. Library Preparation and Sequencing
2.9. Variant Identification and Filtering
2.10. Variant Identification and Filtering
2.11. Plasma Droplet Digital PCR
2.12. Plasma Droplet Digital PCR Data Analysis
2.13. Statistics
3. Results
3.1. Participants and Tissue Variants
3.2. Detected Diagnostic Plasma Variants
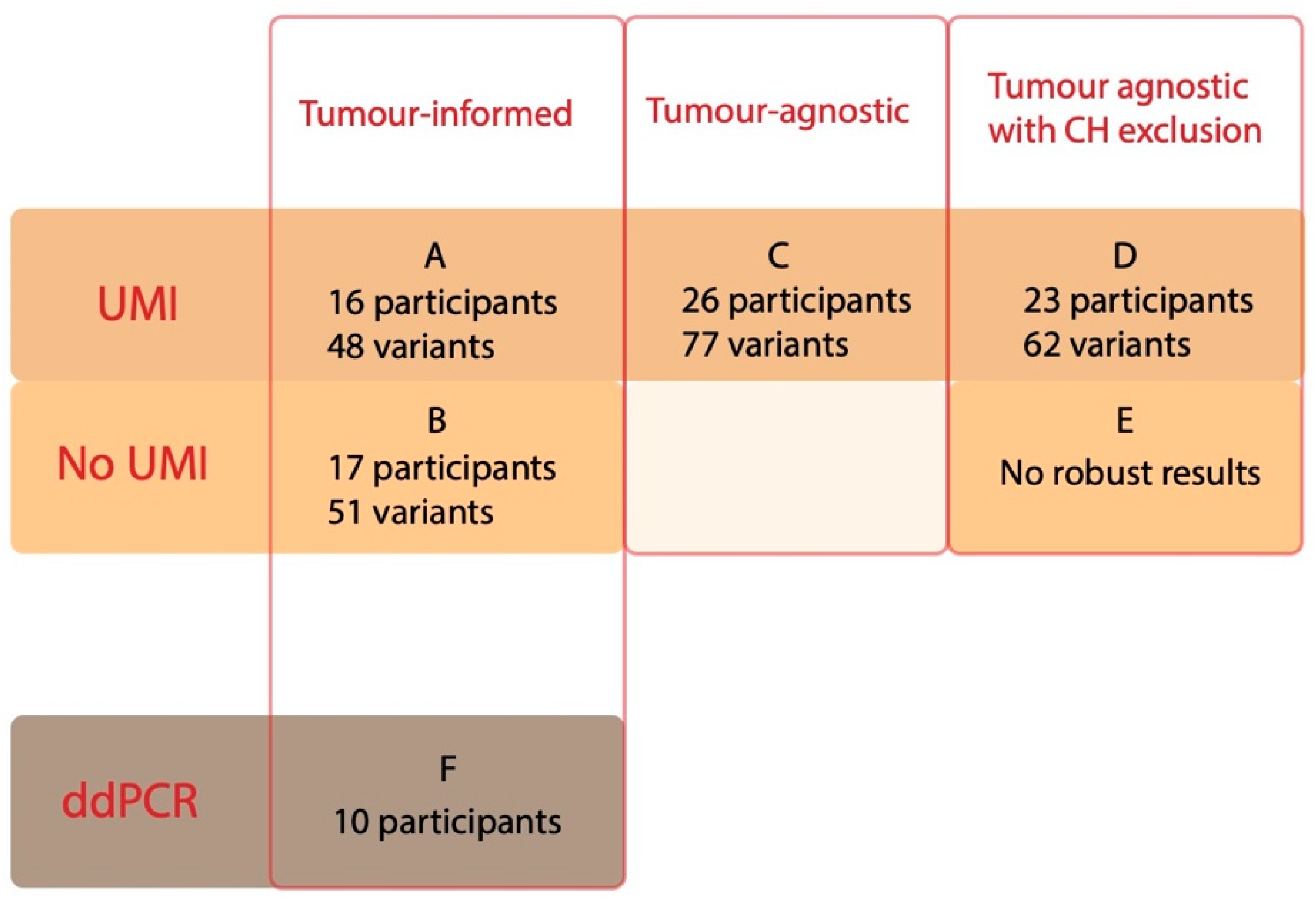
3.2.1. Tumour-Informed Approach
3.2.2. Tumour-Agnostic Approach Using UMIs
3.2.3. Removing CH from the Tumour-Agnostic Approach
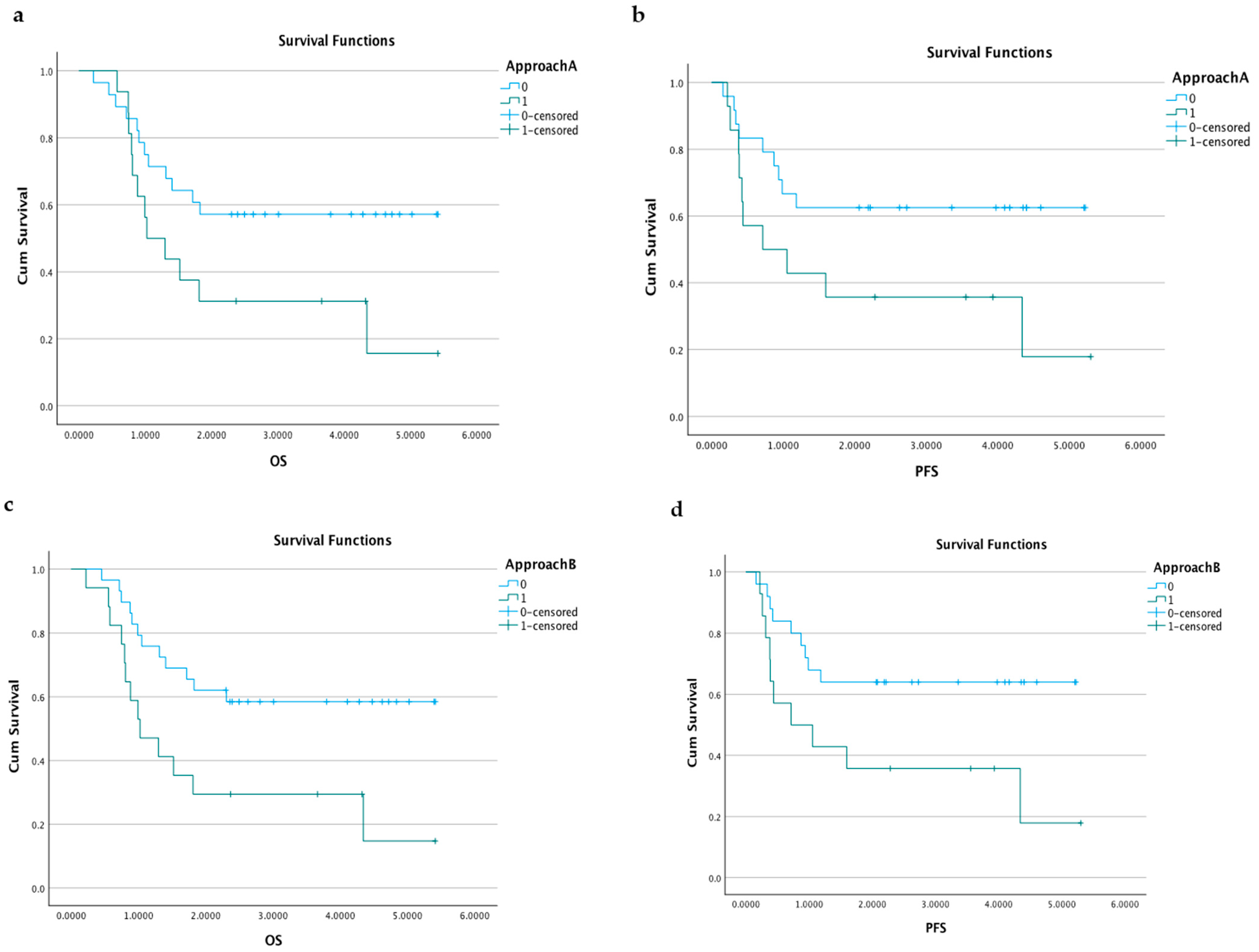
3.2.4. Survival Analyses according to Diagnostic Plasma Samples
3.3. Detected Plasma Variants after Surgery
3.3.1. All Approaches
3.3.2. Survival Predictions according to Plasma Samples after Surgery
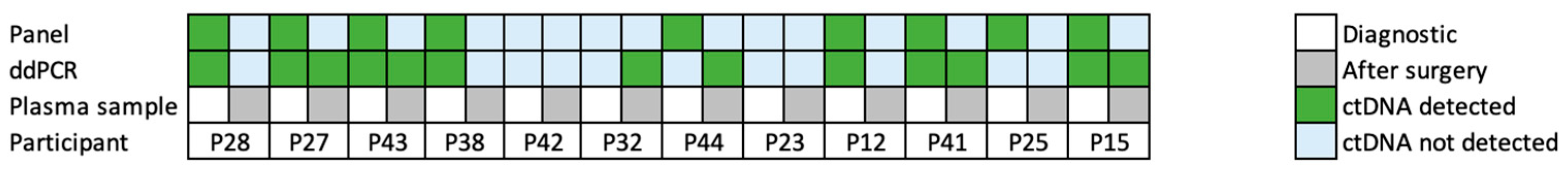
3.4. ddPCR Approach
4. Discussion
4.1. Summary
4.2. Plasma Panel Design
4.3. Plasma Detection Rates
4.4. Increasing the Specificity
4.4.1. Sequencing Error Suppression
4.4.2. Excluding CH Variants
4.5. Plasma Analysis as a Prognostic Biomarker
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.K.; Hannum, G.; Geis, J.; Tynan, J.; Hogg, G.; Zhao, C.; Jensen, T.J.; Mazloom, A.R.; Oeth, P.; Ehrich, M.; et al. Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenat. Diagn. 2015, 35, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Douville, C.; Cohen, J.D.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Schoen, R.E.; Tie, J.; Gibbs, P.; et al. Assessing aneuploidy with repetitive element sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 4858–4863. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.X.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.; van Grieken, N.C.T.; Palsgrove, D.N.; Phallen, J.; Medina, J.E.; Hruban, C.; Broeckaert, M.A.M.; Anagnostou, V.; Adleff, V.; Bruhm, D.C.; et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020, 11, 525. [Google Scholar] [CrossRef]
- Yang, J.; Gong, Y.; Lam, V.K.; Shi, Y.; Guan, Y.; Zhang, Y.; Ji, L.; Chen, Y.; Zhao, Y.; Qian, F.; et al. Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer. Cell Death Dis. 2020, 11, 346. [Google Scholar] [CrossRef]
- Wallander, K.; Eisfeldt, J.; Lindblad, M.; Nilsson, D.; Billiau, K.; Foroughi, H.; Nordenskjöld, M.; Liedén, A.; Tham, E. Cell-free tumour DNA analysis detects copy number alterations in gastro-oesophageal cancer patients. PLoS ONE 2021, 16, e0245488. [Google Scholar] [CrossRef]
- National Cancer Statistics Database, Swedish Social Service. Available online: https://sdb.socialstyrelsen.se/if_can/resultat.aspx (accessed on 14 April 2022).
- Cerami, E.; Gao, J.J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Wickham. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 23 July 2021).
- Lyander, A.; Gellerbring, A.; Hägglund, M.; Elhami, K.; Wirta, V. NGS method for parallel processing of high quality, damaged or fragmented input material using target enrichment. bioRxiv 2023. bioRxiv:2023.01.15.524106. [Google Scholar]
- Foroughi-Asl, H.; Jeggari, A.; Maqbool, K.; Ivanchuk, V.; Elhami, K.; Wirta, V. BALSAMIC: Bioinformatic Analysis Pipeline for SomAtic Mutations in Cancer. Available online: https://zenodo.org/record/7099704/export/hx#.Y2Z5GILMKWB (accessed on 22 November 2021).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic. Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Donald Freed, R.A.; Weber, L.A.; Edwards, J.S. The Sentieon Genomics Tools—A fast and accurate solution to variant calling from next-generation sequence data. bioRxiv 2017, 2017, 115717. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.S.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Freed, D.; Pan, R.; Aldana, R. TNscope: Accurate Detection of Somatic Mutations with Haplotype-based Variant Candidate Detection and Machine Learning Filtering. bioRxiv 2018. bioRxiv:2018:250647. [Google Scholar]
- Andeer, R.; Dalberg, M.; Laaksonen, M.; Magnusson, M.; Nilsson, D.; Rasi, C. Scout, Clinical DNA Variant Visualizer and Browser, Repository Code. Available online: https://github.com/Clinical-Genomics/scout (accessed on 21 November 2021).
- Azad, T.D.; Chaudhuri, A.A.; Fang, P.; Qiao, Y.; Esfahani, M.S.; Chabon, J.J.; Hamilton, E.G.; Yang, Y.D.; Lovejoy, A.; Newman, A.M.; et al. Circulating Tumor DNA Analysis for Detection of Minimal Residual Disease After Chemoradiotherapy for Localized Esophageal Cancer. Gastroenterology 2020, 158, 494–505.e6. [Google Scholar] [CrossRef]
- Openshaw, M.R.; Mohamed, A.A.; Ottolini, B.; Fernandez-Garcia, D.; Richards, C.J.; Page, K.; Guttery, D.S.; Thomas, A.L.; Shaw, J.A. Longitudinal monitoring of circulating tumour DNA improves prognostication and relapse detection in gastroesophageal adenocarcinoma. Br. J. Cancer 2020, 123, 1271–1279. [Google Scholar] [CrossRef]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.K. Clonal Hematopoiesis in Liquid Biopsy: From Biological Noise to Valuable Clinical Implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef] [PubMed]
- Ptashkin, R.N.; Mandelker, D.L.; Coombs, C.C.; Bolton, K.; Yelskaya, Z.; Hyman, D.M.; Solit, D.B.; Baselga, J.; Arcila, M.E.; Ladanyi, M.; et al. Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. JAMA Oncol. 2018, 4, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Koh, Y.; Yamamichi, G.; Yumiba, S.; Tomiyama, E.; Matsushita, M.; Hayashi, Y.; Wang, C.; Ishizuya, Y.; Yamamoto, Y.; et al. Perioperative circulating tumor DNA enables the identification of patients with poor prognosis in upper tract urothelial carcinoma. Cancer Sci. 2022, 113, 1830–1842. [Google Scholar] [CrossRef]
- Varkalaite, G.; Forster, M.; Franke, A.; Kupcinskas, J.; Skieceviciene, J. Liquid Biopsy in Gastric Cancer: Analysis of Somatic Cancer Tissue Mutations in Plasma Cell-Free DNA for Predicting Disease State and Patient Survival. Clin. Transl. Gastroenterol. 2021, 12, e00403. [Google Scholar] [CrossRef]
- Slagter, A.E.; Vollebergh, M.A.; Caspers, I.A.; van Sandick, J.W.; Sikorska, K.; Lind, P.; Nordsmark, M.; Putter, H.; Braak, J.; Meershoek-Klein Kranenbarg, E.; et al. Prognostic value of tumor markers and ctDNA in patients with resectable gastric cancer receiving perioperative treatment: Results from the CRITICS trial. Gastric Cancer 2022, 25, 401–410. [Google Scholar] [CrossRef]
- Konishi, S.; Narita, T.; Hatakeyama, S.; Yoneyama, T.; Yoneyama, M.S.; Tobisawa, Y.; Noro, D.; Sato, T.; Togashi, K.; Okamoto, T.; et al. Utility of total cell-free DNA levels for surgical damage evaluation in patients with urological surgeries. Sci. Rep. 2021, 11, 22103. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Reinert, T.; Christensen, E.; Sethi, H.; Birkenkamp-Demtröder, K.; Gögenur, M.; Gögenur, I.; Zimmermann, B.G.; Dyrskjøt, L.; Andersen, C.L. The effect of surgical trauma on circulating free DNA levels in cancer patients-implications for studies of circulating tumor DNA. Mol. Oncol. 2020, 14, 1670–1679. [Google Scholar] [CrossRef]
- Zhao, Q.; Miao, C.; Lu, Q.; Wu, W.; He, Y.; Wu, S.; Liu, H.; Lian, C. Clinical Significance of Monitoring Circulating Free DNA and Plasma Heat Shock Protein 90alpha in Patients with Esophageal Squamous Cell Carcinoma. Cancer Manag. Res. 2021, 13, 2223–2234. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Participant ID | Cancer Localisation | Age Diagnosis | Sex | Tumour Stage (Based on Pathology Report *) | Overall Survival (Years) | Alive at the End of Study |
|---|---|---|---|---|---|---|
| P01 | Stomach | 71 | Man | IIIC | 1.31 | no |
| P02 | Stomach | 84 | Man | IIB | 5.41 | yes |
| P03 | Stomach | 79 | Man | IIB | 5.41 | yes |
| P04 | Stomach | 79 | Man | IB | 5.39 | yes |
| P05 | Stomach | 78 | Man | IIIC | 0.75 | no |
| P06 | Stomach | 78 | Man | IIIC | 0.90 | no |
| P07 | Stomach | 74 | Man | IIA | 1.81 | no |
| P08 | Stomach | 64 | Man | IB | 4.34 | no |
| P09 | Stomach | 59 | Woman | IIIC | 1.40 | no |
| P10 | Stomach | 55 | Man | IB | 5.02 | yes |
| P11 | Oesophagus | 75 | Man | IIIC | 0.99 | no |
| P12 | Stomach | 65 | Woman | IIIC-IV | 0.81 | no |
| P13 | Stomach | 75 | Woman | IA | 4.62 | yes |
| P14 | Stomach | 57 | Woman | IA | 4.84 | yes |
| P15 | Stomach | 74 | Man | IIIC | 0.79 | no |
| P16 | Stomach | 68 | Woman | IB | 4.72 | yes |
| P17 | Oesophagus | 56 | Woman | IV | 0.99 | no |
| P18 | Stomach | 71 | Man | IIA | 4.32 | yes |
| P19 | Stomach | 77 | Woman | IIB | 4.28 | yes |
| P20 | Stomach | 43 | Man | IIB | 4.48 | yes |
| P21 | Stomach | 76 | Man | IIIC | 0.22 | no |
| P22 | Oesophagus | 48 | Man | IIIC | 1.83 | no |
| P23 | Stomach | 75 | Man | IIA | 4.11 | yes |
| P24 | Stomach | 85 | Man | IIIC | 1.71 | no |
| P25 | Stomach | 76 | Man | IIIC | 0.74 | no |
| P26 | Stomach | 68 | Man | IV | 0.45 | no |
| P27 | Stomach | 81 | Man | IIIB | 0.88 | no |
| P28 | Oesophagus | 81 | Woman | IIIC | 3.66 | yes |
| P29 | Stomach | 74 | Woman | IIIC | 1.05 | no |
| P30 | Oesophagus | 67 | Man | IIIC | 1.02 | no |
| P31 | Oesophagus | 74 | Woman | IIIC | 0.88 | no |
| P32 | Stomach | 80 | Man | IA | 3.79 | yes |
| P33 | Oesophagus | 77 | Man | IIIA | 2.81 | yes |
| P34 | Stomach | 36 | Woman | IIIC | 3.01 | yes |
| P35 | Stomach | 64 | Man | IIIB | 2.63 | yes |
| P36 | Stomach | 62 | Woman | IV | 2.31 | no |
| P37 | Stomach | 81 | Woman | IB | 0.71 | no |
| P38 | Stomach | 77 | Woman | IIA | 2.37 | yes |
| P39 | Oesophagus | 61 | Man | IIA | 2.49 | yes |
| P40 | Stomach | 72 | Man | IB | 2.30 | yes |
| P41 | Stomach | 87 | Man | IIIC | 0.57 | no |
| P42 | Stomach | 61 | Man | IA | 2.31 | yes |
| P43 | Stomach | 79 | Man | IIIC | 1.52 | no |
| P44 | Oesophagus | 83 | Woman | IIIA | 0.55 | no |
| P45 | Oesophagus | 72 | Man | IA | 2.36 | yes |
| P46 | Stomach | 64 | Man | IB | 2.39 | yes |
| P47 | Stomach | 82 | Woman | IIIA | 1.30 | no |
| Participant ID | Expected Variants * | A. Diagnostic | B. Diagnostic | C. Diagnostic | D. Diagnostic | A. Post | B. Post | C. Post | D. Post | Plasma Diagnostic (ng) | Plasma Diagnostic (mL) | Plasma Post (ng) | Plasma Post (mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P01 | N | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 36.9 | 5 | 21.2 | 3 |
| P02 | Y | 1 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 17.9 | 3 | 8.5 | 2 |
| P03 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 26.3 | 4 | 28.3 | 3 |
| P04 | N | 0 | 0 | 4 | 4 | 0 | 0 | 0 | 0 | 42.9 | 4 | 54.5 | 2 |
| P05 | Y | 9 | 9 | 10 | 7 | 4 | 4 | 4 | 1 | 73.5 | 5 | 206 | 3 |
| P06 | N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 250 | 5 | 75 | 3 |
| P07 | Y | 1 | 1 | 2 | 2 | 0 | 0 | 0 | 0 | 33.3 | 3 | 60 | 3 |
| P08 | Y | 2 | 2 | 4 | 4 | 0 | 0 | 0 | 0 | 26.3 | 3 | 81 | 4 |
| P09 | N | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 68 | 3 | 69 | 5 |
| P10 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 25.5 | 5 | 32.2 | 3 |
| P11 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 45.9 | 2 | 139 | 3 |
| P12 | Y | 3 | 3 | 4 | 4 | 0 | 0 | 0 | 0 | 81.5 | 3 | 247 | 3 |
| P13 | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 66 | 5 | 250 | 4 |
| P14 | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 110 | 5 | 250 | 3 |
| P15 | Y | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 236 | 5 | 250 | 3 |
| P16 | N | 0 | 0 | 2 | 1 | 0 | 0 | 1 | 0 | 39.3 | 5 | 231 | 3 |
| P17 | Y | 3 | 3 | 4 | 3 | 2 | 2 | 2 | 2 | 26.7 | 4 | 68.5 | 3 |
| P18 | Y | 15 | 15 | 15 | 15 | 0 | 0 | 0 | 0 | 17.6 | 4 | 28.4 | 4 |
| P19 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 189 | 5 | 47 | 4 |
| P20 | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 47 | 5 | 250 | 3 |
| P21 | Y | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 55.5 | 5 | 250 | 3 |
| P22 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 39.2 | 5 | 29 | 3 |
| P23 | Y | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 20.8 | 4 | 250 | 3 |
| P24 | Y | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 134 | 5 | 158 | 3 |
| P25 | Y | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 48.6 | 4 | 250 | 3 |
| P26 | Y | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 16.8 | 4 | 141 | 3 |
| P27 | Y | 1 | 1 | 3 | 1 | 0 | 0 | 0 | 0 | 222 | 5 | 250 | 4 |
| P28 | Y | 3 | 3 | 3 | 3 | 0 | 0 | 0 | 0 | 52.5 | 3 | 155 | 3 |
| P29 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 106 | 3 | 197 | 3 |
| P30 | Y | 2 | 2 | 3 | 3 | 0 | 0 | 0 | 0 | 19.2 | 4 | 72.5 | 3 |
| P31 | N | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 24.6 | 3 | 250 | 5 |
| P32 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 71.5 | 4 | 250 | 4 |
| P33 | Y | 0 | 0 | 2 | 0 | NA | NA | NA | NA | 116 | 4 | NA | 4 |
| P34 | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 35.8 | 4 | 120 | 3 |
| P35 | N | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 96 | 4 | 149 | 4 |
| P36 | N | NA | NA | NA | NA | 0 | 0 | 0 | NA | NA | 4 | 241 | 4 |
| P37 | Y ** | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 65 | 4 | 250 | 4 |
| P38 | Y | 1 | 1 | 2 | NA | NA | NA | NA | NA | 24 | 4 | NA | 4 |
| P39 | N | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 11.1 | 3 | 168 | 4 |
| P40 | Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 38.7 | 4 | 250 | 4 |
| P41 | Y | 1 | 1 | 2 | 2 | 0 | 0 | 0 | 0 | 47 | 4 | 250 | 4 |
| P42 | Y | NA | NA | NA | NA | 0 | 0 | 0 | NA | NA | 4 | 250 | 4 |
| P43 | Y | 3 | 4 | 4 | 2 | 1 | 1 | 2 | 0 | 98.5 | 4 | 250 | 4 |
| P44 | Y | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 125 | 4 | 250 | 4 |
| P45 | N | NA | NA | NA | NA | 0 | 0 | 0 | NA | NA | NA | 55 | 3 |
| P46 | N | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 33 | 3 | 250 | 4 |
| P47 | Y | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 51.5 | 4 | 250 | 4 |
| Participant | Sample Type and plasma Volume | Target | False-Positive Rate | Total Cell-Free DNA Copies/mL | VAF [%] ddPCR | VAF [%] Gene Panel | ddPCR Result per Variant | ddPCR Result per Patient | Gene Panel Results Tumour-Informed Approaches |
|---|---|---|---|---|---|---|---|---|---|
| P28 | Diagnostic, 3 mL | TP53 p. R273H | 0.03 | 1051 | 0.5 | 0.4 | positive | positive | positive |
| KRAS p. G13D | 0.03 | 0.5 | 0.3 | positive | positive | ||||
| Post-surgery, 3 mL | TP53 p. R273H | 0.03 | 1958 | 0 | 0 | negative | negative | negative | |
| KRAS p. G13D | 0.03 | 0.03 ** | 0 | negative | negative | ||||
| Follow-up-1, 3 mL | TP53 p. R273H | 0.03 | 456 | 0 | NA | negative | negative | NA | |
| KRAS p. G13D | 0.03 | 0 | NA | negative | NA | ||||
| Follow-up-2, 5 mL | TP53 p. R273H | 0.03 | 1002 | 0 | NA | negative | negative | NA | |
| KRAS p. G13D | 0.03 | 0 | NA | negative | NA | ||||
| P27 | Diagnostic, 5 mL | PIK3CA p. H1047R | 0 | 8151 | 0.01 | 0 | positive | positive | negative |
| TP53 p. R273C | 0.05 | 1.32 | 0.6 | positive | positive | ||||
| Post-surgery, 3 mL | PIK3CA p. H1047R | 0 | 27,247 | 0.006 | 0 | negative | positive | negative | |
| TP53 p. R273C | 0.05 | 0.1 | 0 | positive | negative | ||||
| Follow-up-1, 3 mL | PIK3CA p. H1047R | 0 | 1156 | 0 | NA | negative | negative | NA | |
| TP53 p. R273C | 0.05 | 0 | NA | negative | NA | ||||
| P43 | Diagnostic, 4 mL | KRAS p. G12D | 0.06 | 2154 | 0.32 | 0.5 | positive | positive | positive |
| Post-surgery, 3 mL | KRAS p. G12D | 0.06 | 5348 | 0.24 | 0 | positive | positive | negative | |
| Follow-up-1, 5 mL | KRAS p. G12D | 0.06 | 2183 | 0.6 | NA | positive | positive | NA | |
| P38 | Diagnostic, 4 mL | TP53 p. R248W | 0.01 | 1119 | 0.19 | 0.3 | positive | positive | positive |
| Post-surgery, 5 mL | TP53 p. R248W | 0.01 | 7111 | 0,01 | 0 | negative | negative | negative | |
| P41 | Diagnostic, 4 mL | RB1 p. R358 * | 0.04 | 933 | 14.7 | 0 | positive | positive | negative |
| TP53 p. R273C | 0.03 | 6.8 | 17 | positive | positive | ||||
| Post-surgery, 4 mL | RB1 p. R358 * | 0.04 | 28,862 | 0.17 | 0 | positive | positive | negative | |
| TP53 p. R273C | 0.03 | 0.27 | 0 | positive | negative | ||||
| Follow-up-1, 4 mL | RB1 p. R358 * | 0.04 | 1376 | 3.15 | NA | positive | positive | NA | |
| TP53 p. R273C | 0.03 | 1.42 | NA | positive | NA | ||||
| P25 | Diagnostic, 4 mL | TP53 p. R196 *, c.586C>T | 0.02 | 5080 | 0.02 | 0.07 | negative | negative | positive |
| Post-surgery, 3 mL | TP53 p. R196 *, c.586C>T | 0.02 | 131,322 | 0.0003 | 0 | negative | negative | negative | |
| Follow-up-1, 3 mL | TP53 p. R196 *, c.586C>T | 0.02 | 1898 | 0.02 | NA | negative | negative | NA | |
| Follow-up-2, 3 mL | TP53 p. R196 *, c.586C>T | 0.02 | 2928 | 0.3 | NA | positive | positive | NA | |
| P12 | Diagnostic, 3 mL | APC p. R823 * | 0.04 | 3597 | 0.59 | 0,7 | positive | positive | positive |
| TP53 c.720_766del | 0 | 0.98 | 0,3 | positive | positive | ||||
| Chemo-follow1, 3 mL | APC p. R823 * | 0.04 | 2776 | 0 | NA | negative | negative | NA | |
| TP53 c.720_766del | 0 | 0 | NA | negative | NA | ||||
| Chemo-follow2, 3 mL | APC p. R823 * | 0.04 | 2180 | 0 | NA | negative | negative | NA | |
| TP53 c.720_766del | 0 | 0 | NA | negative | NA | ||||
| Chemo-follow3, 3 mL | APC p. R823 * | 0.04 | 897 | 0 | NA | negative | negative | NA | |
| TP53 c.720_766del | 0 | 0 | NA | negative | NA | ||||
| Chemo-follow4, 3 mL | APC p. R823 * | 0.04 | 2748 | 0.16 | NA | positive | positive | NA | |
| TP53 c.720_766del | 0 | 0.68 | NA | positive | NA | ||||
| Post-surgery, 3 mL | APC p. R823 * | 0.04 | 12,596 | 0.02 | 0 | negative | negative | negative | |
| TP53 c.720_766del | 0 | 0 | 0 | negative | negative | negative | |||
| P15 | Diagnostic, 3 mL | TP53 p. R248Q, c.743G>A | 0.04 | 10,437 | 0.27 | 0,23 | positive | positive | positive |
| ERBB2 p. S310Y, c.929C>A | 0 | 0.19 | 0 | positive | negative | ||||
| Post-surgery, 3 mL | TP53 p. R248Q, c.743G>A | 0.04 | 13,968 | 0.04 | 0 | negative | positive | negative | |
| ERBB2 p. S310Y, c.929C>A | 0 | 0,01 | 0 | positive | negative | ||||
| Follow-up1, 3 mL | TP53 p. R248Q, c.743G>A | 0.04 | 7323 | 3.71 | NA | positive | positive | NA | |
| ERBB2 p. S310Y, c.929C>A | 0 | 4.0 | NA | positive | NA | ||||
| P44 | Diagnostic, 3 mL | TP53 p. R273H | 0.03 | 576 | 0.25 ** | 0,26 | negative | negative | positive |
| KRAS p. G13D | 0.03 | 0 | 0 | negative | negative | ||||
| Post-surgery, 4 mL | TP53 p. R273H | 0.03 | 4879 | 0.1 | 0 | positive | positive | negative | |
| KRAS p. G13D | 0.03 | 0.07 | 0 | positive | negative | ||||
| P42 | Diagnostic, 4 mL | TP53 p. R158fs *12, c.472del | 0.02 | 5721 | 0.006 | 0 | negative | negative | NA |
| Post-surgery, 4 mL | TP53 p. R158fs *12, c.472del | 0.02 | 38,515 | 0 | 0 | negative | negative | negative | |
| P32 | Diagnostic, 4 mL | TP53 p. R248W | 0.02 | 2455 | 0 | 0 | negative | negative | negative |
| Post-surgery, 5 mL | TP53 p. R248W | 0.02 | 100,726 | 0.04 | 0 | positive | positive | negative | |
| Follow-up-1, 3 mL | TP53 p. R248W | 0.02 | 1227 | 0 | NA | negative | negative | NA | |
| Follow-up-2, 5 mL | TP53 p. R248W | 0.02 | 1944 | 0 | NA | negative | negative | NA | |
| P23 | Diagnostic, 4 mL | TP53 p. R273H | 0.03 | 560 | 0 | 0 | negative | negative | negative |
| KRAS p. G13D | 0.03 | 0.2 ** | 0 | negative | negative | ||||
| Pre-surgery-2, 4 mL | TP53 p. R273H | 0.03 | 1054 | 0 | NA | negative | negative | NA | |
| KRAS p. G13D | 0.03 | 0 | NA | negative | NA | ||||
| Post-surgery, 3 mL | TP53 p. R273H | 0.03 | 542,312 | 0 | 0 | negative | negative | negative | |
| KRAS p. G13D | 0.03 | 0 | 0 | negative | negative | ||||
| Follow-up-1, 3 mL | TP53 p. R273H | 0.03 | 8043 | 0.01 | NA | negative | negative | NA | |
| KRAS p. G13D | 0.03 | 0.03 | NA | negative | NA | ||||
| Follow-up-2, 3 mL | TP53 p. R273H | 0.03 | 766 | 0 | NA | negative | negative | NA | |
| KRAS p. G13D | 0.03 | 0 | NA | negative | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallander, K.; Haider, Z.; Jeggari, A.; Foroughi-Asl, H.; Gellerbring, A.; Lyander, A.; Chozhan, A.; Cuba Gyllensten, O.; Hägglund, M.; Wirta, V.; et al. Sensitive Detection of Cell-Free Tumour DNA Using Optimised Targeted Sequencing Can Predict Prognosis in Gastro-Oesophageal Cancer. Cancers 2023, 15, 1160. https://doi.org/10.3390/cancers15041160
Wallander K, Haider Z, Jeggari A, Foroughi-Asl H, Gellerbring A, Lyander A, Chozhan A, Cuba Gyllensten O, Hägglund M, Wirta V, et al. Sensitive Detection of Cell-Free Tumour DNA Using Optimised Targeted Sequencing Can Predict Prognosis in Gastro-Oesophageal Cancer. Cancers. 2023; 15(4):1160. https://doi.org/10.3390/cancers15041160
Chicago/Turabian StyleWallander, Karin, Zahra Haider, Ashwini Jeggari, Hassan Foroughi-Asl, Anna Gellerbring, Anna Lyander, Athithyan Chozhan, Ollanta Cuba Gyllensten, Moa Hägglund, Valtteri Wirta, and et al. 2023. "Sensitive Detection of Cell-Free Tumour DNA Using Optimised Targeted Sequencing Can Predict Prognosis in Gastro-Oesophageal Cancer" Cancers 15, no. 4: 1160. https://doi.org/10.3390/cancers15041160
APA StyleWallander, K., Haider, Z., Jeggari, A., Foroughi-Asl, H., Gellerbring, A., Lyander, A., Chozhan, A., Cuba Gyllensten, O., Hägglund, M., Wirta, V., Nordenskjöld, M., Lindblad, M., & Tham, E. (2023). Sensitive Detection of Cell-Free Tumour DNA Using Optimised Targeted Sequencing Can Predict Prognosis in Gastro-Oesophageal Cancer. Cancers, 15(4), 1160. https://doi.org/10.3390/cancers15041160