
Genotoxins: The Mechanistic Links between Escherichia coli and Colorectal Cancer

Abstract
Simple Summary
Abstract
1. Introduction
2. Cytolethal Distending Toxin
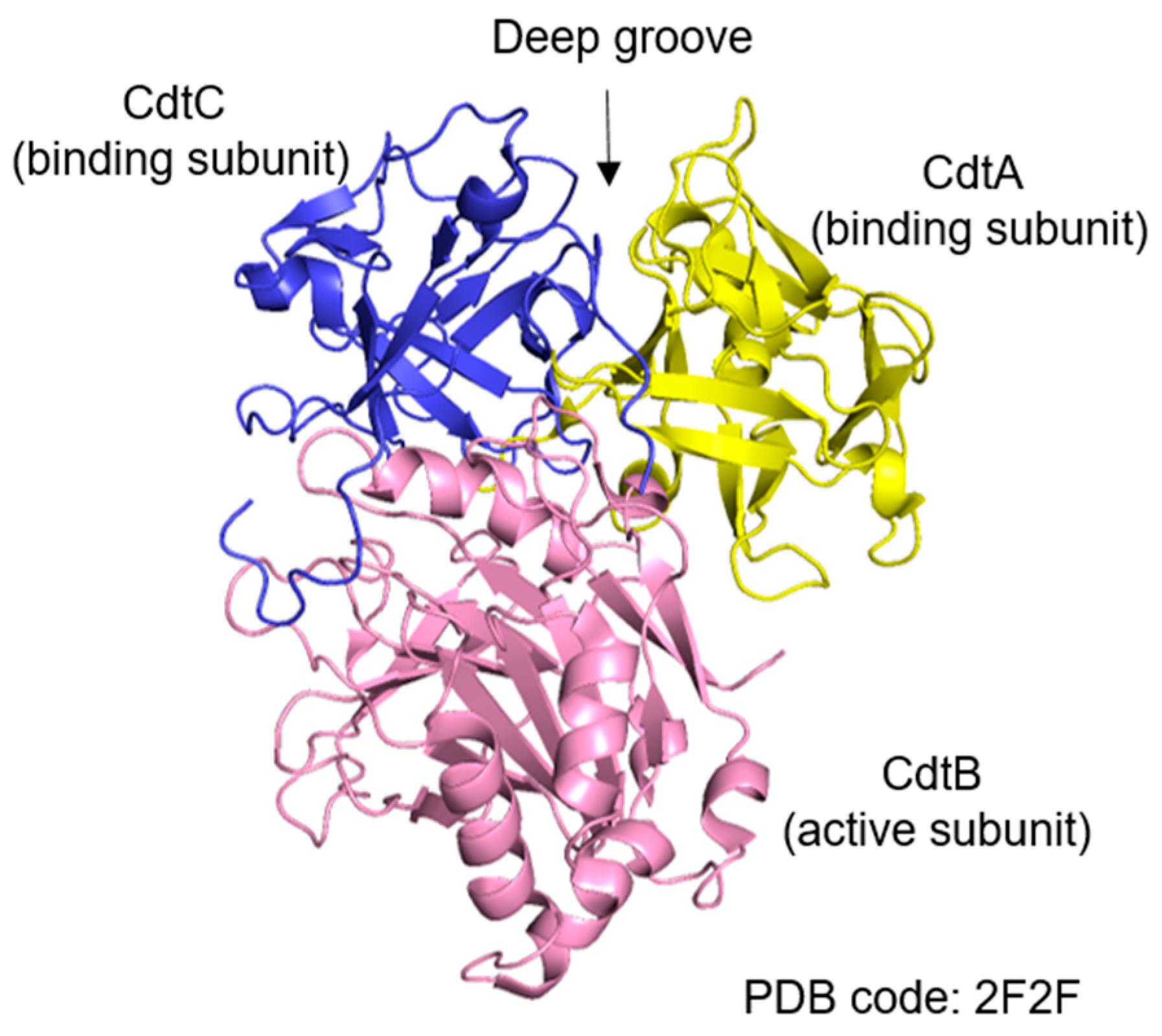
2.1. Structural Features and Subunits of Cytolethal Distending Toxin
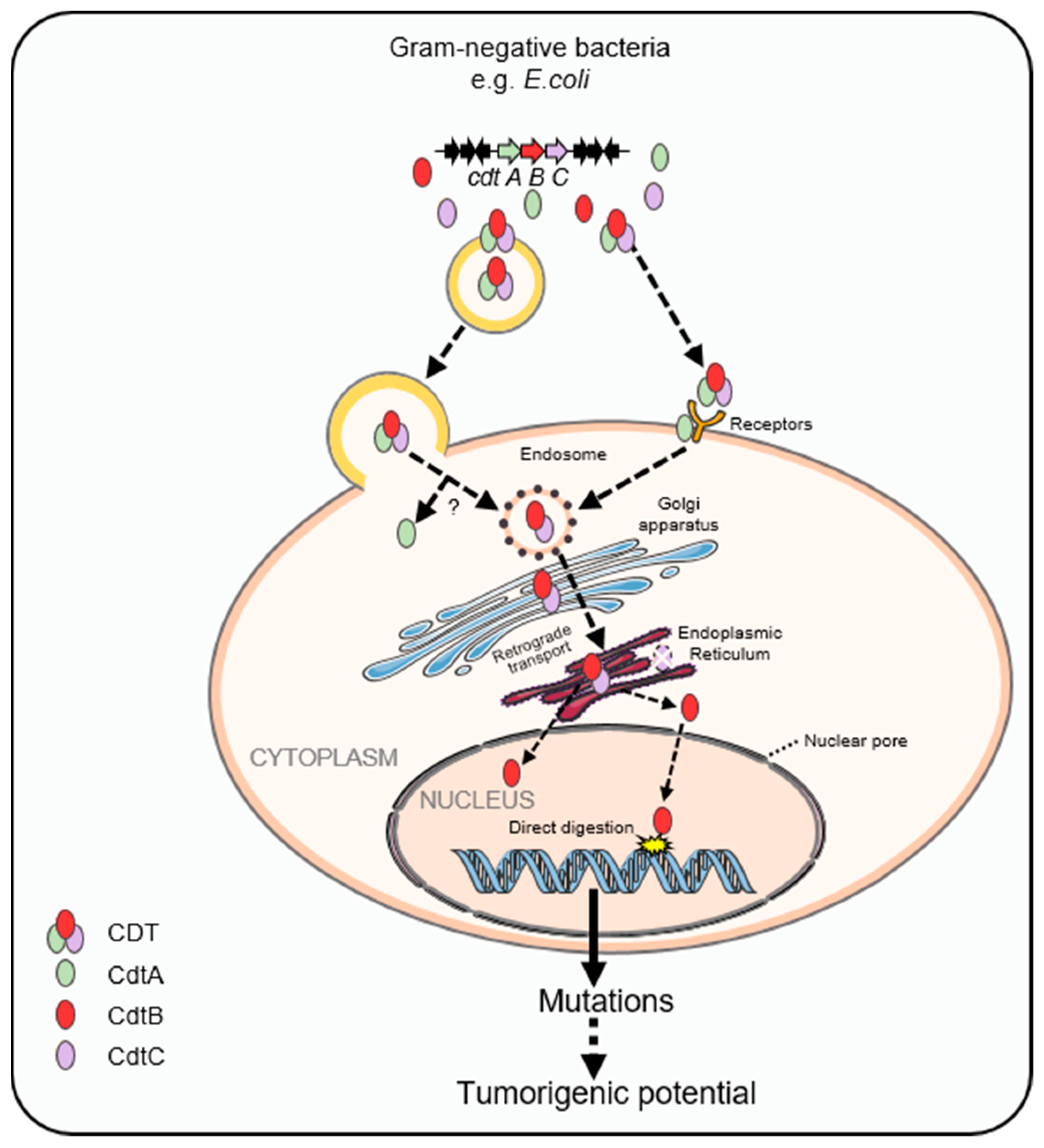
2.2. Carcinogenic Effects of Cytolethal Distending Toxin
3. Colibactin
3.1. Structural Features of Colibactin
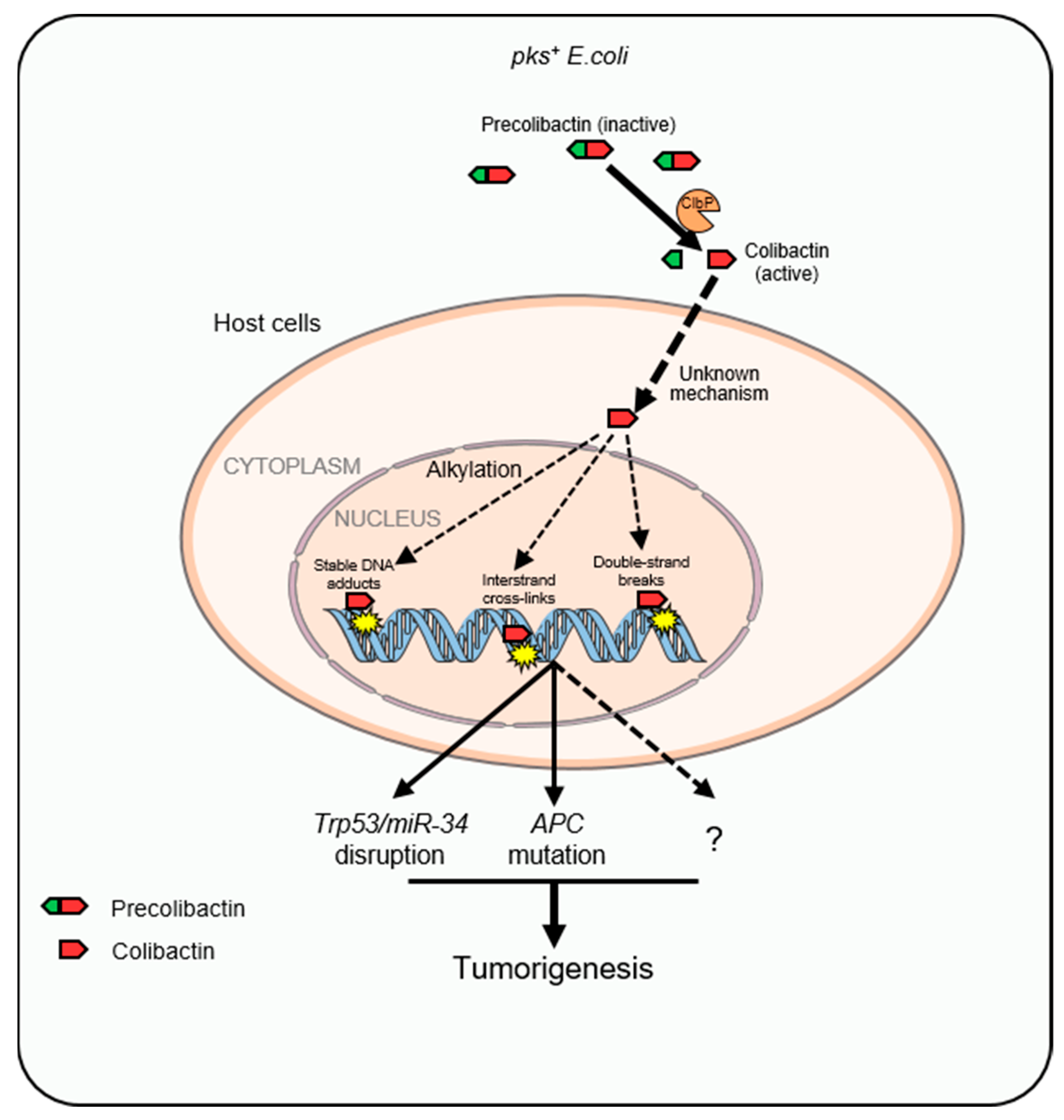
3.2. Colibactin-Induced Genotoxicity and CRC
3.3. Unique Mutational Signatures Induced by Colibactin
4. UshA
4.1. Biological Functions of UshA
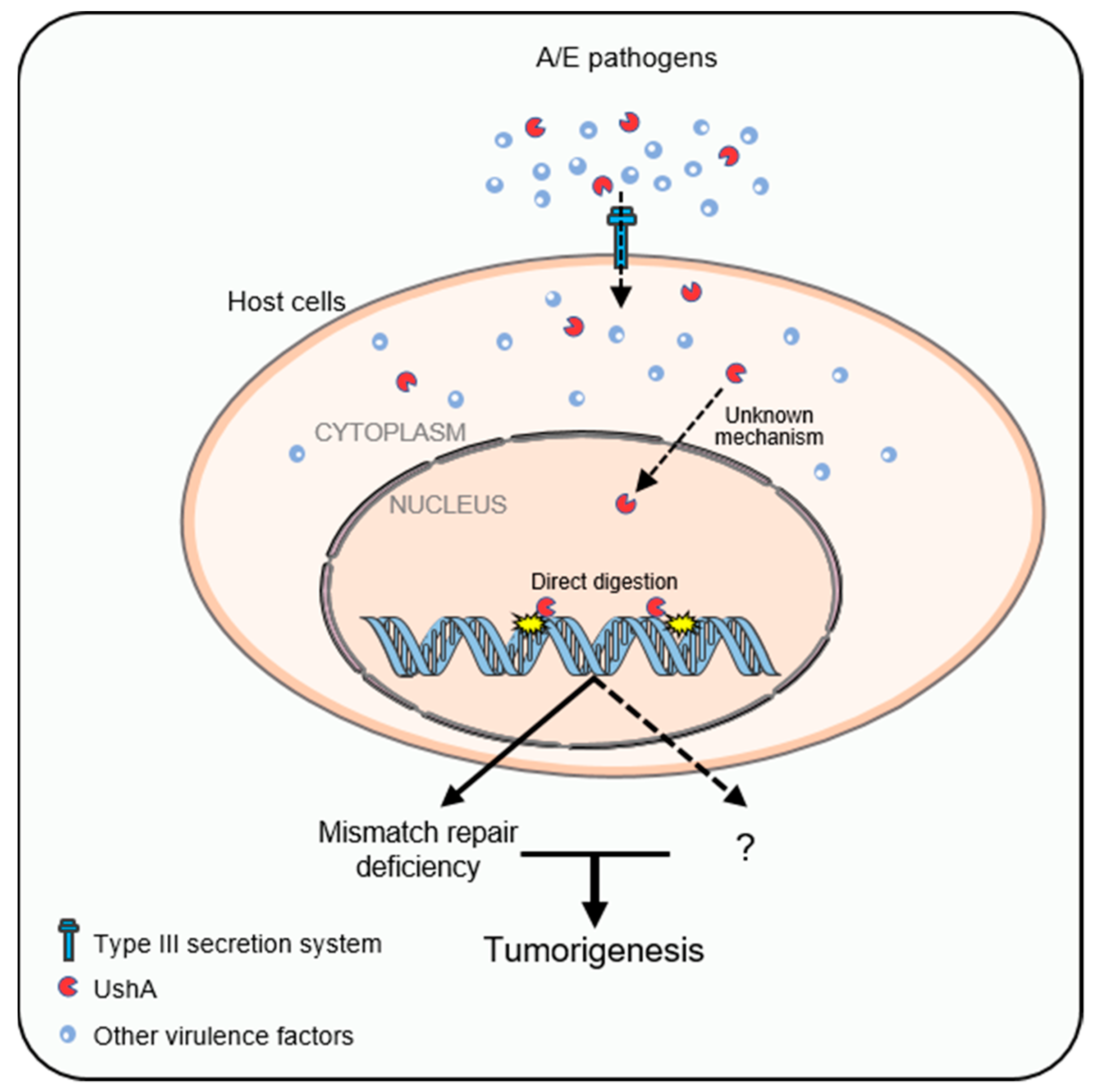
4.2. Attaching and Effacing (A/E) Pathogens and Their Tumorigenic Role in CRC
4.3. UshA as a Newly Identified T3SS-Dependent Genotoxin Related to CRC
5. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jiang, Y.; Yuan, H.; Li, Z.; Ji, X.; Shen, Q.; Tuo, J.; Bi, J.; Li, H.; Xiang, Y. Global pattern and trends of colorectal cancer survival: A systematic review of population-based registration data. Cancer Biol. Med. 2021, 19, 175–186. [Google Scholar] [CrossRef]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef]
- Narayan, S.; Sharma, R. Molecular mechanism of adenomatous polyposis coli-induced blockade of base excision repair pathway in colorectal carcinogenesis. Life Sci. 2015, 139, 145–152. [Google Scholar] [CrossRef]
- Win, A.K.; Lindor, N.M.; Young, J.P.; Macrae, F.A.; Young, G.P.; Williamson, E.; Parry, S.; Goldblatt, J.; Lipton, L.; Winship, I.; et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J. Natl. Cancer Inst. 2012, 104, 1363–1372. [Google Scholar] [CrossRef]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef]
- Bazieva, N.T.; Varlamova, T.M. Several problems in forecasting constitutionally tall stature. Pediatriia 1989, 11, 83–87. [Google Scholar]
- Van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.N.; Fang, J.Y. Gut Microbiota and Colorectal Cancer. Gastrointest. Tumors 2015, 2, 26–32. [Google Scholar] [CrossRef]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Boudeau, J.; Glasser, A.L.; Masseret, E.; Joly, B.; Darfeuille-Michaud, A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect. Immun. 1999, 67, 4499–4509. [Google Scholar] [CrossRef]
- Picard, B.; Garcia, J.S.; Gouriou, S.; Duriez, P.; Brahimi, N.; Bingen, E.; Elion, J.; Denamur, E. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 1999, 67, 546–553. [Google Scholar] [CrossRef]
- Tivendale, K.A.; Logue, C.M.; Kariyawasam, S.; Jordan, D.; Hussein, A.; Li, G.; Wannemuehler, Y.; Nolan, L.K. Avian-pathogenic Escherichia coli strains are similar to neonatal meningitis E. coli strains and are able to cause meningitis in the rat model of human disease. Infect. Immun. 2010, 78, 3412–3419. [Google Scholar] [CrossRef]
- Köhler, C.D.; Dobrindt, U. What defines extraintestinal pathogenic Escherichia coli? Int. J. Med. Microbiol. 2011, 301, 642–647. [Google Scholar] [CrossRef]
- Mulvey, M.A.; Lopez-Boado, Y.S.; Wilson, C.L.; Roth, R.; Parks, W.C.; Heuser, J.; Hultgren, S.J. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 1998, 282, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.D.; Herbelin, C.J.; Bumbaugh, A.C.; Selander, R.K.; Whittam, T.S. Parallel evolution of virulence in pathogenic Escherichia coli. Nature 2000, 406, 64–67. [Google Scholar] [CrossRef]
- Phillips, D.H.; Arlt, V.M. Genotoxicity: Damage to DNA and its consequences. Exs 2009, 99, 87–110. [Google Scholar] [CrossRef] [PubMed]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Déchelotte, P.; Bonnet, R.; Pezet, D.; Darfeuille-Michaud, A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fu, K.; Wier, E.M.; Lei, Y.; Hodgson, A.; Xu, D.; Xia, X.; Zheng, D.; Ding, H.; Sears, C.L.; et al. Bacterial Genotoxin Accelerates Transient Infection-Driven Murine Colon Tumorigenesis. Cancer Discov. 2022, 12, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Lior, H. Response of Chinese hamster ovary cells to a cytolethal distending toxin (CDT) of Escherichia coli and possible misinterpretation as heat-labile (LT) enterotoxin. FEMS Microbiol. Lett. 1987, 43, 19–23. [Google Scholar] [CrossRef]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875. [Google Scholar] [CrossRef] [PubMed]
- Pons, B.J.; Vignard, J.; Mirey, G. Cytolethal Distending Toxin Subunit B: A Review of Structure-Function Relationship. Toxins 2019, 11, 595. [Google Scholar] [CrossRef]
- Yamada, T.; Komoto, J.; Saiki, K.; Konishi, K.; Takusagawa, F. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): Crystal structure of CDT from Actinobacillus actinomycetemcomitans. Protein Sci. A Publ. Protein Soc. 2006, 15, 362–372. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Lara-Tejero, M.; Galán, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef]
- Nesić, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Cortes-Bratti, X.; Guidi, R.; Frisan, T. The biology of the cytolethal distending toxins. Toxins 2011, 3, 172–190. [Google Scholar] [CrossRef]
- Carette, J.E.; Guimaraes, C.P.; Wuethrich, I.; Blomen, V.A.; Varadarajan, M.; Sun, C.; Bell, G.; Yuan, B.; Muellner, M.K.; Nijman, S.M.; et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat. Biotechnol. 2011, 29, 542–546. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Jang, J.Y.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Localization of Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunits during intoxication of live cells. Infect. Immun. 2012, 80, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Taieb, F.; Petit, C.; Nougayrède, J.P.; Oswald, E. The Enterobacterial Genotoxins: Cytolethal Distending Toxin and Colibactin. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef]
- Kuehn, M.J.; Kesty, N.C. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005, 19, 2645–2655. [Google Scholar] [CrossRef]
- DiRienzo, J.M. Uptake and processing of the cytolethal distending toxin by mammalian cells. Toxins 2014, 6, 3098–3116. [Google Scholar] [CrossRef]
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlöw, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell. Microbiol. 2005, 7, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon. Off. J. Int. Soc. Toxinol. 2001, 39, 1729–1736. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J. Biol. Chem. 2001, 276, 5296–5302. [Google Scholar] [CrossRef] [PubMed]
- Frisan, T.; Cortes-Bratti, X.; Chaves-Olarte, E.; Stenerlöw, B.; Thelestam, M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Fedor, Y.; Vignard, J.; Nicolau-Travers, M.L.; Boutet-Robinet, E.; Watrin, C.; Salles, B.; Mirey, G. From single-strand breaks to double-strand breaks during S-phase: A new mode of action of the Escherichia coli Cytolethal Distending Toxin. Cell. Microbiol. 2013, 15, 1–15. [Google Scholar] [CrossRef]
- Graillot, V.; Dormoy, I.; Dupuy, J.; Shay, J.W.; Huc, L.; Mirey, G.; Vignard, J. Genotoxicity of Cytolethal Distending Toxin (CDT) on Isogenic Human Colorectal Cell Lines: Potential Promoting Effects for Colorectal Carcinogenesis. Front. Cell. Infect. Microbiol. 2016, 6, 34. [Google Scholar] [CrossRef]
- Guidi, R.; Guerra, L.; Levi, L.; Stenerlöw, B.; Fox, J.G.; Josenhans, C.; Masucci, M.G.; Frisan, T. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell. Microbiol. 2013, 15, 98–113. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Feng, Y.; Ge, L.; Parry, N.; Muthupalani, S.; Fox, J.G. Helicobacter hepaticus cytolethal distending toxin promotes intestinal carcinogenesis in 129Rag2-deficient mice. Cell. Microbiol. 2017, 19, e12728. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Rogers, A.B.; Feng, Y.; Lee, A.; Xu, S.; Taylor, N.S.; Fox, J.G. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007, 9, 2070–2080. [Google Scholar] [CrossRef]
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef]
- Desvaux, M.; Dalmasso, G.; Beyrouthy, R.; Barnich, N.; Delmas, J.; Bonnet, R. Pathogenicity Factors of Genomic Islands in Intestinal and Extraintestinal Escherichia coli. Front. Microbiol. 2020, 11, 2065. [Google Scholar] [CrossRef]
- Johnson, J.R.; Johnston, B.; Kuskowski, M.A.; Nougayrede, J.P.; Oswald, E. Molecular epidemiology and phylogenetic distribution of the Escherichia coli pks genomic island. J. Clin. Microbiol. 2008, 46, 3906–3911. [Google Scholar] [CrossRef]
- Nougayrède, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Dubois, D.; Baron, O.; Cougnoux, A.; Delmas, J.; Pradel, N.; Boury, M.; Bouchon, B.; Bringer, M.A.; Nougayrède, J.P.; Oswald, E.; et al. ClbP is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J. Biol. Chem. 2011, 286, 35562–35570. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Gibold, L.; Robin, F.; Dubois, D.; Pradel, N.; Darfeuille-Michaud, A.; Dalmasso, G.; Delmas, J.; Bonnet, R. Analysis of structure-function relationships in the colibactin-maturating enzyme ClbP. J. Mol. Biol. 2012, 424, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Kim, C.S.; Healy, A.R.; Wernke, K.M.; Wang, Z.; Frischling, M.C.; Shine, E.E.; Wang, W.; Herzon, S.B.; Crawford, J.M. Structure elucidation of colibactin and its DNA cross-links. Science 2019, 365, eaax2685. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carra, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, 689. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef]
- Arthur, J.C. Microbiota and colorectal cancer: Colibactin makes its mark. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 317–318. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrède, J.P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 2018, 9, e02393-17. [Google Scholar] [CrossRef]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Healy, A.R.; Herzon, S.B. Molecular Basis of Gut Microbiome-Associated Colorectal Cancer: A Synthetic Perspective. J. Am. Chem. Soc. 2017, 139, 14817–14824. [Google Scholar] [CrossRef] [PubMed]
- Olier, M.; Marcq, I.; Salvador-Cartier, C.; Secher, T.; Dobrindt, U.; Boury, M.; Bacquié, V.; Pénary, M.; Gaultier, E.; Nougayrède, J.P.; et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes 2012, 3, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12, 1003. [Google Scholar] [CrossRef] [PubMed]
- Zakataeva, N.P. Microbial 5′-nucleotidases: Their characteristics, roles in cellular metabolism, and possible practical applications. Appl. Microbiol. Biotechnol. 2021, 105, 7661–7681. [Google Scholar] [CrossRef]
- Burns, D.M.; Beacham, I.R. Nucleotide sequence and transcriptional analysis of the E. coli ushA gene, encoding periplasmic UDP-sugar hydrolase (5′-nucleotidase): Regulation of the ushA gene, and the signal sequence of its encoded protein product. Nucleic Acids Res. 1986, 14, 4325–4342. [Google Scholar] [CrossRef]
- Zakataeva, N.P.; Romanenkov, D.V.; Yusupova, Y.R.; Skripnikova, V.S.; Asahara, T.; Gronskiy, S.V. Identification, Heterologous Expression, and Functional Characterization of Bacillus subtilis YutF, a HAD Superfamily 5′-Nucleotidase with Broad Substrate Specificity. PLoS ONE 2016, 11, e0167580. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Y.J.; Ji, D.; Lin, X.; Liu, Y.; Zhang, Y.; Liu, W.; Zhao, Z.K. Identification of UshA as a major enzyme for NAD degradation in Escherichia coli. Enzym. Microb. Technol. 2014, 58–59, 75–79. [Google Scholar] [CrossRef]
- Krug, U.; Patzschke, R.; Zebisch, M.; Balbach, J.; Sträter, N. Contribution of the two domains of E. coli 5′-nucleotidase to substrate specificity and catalysis. FEBS Lett. 2013, 587, 460–466. [Google Scholar] [CrossRef]
- Cabezas, A.; Lopez-Villamizar, I.; Costas, M.J.; Cameselle, J.C.; Ribeiro, J.M. Substrate Specificity of Chimeric Enzymes Formed by Interchange of the Catalytic and Specificity Domains of the 5′-Nucleotidase UshA and the 3′-Nucleotidase CpdB. Molecules 2021, 26, 2307. [Google Scholar] [CrossRef]
- Knöfel, T.; Sträter, N. E. coli 5′-nucleotidase undergoes a hinge-bending domain rotation resembling a ball-and-socket motion. J. Mol. Biol. 2001, 309, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Döpfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001921. [Google Scholar] [CrossRef]
- Chen, H.D.; Frankel, G. Enteropathogenic Escherichia coli: Unravelling pathogenesis. FEMS Microbiol. Rev. 2005, 29, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.W.; Remis, R.S.; Helgerson, S.D.; McGee, H.B.; Wells, J.G.; Davis, B.R.; Hebert, R.J.; Olcott, E.S.; Johnson, L.M.; Hargrett, N.T.; et al. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 1983, 308, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Ameer, M.A.; Wasey, A.; Salen, P. Escherichia coli (E. coli 0157 H7). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Xia, X.; Liu, Y.; Hodgson, A.; Xu, D.; Guo, W.; Yu, H.; She, W.; Zhou, C.; Lan, L.; Fu, K.; et al. EspF is crucial for Citrobacter rodentium-induced tight junction disruption and lethality in immunocompromised animals. PLoS Pathog. 2019, 15, e1007898. [Google Scholar] [CrossRef]
- Goosney, D.L.; Gruenheid, S.; Finlay, B.B. Gut feelings: Enteropathogenic E. coli (EPEC) interactions with the host. Annu. Rev. Cell Dev. Biol. 2000, 16, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.W.; Keeney, K.M.; Crepin, V.F.; Rathinam, V.A.; Fitzgerald, K.A.; Finlay, B.B.; Frankel, G. Citrobacter rodentium: Infection, inflammation and the microbiota. Nat. Rev. Microbiol. 2014, 12, 612–623. [Google Scholar] [CrossRef]
- Moon, H.W.; Whipp, S.C.; Argenzio, R.A.; Levine, M.M.; Giannella, R.A. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 1983, 41, 1340–1351. [Google Scholar] [CrossRef]
- Knutton, S.; Baldwin, T.; Williams, P.H.; McNeish, A.S. Actin accumulation at sites of bacterial adhesion to tissue culture cells: Basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 1989, 57, 1290–1298. [Google Scholar] [CrossRef]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef]
- Donnenberg, M.S.; Kaper, J.B. Enteropathogenic Escherichia coli. Infect. Immun. 1992, 60, 3953–3961. [Google Scholar] [CrossRef] [PubMed]
- Tobe, T.; Beatson, S.A.; Taniguchi, H.; Abe, H.; Bailey, C.M.; Fivian, A.; Younis, R.; Matthews, S.; Marches, O.; Frankel, G.; et al. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. USA 2006, 103, 14941–14946. [Google Scholar] [CrossRef]
- Deng, W.; de Hoog, C.L.; Yu, H.B.; Li, Y.; Croxen, M.A.; Thomas, N.A.; Puente, J.L.; Foster, L.J.; Finlay, B.B. A comprehensive proteomic analysis of the type III secretome of Citrobacter rodentium. J. Biol. Chem. 2010, 285, 6790–6800. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Yu, H.B.; de Hoog, C.L.; Stoynov, N.; Li, Y.; Foster, L.J.; Finlay, B.B. Quantitative proteomic analysis of type III secretome of enteropathogenic Escherichia coli reveals an expanded effector repertoire for attaching/effacing bacterial pathogens. Mol. Cell. Proteom. MCP 2012, 11, 692–709. [Google Scholar] [CrossRef] [PubMed]
- Petty, N.K.; Bulgin, R.; Crepin, V.F.; Cerdeno-Tarraga, A.M.; Schroeder, G.N.; Quail, M.A.; Lennard, N.; Corton, C.; Barron, A.; Clark, L.; et al. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J. Bacteriol. 2010, 192, 525–538. [Google Scholar] [CrossRef]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef]
- Swidsinski, A.; Khilkin, M.; Kerjaschki, D.; Schreiber, S.; Ortner, M.; Weber, J.; Lochs, H. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology 1998, 115, 281–286. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Scanlon, K.M.; Donnenberg, M.S. An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. mBio 2013, 4, e00152-13. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Short, A.J.; Donnenberg, M.S.; Bader, S.; Harrison, D.J. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS ONE 2009, 4, e5517. [Google Scholar] [CrossRef]
- Bürtin, F.; Mullins, C.S.; Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 2020, 26, 1394–1426. [Google Scholar] [CrossRef]
- Newman, J.V.; Kosaka, T.; Sheppard, B.J.; Fox, J.G.; Schauer, D.B. Bacterial infection promotes colon tumorigenesis in Apc(Min/+) mice. J. Infect. Dis. 2001, 184, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J.C.; Meckbach, R.; Jacob, P. Genomic instability and radiation risk in molecular pathways to colon cancer. PLoS ONE 2014, 9, e111024. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Puente, J.L.; Gruenheid, S.; Li, Y.; Vallance, B.A.; Vázquez, A.; Barba, J.; Ibarra, J.A.; O’Donnell, P.; Metalnikov, P.; et al. Dissecting virulence: Systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. USA 2004, 101, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; Miller, B.M.; Rivera-Chávez, F.; Velazquez, E.M.; Byndloss, M.X.; Chávez-Arroyo, A.; Lokken, K.L.; Tsolis, R.M.; Winter, S.E.; Bäumler, A.J. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science 2016, 353, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Knöfel, T.; Sträter, N. X-ray structure of the Escherichia coli periplasmic 5′-nucleotidase containing a dimetal catalytic site. Nat. Struct. Biol. 1999, 6, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDT-Producing Species | Related Cancer | Animal Model | Possible Contribution of CDT |
|---|---|---|---|
| C. jejuni | CRC [46] | Germ-free ApcMin/+mice | Promotes CRC and alters microbial composition and transcriptomic responses |
| H. hepaticus | Intestinal carcinoma [47] | 129/SvEv Rag2−/− mice | Promotes intestinal carcinogenesis through inducing increased DSBs and activating the Stat3 signaling pathway |
| H. hepaticus | Hepatocarcinogenesis [48] | A/JCr mice | Promotes hepatocarcinogenesis by the activation of the NF-κB pathway |
| Name of Genotoxin | Constitute | Function in DNA Damage | How to Get into Host Cells |
|---|---|---|---|
| CDT | CDT family are AB2 heterotrimers consisting of the active subunit CdtB (A) and two binding moieties, CdtA and CdtC (B) [25,26] | nicking or relaxation activities [30,39] DSBs [41,42] SSBs [43] | CDT binds to the receptor on the surface of host cell or is wrapped in outer membrane vesicles that can transport the toxin into the cell by fusion with the plasma membrane [28,29,30] |
| Colibactin | Colibactin is a polyketide-nonribosomal peptide [52,53] | stable DNA adducts [55,56] DSBs [52,57,58] replication stress [60] interstrand cross-links [60] | Unknown |
| UshA | UshA is a miltiple-functional enzyme [23,66,68] | DSBs +SSBs [23] | T3SS [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Fu, K. Genotoxins: The Mechanistic Links between Escherichia coli and Colorectal Cancer. Cancers 2023, 15, 1152. https://doi.org/10.3390/cancers15041152
Wang Y, Fu K. Genotoxins: The Mechanistic Links between Escherichia coli and Colorectal Cancer. Cancers. 2023; 15(4):1152. https://doi.org/10.3390/cancers15041152
Chicago/Turabian StyleWang, Ya, and Kai Fu. 2023. "Genotoxins: The Mechanistic Links between Escherichia coli and Colorectal Cancer" Cancers 15, no. 4: 1152. https://doi.org/10.3390/cancers15041152
APA StyleWang, Y., & Fu, K. (2023). Genotoxins: The Mechanistic Links between Escherichia coli and Colorectal Cancer. Cancers, 15(4), 1152. https://doi.org/10.3390/cancers15041152