
Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management
 ,
, 
Abstract
Simple Summary
Abstract
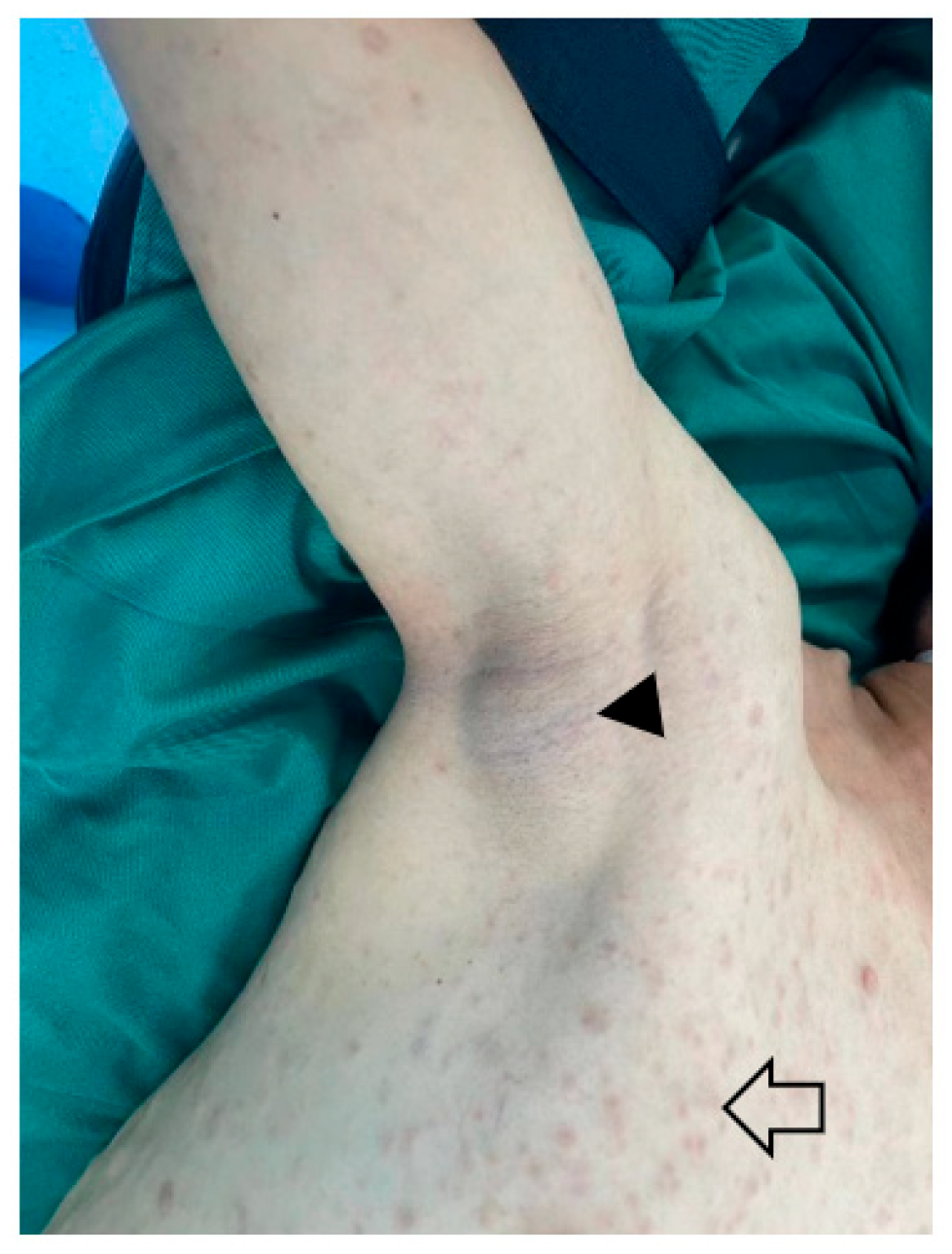
1. Definition and Epidemiology
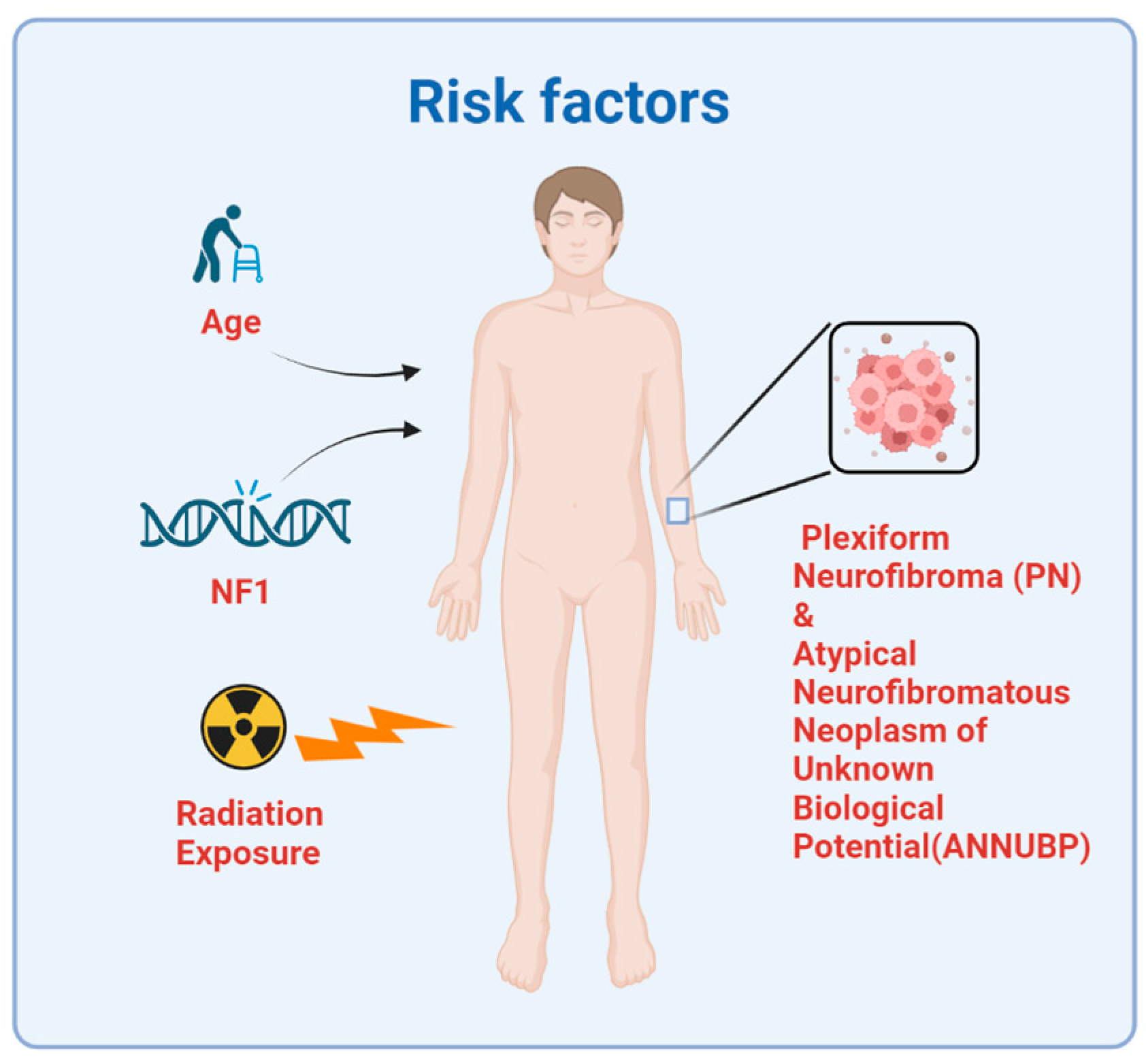
2. Etiology and Risk Factors
2.1. Neurofibromatosis Type 1
2.2. Radiation
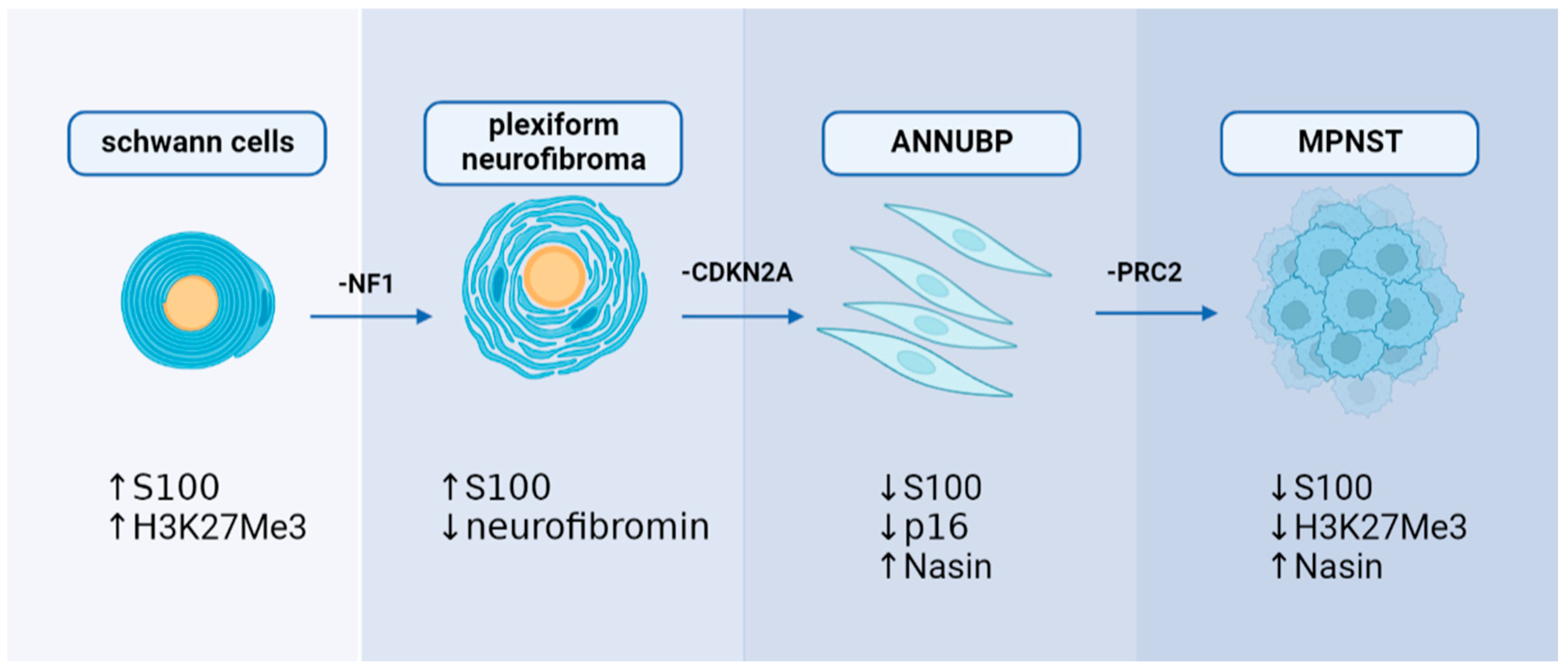
2.3. Plexiform Neurofibroma and Atypical Neurofibromatous Neoplasm of Unknown Biological Potential
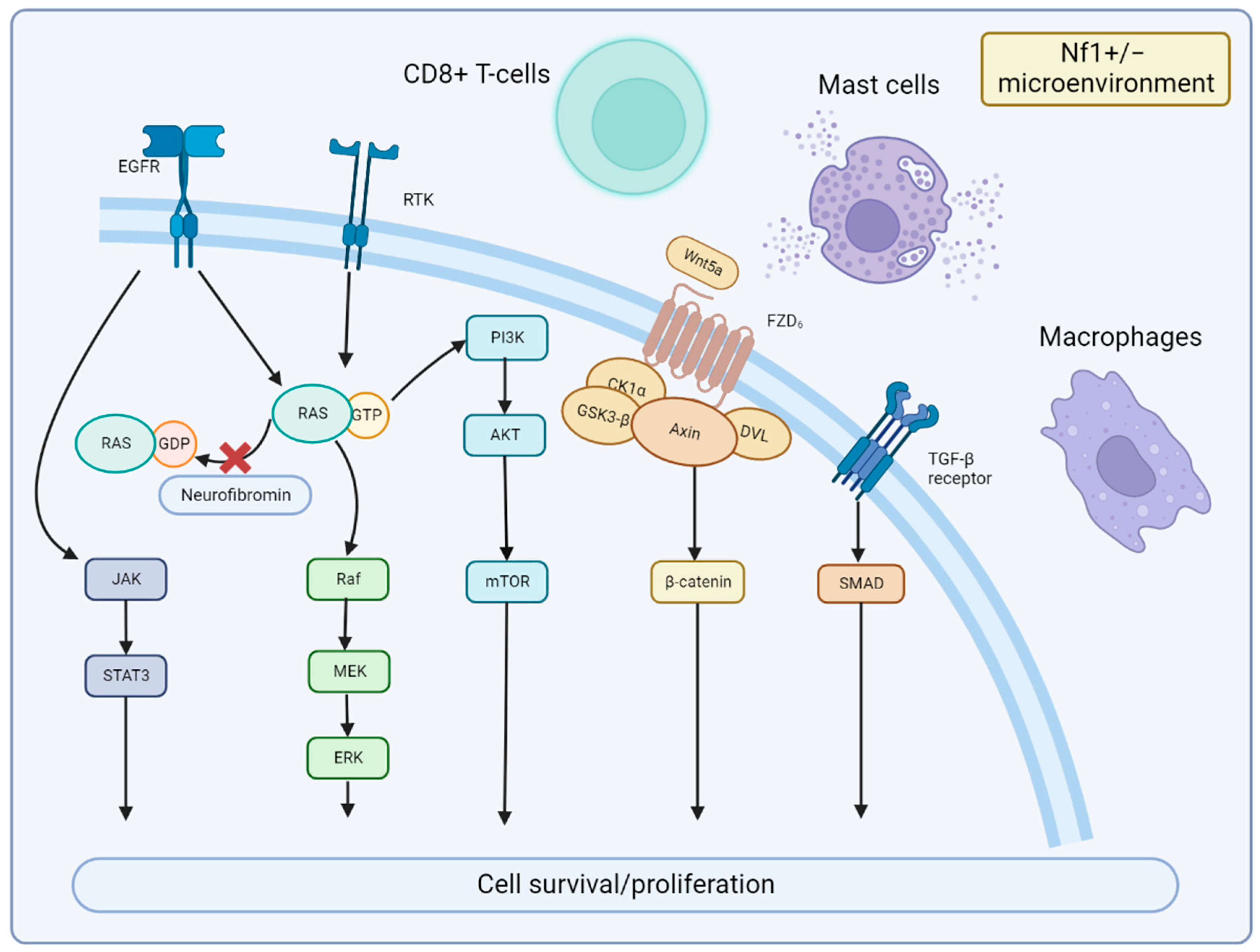
3. Mechanisms of MPNST Pathogenesis
3.1. Genetic Mechanism
3.2. Signaling Pathway and Microenvironment
4. Diagnosis
4.1. Imaging
4.2. Biopsy
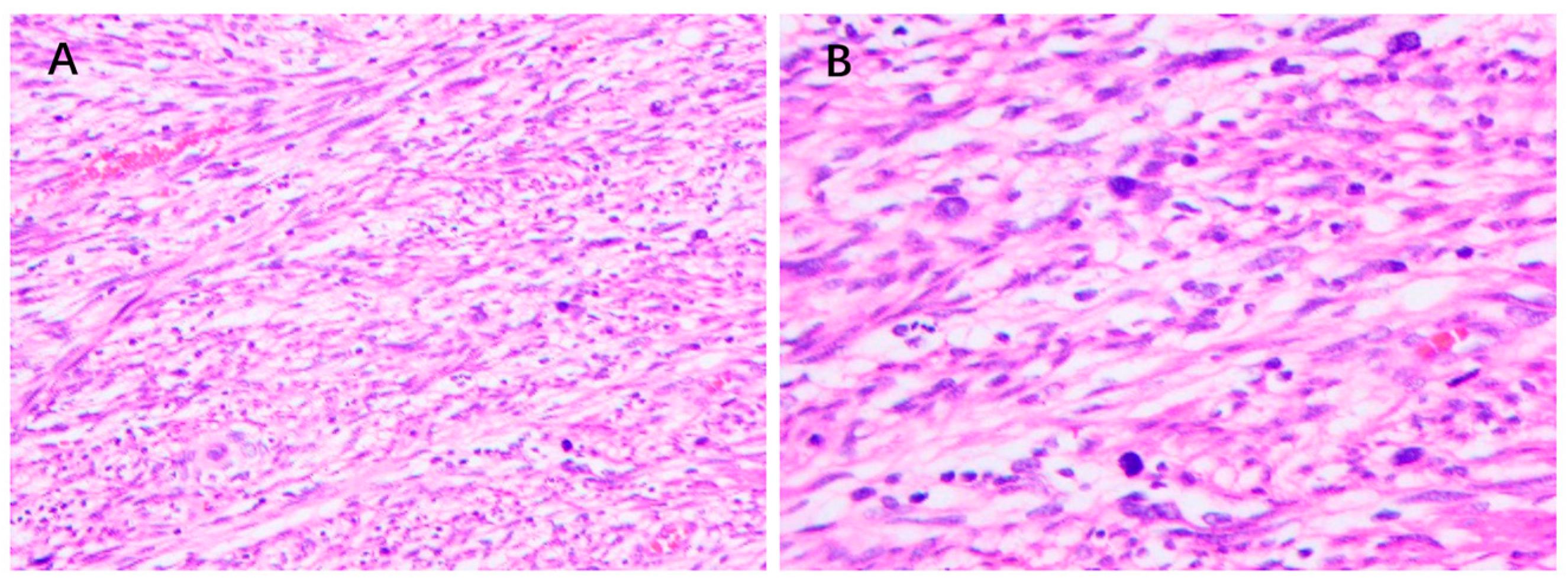
4.3. Pathology
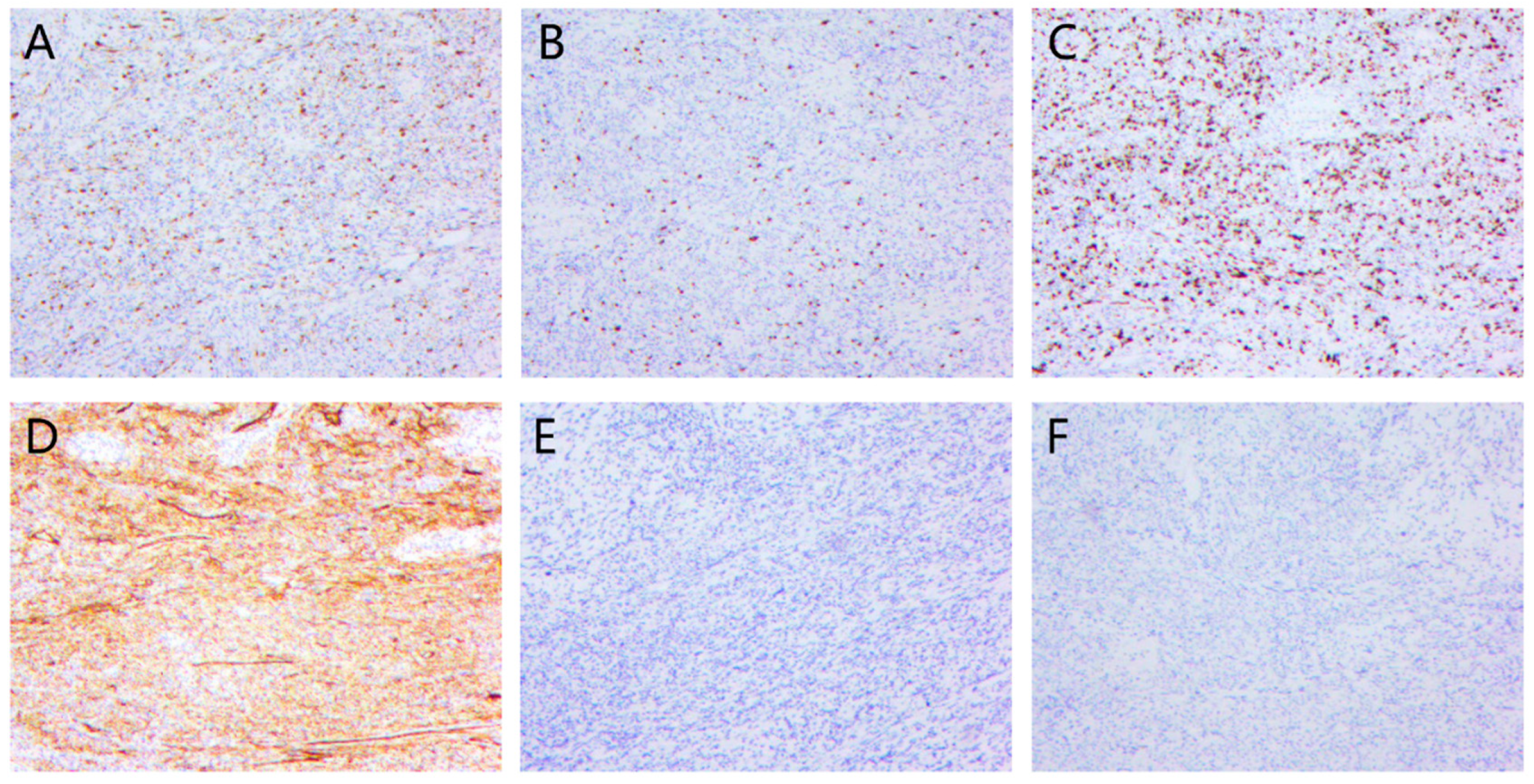
4.4. Immunohistochemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5. Treatment
5.1. Surgery
5.2. Chemotherapy
5.3. Radiation Therapy
5.4. Targeted Therapies
| Drug | Target | Phase | n | Result | Reference |
|---|---|---|---|---|---|
| Erlotinib | EGFR | II | 24 | no responses, one stable disease | [107] |
| Sorafenib | RAF VEGFR/c-KIT | II | 12 | no responses | [108] |
| Imatinib | c-KIT PDGFR VEGFR | II | 7 | no responses, one stable disease | [109] |
| Dasatinib | c-KIT c-SRC | II | 14 | no responses | [110] |
| Bevacizumab/Everolimus | VEGF mTOR | II | 25 | CBR 12% (two stable disease, one partial response) | [112] |
| Ganetespib/Everolimus | Hsp90 mTOR | I/II | 20 | no responses | [113] |
| Selumetinib/Sirolimus | MEK/mTOR | II | 21 | enrolling | N/A |
6. Prognosis
7. Challenges and Prospects
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cai, Z.; Tang, X.; Liang, H.; Yang, R.; Yan, T.; Guo, W. Prognosis and risk factors for malignant peripheral nerve sheath tumor: A systematic review and meta-analysis. World J. Surg. Oncol. 2020, 18, 257. [Google Scholar] [CrossRef] [PubMed]
- Vauthey, J.N.; Woodruff, J.M.; Brennan, M.F. Extremity malignant peripheral nerve sheath tumors (neurogenic sarcomas): A 10-year experience. Ann. Surg. Oncol. 1995, 2, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Baehring, J.M.; Betensky, R.A.; Batchelor, T.T. Malignant peripheral nerve sheath tumor: The clinical spectrum and outcome of treatment. Neurology 2003, 61, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.; Lazar, A.; Lev, D. Malignant peripheral nerve sheath tumour (MPNST): The clinical implications of cellular signalling pathways. Expert Rev. Mol. Med. 2009, 11, e30. [Google Scholar] [CrossRef]
- Stucky, C.C.; Johnson, K.N.; Gray, R.J.; Pockaj, B.A.; Ocal, I.T.; Rose, P.S.; Wasif, N. Malignant peripheral nerve sheath tumors (MPNST): The Mayo Clinic experience. Ann. Surg. Oncol. 2012, 19, 878–885. [Google Scholar] [CrossRef]
- Ducatman, B.S.; Scheithauer, B.W.; Piepgras, D.G.; Reiman, H.M.; Ilstrup, D.M. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986, 57, 2006–2021. [Google Scholar] [CrossRef]
- Mowery, A.; Clayburgh, D. Malignant peripheral nerve sheath tumors: Analysis of the national cancer database. Oral. Oncol. 2019, 98, 13–19. [Google Scholar] [CrossRef]
- Yan, P.; Huang, R.; Hu, P.; Liu, F.; Zhu, X.; Hu, P.; Yin, H.; Zhang, J.; Meng, T.; Huang, Z. Nomograms for predicting the overall and cause-specific survival in patients with malignant peripheral nerve sheath tumor: A population-based study. J. Neurooncol. 2019, 143, 495–503. [Google Scholar] [CrossRef]
- Le Guellec, S.; Decouvelaere, A.V.; Filleron, T.; Valo, I.; Charon-Barra, C.; Robin, Y.M.; Terrier, P.; Chevreau, C.; Coindre, J.M. Malignant Peripheral Nerve Sheath Tumor Is a Challenging Diagnosis: A Systematic Pathology Review, Immunohistochemistry, and Molecular Analysis in 160 Patients From the French Sarcoma Group Database. Am. J. Surg. Pathol. 2016, 40, 896–908. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Jiang, S.; Ma, F.X.; Lu, H. Peripheral nerve tumors of the hand: Clinical features, diagnosis, and treatment. World J. Clin. Cases 2020, 8, 5086–5098. [Google Scholar] [CrossRef]
- Domingues, A.M.; Moertel, C.L.; Marcotte, E.L. The Role of Socioeconomic Status and Race/Ethnicity in Malignant Peripheral Nerve Sheath Tumor Survival: A Surveillance, Epidemiology, and End Results-Based Analysis. Cancer Epidemiol. Biomark. Prev. 2022, 31, 1830–1838. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, 17004. [Google Scholar] [CrossRef]
- Uusitalo, E.; Leppavirta, J.; Koffert, A.; Suominen, S.; Vahtera, J.; Vahlberg, T.; Poyhonen, M.; Peltonen, J.; Peltonen, S. Incidence and mortality of neurofibromatosis: A total population study in Finland. J. Investig. Derm. 2015, 135, 904–906. [Google Scholar] [CrossRef]
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992, 356, 713–715. [Google Scholar] [CrossRef]
- Dasgupta, B.; Yi, Y.; Chen, D.Y.; Weber, J.D.; Gutmann, D.H. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1–associated human and mouse brain tumors. Cancer Res. 2005, 65, 2755–2760. [Google Scholar] [CrossRef]
- De Luca, A.; Bottillo, I.; Dasdia, M.C.; Morella, A.; Lanari, V.; Bernardini, L.; Divona, L.; Giustini, S.; Sinibaldi, L.; Novelli, A.; et al. Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J. Med Genet. 2007, 44, 800–808. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Anderson, J.L.; Gutmann, D.H. Neurofibromatosis type 1. Handb. Clin. Neurol. 2015, 132, 75–86. [Google Scholar] [CrossRef]
- Evans, D.G.; O’Hara, C.; Wilding, A.; Ingham, S.L.; Howard, E.; Dawson, J.; Moran, A.; Scott-Kitching, V.; Holt, F.; Huson, S.M. Mortality in neurofibromatosis 1: In North West England: An assessment of actuarial survival in a region of the UK since 1989. Eur. J. Hum. Genet. 2011, 19, 1187–1191. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Cooper, D.N. Classification of NF1 microdeletions and its importance for establishing genotype/phenotype correlations in patients with NF1 microdeletions. Hum. Genet. 2021, 140, 1635–1649. [Google Scholar] [CrossRef]
- Mavrogenis, A.F.; Pala, E.; Guerra, G.; Ruggieri, P. Post-radiation sarcomas. Clinical outcome of 52 Patients. J. Surg. Oncol. 2012, 105, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Hayano, A. Radiation-Induced Malignant Peripheral Nerve Sheath Tumors: A Systematic Review. World Neurosurg. 2017, 105, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.Y.; Wang, H.T.; Jacobson, A.; Lietz, A.P.; Choy, E.; Raskin, K.A.; Schwab, J.H.; Deshpande, V.; Nielsen, G.P.; DeLaney, T.F.; et al. Radiation-induced and neurofibromatosis-associated malignant peripheral nerve sheath tumors (MPNST) have worse outcomes than sporadic MPNST. Radiother. Oncol. 2019, 137, 61–70. [Google Scholar] [CrossRef]
- Prieto-Granada, C.N.; Wiesner, T.; Messina, J.L.; Jungbluth, A.A.; Chi, P.; Antonescu, C.R. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am. J. Surg. Pathol. 2016, 40, 479–489. [Google Scholar] [CrossRef]
- Mautner, V.F.; Asuagbor, F.A.; Dombi, E.; Funsterer, C.; Kluwe, L.; Wenzel, R.; Widemann, B.C.; Friedman, J.M. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro Oncol. 2008, 10, 593–598. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Spurlock, G.; Monem, B.; Thomas, N.; Friedrich, R.E.; Kluwe, L.; Mautner, V. Germline and somatic NF1 gene mutations in plexiform neurofibromas. Hum. Mutat. 2008, 29, E103–E111. [Google Scholar] [CrossRef]
- Dombi, E.; Solomon, J.; Gillespie, A.J.; Fox, E.; Balis, F.M.; Patronas, N.; Korf, B.R.; Babovic-Vuksanovic, D.; Packer, R.J.; Belasco, J.; et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: Relationship to age and body weight. Neurology 2007, 68, 643–647. [Google Scholar] [CrossRef]
- Nguyen, R.; Kluwe, L.; Fuensterer, C.; Kentsch, M.; Friedrich, R.E.; Mautner, V.F. Plexiform neurofibromas in children with neurofibromatosis type 1: Frequency and associated clinical deficits. J. Pediatr. 2011, 159, 652–655.e2. [Google Scholar] [CrossRef]
- Mautner, V.F.; Hartmann, M.; Kluwe, L.; Friedrich, R.E.; Funsterer, C. MRI growth patterns of plexiform neurofibromas in patients with neurofibromatosis type 1. Neuroradiology 2006, 48, 160–165. [Google Scholar] [CrossRef]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—A consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef]
- Mow, T.C.; Navadgi, S.; Jackett, L.; Galloway, S.; Banting, S. Malignant peripheral nerve sheath tumour arising de novo from ganglioneuroma. Pathology 2015, 47, 595–598. [Google Scholar] [CrossRef]
- McMenamin, M.E.; Fletcher, C.D. Expanding the spectrum of malignant change in schwannomas: Epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: A study of 17 cases. Am. J. Surg. Pathol. 2001, 25, 13–25. [Google Scholar] [CrossRef]
- Wu, J.; Williams, J.P.; Rizvi, T.A.; Kordich, J.J.; Witte, D.; Meijer, D.; Stemmer-Rachamimov, A.O.; Cancelas, J.A.; Ratner, N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 2008, 13, 105–116. [Google Scholar] [CrossRef]
- Brossier, N.M.; Carroll, S.L. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain Res. Bull. 2012, 88, 58–71. [Google Scholar] [CrossRef]
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.C.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol. 2019, 21, 981–992. [Google Scholar] [CrossRef]
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of human malignant peripheral nerve sheath tumors. Neuro-Oncol. Adv. 2020, 2, i50–i61. [Google Scholar] [CrossRef]
- Carrió, M.; Gel, B.; Terribas, E.; Zucchiatti, A.C.; Moliné, T.; Rosas, I.; Teulé, Á.; Cajal, S.R.Y.; López-Gutiérrez, J.C.; Blanco, I.; et al. Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: Correlating histological and genomic findings. Hum. Mutat. 2018, 39, 1112–1125. [Google Scholar] [CrossRef]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum. Mol. Genet. 2019, 28, 2752–2762. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Ma, K.H.; Duong, P.; Moran, J.J.; Junaidi, N.; Svaren, J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 2018, 66, 2487–2502. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Dahiya, S.; Friedmann-Morvinski, D.; Verma, I.M.; Clapp, D.W.; Gutmann, D.H. Spatially- and temporally-controlled postnatal p53 knockdown cooperates with embryonic Schwann cell precursor Nf1 gene loss to promote malignant peripheral nerve sheath tumor formation. Oncotarget 2016, 7, 7403–7414. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, N.; Malzer, E.; Zietsch, J.; Okuducu, A.F.; Mucha, J.; Mawrin, C.; Mautner, V.F.; Schildhaus, H.U.; von Deimling, A. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008, 10, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Bottillo, I.; Ahiquist, T.; Brekke, H.; Danielsen, S.A.; van den Berg, E.; Mertens, F.; Lothe, R.A.; Dallapiccola, B. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 2009, 217, 693–701. [Google Scholar] [CrossRef]
- Kao, E.Y.; Wakeman, K.M.; Wu, Y.; Gross, J.M.; Chen, E.Y.; Ricciotti, R.W.; Liu, Y.J.; Mantilla, J.G. Prevalence and detection of actionable BRAF V600 and NRAS Q61 mutations in malignant peripheral nerve sheath tumor by droplet digital PCR. Hum. Pathol. 2022, 129, 90–97. [Google Scholar] [CrossRef]
- Kaplan, H.G.; Rostad, S.; Ross, J.S.; Ali, S.M.; Millis, S.Z. Genomic Profiling in Patients With Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways With Targetable Mutations. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 967–974. [Google Scholar] [CrossRef]
- Plaat, B.E.C.; Molenaar, W.M.; Mastik, M.F.; Hoekstra, H.J.; Meerman, G.J.T.; van den Berg, E. Computer-assisted cytogenetic analysis of 51 malignant peripheral-nerve-sheath tumors: Sporadic vs. neurofibromatosis-type-1-associated malignant schwannomas. Int. J. Cancer 1999, 83, 171–178. [Google Scholar] [CrossRef]
- Ambrosini, G.; Cheema, H.S.; Seelman, S.; Teed, A.; Sambol, E.B.; Singer, S.; Schwartz, G.K. Sorafenib inhibits growth and mitogen-activated protein kinase signaling in malignant peripheral nerve sheath cells. Mol. Cancer Ther. 2008, 7, 890–896. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of selumetinib in neurofibromatosis type 1–related plexiform neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- Nagabushan, S.; Lau, L.M.S.; Barahona, P.; Wong, M.; Sherstyuk, A.; Marshall, G.M.; Tyrrell, V.; Wegner, E.A.; Ekert, P.G.; Cowley, M.J.; et al. Efficacy of MEK inhibition in a recurrent malignant peripheral nerve sheath tumor. NPJ Precis. Oncol. 2021, 5, 9. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Johansson, G.; Mahller, Y.Y.; Collins, M.H.; Kim, M.O.; Nobukuni, T.; Perentesis, J.; Cripe, T.P.; Lane, H.A.; Kozma, S.C.; Thomas, G.; et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol. Cancer Ther. 2008, 7, 1237–1245. [Google Scholar] [CrossRef]
- Widemann, B.C.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Van Tine, B.A.; Kim, A.; Turpin, B.; Dombi, E.; Jayaprakash, N. SARC016: Phase II Study of Everolimus in Combination with Bevacizumab in Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST); American Society of Clinical Oncology: Alexandria, VA, USA, 2016. [Google Scholar]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Luscan, A.; Shackleford, G.G.; Masliah-Planchon, J.; Laurendeau, I.; Ortonne, N.; Varin, J.; Lallemand, F.; Leroy, K.; Dumaine, V.; Hivelin, M.; et al. The Activation of the WNT Signaling Pathway Is a Hallmark in Neurofibromatosis Type 1 TumorigenesisWnt Pathway Activation in NF1 Tumorigenesis. Clin. Cancer Res. 2014, 20, 358–371. [Google Scholar] [CrossRef]
- Stonecypher, M.S.; Byer, S.J.; Grizzle, W.E.; Carroll, S.L. Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene 2005, 24, 5589–5605. [Google Scholar] [CrossRef]
- Wu, J.; Patmore, D.M.; Jousma, E.; Eaves, D.W.; Breving, K.; Patel, A.V.; Schwartz, E.B.; Fuchs, J.R.; Cripe, T.P.; Stemmer-O Stemmer-Rachamimov, A.; et al. EGFR–STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 2014, 33, 173–180. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Le, L.Q. Heterozygous Tumor Suppressor Microenvironment in Cancer Development. Trends. Cancer 2019, 5, 541–546. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Liao, C.P.; Wang, Y.; Ramani, V.; Vandergriff, T.; Lee, M.; Patel, A.; Ariizumi, K.; Le, L.Q. NF1 heterozygosity fosters de novo tumorigenesis but impairs malignant transformation. Nat. Commun. 2018, 9, 5014. [Google Scholar] [CrossRef]
- Yang, F.-C.; Ingram, D.A.; Chen, S.; Zhu, Y.; Yuan, J.; Li, X.; Yang, X.; Knowles, S.; Horn, W.; Li, Y.; et al. Nf1-dependent tumors require a microenvironment containing Nf1+/−-and c-kit-dependent bone marrow. Cell 2008, 135, 437–448. [Google Scholar] [CrossRef]
- Robertson, K.A.; Nalepa, G.; Yang, F.C.; Bowers, D.C.; Ho, C.Y.; Hutchins, G.D.; Croop, J.M.; Vik, T.A.; Denne, S.C.; Parada, L.F.; et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet Oncol. 2012, 13, 1218–1224. [Google Scholar] [CrossRef]
- Prada, C.E.; Jousma, E.; Rizvi, T.A.; Wu, J.; Dunn, R.S.; Mayes, D.A.; Cancelas, J.A.; Dombi, E.; Kim, M.O.; West, B.L.; et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 2013, 125, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.P.; Katlariwala, P.; Low, G.; Murad, M.H.; McInnes, M.D.F.; Jacques, L.; Jack, A.S. Diagnostic Accuracy of MRI for the Detection of Malignant Peripheral Nerve Sheath Tumors: A Systematic Review and Meta-Analysis. AJR Am. J. Roentgenol 2021, 217, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Gupta, K.; Jaiswal, R.; Vashisht, H. MPNST-A Diagnostic Dilemma. Indian J. Dent. Sci. 2013, 5, 126–129. [Google Scholar]
- Broski, S.M.; Johnson, G.B.; Howe, B.M.; Nathan, M.A.; Wenger, D.E.; Spinner, R.J.; Amrami, K.K. Evaluation of 18F-FDG PET and MRI in differentiating benign and malignant peripheral nerve sheath tumors. Skelet. Radiol. 2016, 45, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Winfree, C.J. 173 A Clinical and Radiographic Score to Assess Malignant Potential of Peripheral Nerve Sheath Tumors. Neurosurgery 2016, 63, 169. [Google Scholar] [CrossRef]
- Ferner, R.; Golding, J.F.; Smith, M.; Calonje, E.; Jan, W.; Sanjayanathan, V.; O’Doherty, M. [18F] 2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) as a diagnostic tool for neurofibromatosis 1 (NF1) associated malignant peripheral nerve sheath tumours (MPNSTs): A long-term clinical study. Ann. Oncol. 2008, 19, 390–394. [Google Scholar] [CrossRef]
- Tsai, L.L.; Drubach, L.; Fahey, F.; Irons, M.; Voss, S.; Ullrich, N.J. [18F]-Fluorodeoxyglucose positron emission tomography in children with neurofibromatosis type 1 and plexiform neurofibromas: Correlation with malignant transformation. J. Neurooncol. 2012, 108, 469–475. [Google Scholar] [CrossRef]
- Benz, M.R.; Czernin, J.; Dry, S.M.; Tap, W.D.; Allen-Auerbach, M.S.; Elashoff, D.; Phelps, M.E.; Weber, W.A.; Eilber, F.C. Quantitative F18-fluorodeoxyglucose positron emission tomography accurately characterizes peripheral nerve sheath tumors as malignant or benign. Cancer 2010, 116, 451–458. [Google Scholar] [CrossRef]
- Treglia, G.; Taralli, S.; Bertagna, F.; Salsano, M.; Muoio, B.; Novellis, P.; Vita, M.L.; Maggi, F.; Giordano, A. Usefulness of whole-body fluorine-18-fluorodeoxyglucose positron emission tomography in patients with neurofibromatosis type 1: A systematic review. Radiol. Res. Pr. 2012, 2012, 431029. [Google Scholar] [CrossRef]
- Brahmi, M.; Thiesse, P.; Ranchere, D.; Mognetti, T.; Pinson, S.; Renard, C.; Decouvelaere, A.V.; Blay, J.Y.; Combemale, P. Diagnostic Accuracy of PET/CT-Guided Percutaneous Biopsies for Malignant Peripheral Nerve Sheath Tumors in Neurofibromatosis Type 1 Patients. PLoS ONE 2015, 10, e0138386. [Google Scholar] [CrossRef]
- Reinert, C.P.; Schuhmann, M.U.; Bender, B.; Gugel, I.; la Fougere, C.; Schafer, J.; Gatidis, S. Comprehensive anatomical and functional imaging in patients with type I neurofibromatosis using simultaneous FDG-PET/MRI. Eur. J. Nucl. Med Mol. Imaging 2019, 46, 776–787. [Google Scholar] [CrossRef]
- Bailey, D.L.; Antoch, G.; Bartenstein, P.; Barthel, H.; Beer, A.J.; Bisdas, S.; Bluemke, D.A.; Boellaard, R.; Claussen, C.D.; Franzius, C.; et al. Combined PET/MR: The Real Work Has Just Started. Summary Report of the Third International Workshop on PET/MR Imaging; February 17-21, 2014, Tubingen, Germany. Mol. Imaging Biol. 2015, 17, 297–312. [Google Scholar] [CrossRef]
- Amanullah, A.; Alavi, A.; Revheim, M.-E.; Werner, T.; Saboury, B. Can FDG PET/MRI be used to identify plexiform neurofibromas at high risk of malignant transformation to peripheral nerve sheath tumor? Soc. Nuclear Med. 2022, 63, 2650. [Google Scholar]
- Graham, D.S.; Russell, T.A.; Eckardt, M.A.; Motamedi, K.; Seeger, L.L.; Singh, A.S.; Bernthal, N.M.; Kalbasi, A.; Dry, S.M.; Nelson, S.D.; et al. Oncologic accuracy of image-guided percutaneous core-needle biopsy of peripheral nerve sheath tumors at a high-volume sarcoma center. Am. J. Clin. Oncol. 2019, 42, 739–743. [Google Scholar] [CrossRef]
- Pianta, M.; Chock, E.; Schlicht, S.; McCombe, D. Accuracy and complications of CT-guided core needle biopsy of peripheral nerve sheath tumours. Skelet. Radiol. 2015, 44, 1341–1349. [Google Scholar] [CrossRef]
- McGee, R.S., Jr.; Ward, W.G.; Kilpatrick, S.E. Malignant peripheral nerve sheath tumor: A fine-needle aspiration biopsy study. Diagn. Cytopathol. 1997, 17, 298–305. [Google Scholar] [CrossRef]
- Mito, J.K.; Qian, X.; Doyle, L.A.; Hornick, J.L.; Jo, V.Y. Role of histone H3K27 trimethylation loss as a marker for malignant peripheral nerve sheath tumor in fine-needle aspiration and small biopsy specimens. Am. J. Clin. Pathol. 2017, 148, 179–189. [Google Scholar] [CrossRef]
- Prudner, B.C.; Ball, T.; Rathore, R.; Hirbe, A.C. Diagnosis and management of malignant peripheral nerve sheath tumors: Current practice and future perspectives. Neuro-Oncol. Adv. 2020, 2, i40–i49. [Google Scholar] [CrossRef]
- Thway, K.; Fisher, C. Malignant peripheral nerve sheath tumor: Pathology and genetics. Ann. Diagn. Pathol. 2014, 18, 109–116. [Google Scholar] [CrossRef]
- Coindre, J.M.; Terrier, P.; Guillou, L.; Le Doussal, V.; Collin, F.; Ranchere, D.; Sastre, X.; Vilain, M.O.; Bonichon, F.; N’Guyen Bui, B. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: A study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001, 91, 1914–1926. [Google Scholar] [CrossRef]
- Gonzalez-Martinez, T.; Perez-Pinera, P.; Diaz-Esnal, B.; Vega, J. S-100 proteins in the human peripheral nervous system. Microsc. Res. Tech. 2003, 60, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Smith, K.D.; Liu, J.; Lahat, G.; Myers, S.; Wang, W.L.; Zhang, W.; McCutcheon, I.E.; Slopis, J.M.; Lazar, A.J.; et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann. Surg. 2009, 249, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Karamchandani, J.R.; Nielsen, T.O.; van de Rijn, M.; West, R.B. Sox10 and S100 in the diagnosis of soft-tissue neoplasms. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.W.; Nickoloff, B.J. CD-34 is expressed by a distinctive cell population in peripheral nerve, nerve sheath tumors, and related lesions. Am. J. Surg. Pathol. 1993, 17, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Oda, Y.; Tamiya, S.; Masuda, K.; Tsuneyoshi, M. Malignant peripheral nerve sheath tumour arising within neurofibroma. An immunohistochemical analysis in the comparison between benign and malignant components. J. Clin. Pathol. 2001, 54, 631–636. [Google Scholar] [CrossRef]
- Shimada, S.; Tsuzuki, T.; Kuroda, M.; Nagasaka, T.; Hara, K.; Takahashi, E.; Hayakawa, S.; Ono, K.; Maeda, N.; Mori, N.; et al. Nestin expression as a new marker in malignant peripheral nerve sheath tumors. Pathol. Int. 2007, 57, 60–67. [Google Scholar] [CrossRef]
- Verdijk, R.M.; den Bakker, M.A.; Dubbink, H.J.; Hop, W.C.; Dinjens, W.N.; Kros, J.M. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J. Neuropathol. Exp. Neurol. 2010, 69, 16–26. [Google Scholar] [CrossRef]
- Pekmezci, M.; Reuss, D.E.; Hirbe, A.C.; Dahiya, S.; Gutmann, D.H.; von Deimling, A.; Horvai, A.E.; Perry, A. Morphologic and immunohistochemical features of malignant peripheral nerve sheath tumors and cellular schwannomas. Mod. Pathol. 2015, 28, 187–200. [Google Scholar] [CrossRef]
- Zhou, H.; Coffin, C.M.; Perkins, S.L.; Tripp, S.R.; Liew, M.; Viskochil, D.H. Malignant peripheral nerve sheath tumor: A comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am. J. Surg. Pathol. 2003, 27, 1337–1345. [Google Scholar] [CrossRef]
- Cleven, A.H.; Al Sannaa, G.A.; Briaire-de Bruijn, I.; Ingram, D.R.; Van De Rijn, M.; Rubin, B.P.; De Vries, M.W.; Watson, K.L.; Torres, K.E.; Wang, W.-L.; et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016, 29, 582–590. [Google Scholar] [CrossRef]
- Röhrich, M.; Koelsche, C.; Schrimpf, D.; Capper, D.; Sahm, F.; Kratz, A.; Reuss, J.; Hovestadt, V.; Jones, D.T.; Bewerunge-Hudler, M.; et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016, 131, 877–887. [Google Scholar] [CrossRef]
- Perry, A.; Kunz, S.N.; Fuller, C.E.; Banerjee, R.; Marley, E.F.; Liapis, H.; Watson, M.A.; Gutmann, D.H. Differential NF1, p16, and EGFR patterns by interphase cytogenetics (FISH) in malignant peripheral nerve sheath tumor (MPNST) and morphologically similar spindle cell neoplasms. J. Neuropathol. Exp. Neurol. 2002, 61, 702–709. [Google Scholar] [CrossRef]
- Dunn, G.P.; Spiliopoulos, K.; Plotkin, S.R.; Hornicek, F.J.; Harmon, D.C.; Delaney, T.F.; Williams, Z. Role of resection of malignant peripheral nerve sheath tumors in patients with neurofibromatosis type 1. J. Neurosurg. 2013, 118, 142–148. [Google Scholar] [CrossRef]
- Higham, C.S.; Steinberg, S.M.; Dombi, E.; Perry, A.; Helman, L.J.; Schuetze, S.M.; Ludwig, J.A.; Staddon, A.; Milhem, M.M.; Rushing, D.; et al. SARC006: Phase II Trial of Chemotherapy in Sporadic and Neurofibromatosis Type 1 Associated Chemotherapy-Naive Malignant Peripheral Nerve Sheath Tumors. Sarcoma 2017, 2017, 8685638. [Google Scholar] [CrossRef]
- Kroep, J.R.; Ouali, M.; Gelderblom, H.; Le Cesne, A.; Dekker, T.J.A.; Van Glabbeke, M.; Hogendoorn, P.C.W.; Hohenberger, P. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: An EORTC soft tissue and bone sarcoma group study. Ann. Oncol. 2011, 22, 207–214. [Google Scholar] [CrossRef]
- Zehou, O.; Fabre, E.; Zelek, L.; Sbidian, E.; Ortonne, N.; Banu, E.; Wolkenstein, P.; Valeyrie-Allanore, L. Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1: A 10-year institutional review. Orphanet. J. Rare Dis. 2013, 8, 127. [Google Scholar] [CrossRef]
- Kolberg, M.; Holand, M.; Agesen, T.H.; Brekke, H.R.; Liestol, K.; Hall, K.S.; Mertens, F.; Picci, P.; Smeland, S.; Lothe, R.A. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol. 2013, 15, 135–147. [Google Scholar] [CrossRef]
- Frustaci, S.; Gherlinzoni, F.; De Paoli, A.; Bonetti, M.; Azzarelli, A.; Comandone, A.; Olmi, P.; Buonadonna, A.; Pignatti, G.; Barbieri, E.; et al. Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: Results of the Italian randomized cooperative trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 1238–1247. [Google Scholar] [CrossRef]
- Ferner, R.E.; Gutmann, D.H. International Consensus Statement on Malignant Peripheral Nerve Sheath Tumors in Neurofibromatosis; AACR: Philadelphia, PA, USA, 2002. [Google Scholar]
- Sloan, L.; Terezakis, S.A.; Blakeley, J.O.; Slobogean, B.; Kleinberg, L.R. Long-Term Outcomes of Radiation Therapy (RT) in the Management of Malignant Peripheral Nerve Sheath Tumors (MPNST) in Patients with Neurofibromatosis Type 1 (NF1). Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, E474–E475. [Google Scholar] [CrossRef]
- Kahn, J.; Gillespie, A.; Tsokos, M.; Ondos, J.; Dombi, E.; Camphausen, K.; Widemann, B.C.; Kaushal, A. Radiation therapy in management of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Front. Oncol. 2014, 4, 324. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Q.; Eisenberg, B.L.; Kane, J.M.; Li, X.A.; Lucas, D.; Petersen, I.A.; DeLaney, T.F.; Freeman, C.R.; Finkelstein, S.E.; et al. Significant Reduction of Late Toxicities in Patients With Extremity Sarcoma Treated With Image-Guided Radiation Therapy to a Reduced Target Volume: Results of Radiation Therapy Oncology Group RTOG-0630 Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Hirose, T.; Scheithauer, B.W.; Schild, S.E.; Gunderson, L.L. Malignant peripheral nerve sheath tumor: Analysis of treatment outcome. Int. J. Radiat. Oncol. Biol. Phys 1998, 42, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Pisters, P.W.; Harrison, L.B.; Leung, D.H.; Woodruff, J.M.; Casper, E.S.; Brennan, M.F. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1996, 14, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Velasco-Miguel, S.; Vass, W.C.; Parada, L.F.; DeClue, J.E. Epidermal growth factor receptor signaling pathways are associated with tumorigenesis in the Nf1:p53 mouse tumor model. Cancer Res. 2002, 62, 4507–4513. [Google Scholar] [PubMed]
- Albritton, K.H.; Rankin, C.; Coffin, C.M.; Ratner, N.; Budd, G.T.; Schuetze, S.M.; Randall, R.L.; Declue, J.E.; Borden, E.C. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST). J. Clin. Oncol. 2006, 24, 524s. [Google Scholar] [CrossRef]
- Maki, R.G.; D’Adamo, D.R.; Keohan, M.L.; Saulle, M.; Schuetze, S.M.; Undevia, S.D.; Livingston, M.B.; Cooney, M.M.; Hensley, M.L.; Mita, M.M.; et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3133–3140. [Google Scholar] [CrossRef]
- Chugh, R.; Wathen, J.K.; Maki, R.G.; Benjamin, R.S.; Patel, S.R.; Meyers, P.A.; Priebat, D.A.; Reinke, D.K.; Thomas, D.G.; Keohan, M.L.; et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3148–3153. [Google Scholar] [CrossRef]
- Schuetze, S.; Wathen, K.; Choy, E.; Samuels, B.L.; Ganjoo, K.N.; Staddon, A.P.; von Mehren, M.; Chow, W.A.; Trent, J.C.; Baker, L.H. Results of a Sarcoma Alliance for Research through Collaboration (SARC) phase II trial of dasatinib in previously treated, high-grade, advanced sarcoma. J. Clin. Oncol. 2010, 28, 10009. [Google Scholar] [CrossRef]
- Endo, M.; Yamamoto, H.; Setsu, N.; Kohashi, K.; Takahashi, Y.; Ishii, T.; Iida, K.-I.; Matsumoto, Y.; Hakozaki, M.; Aoki, M.; et al. Prognostic Significance of AKT/mTOR and MAPK Pathways and Antitumor Effect of mTOR Inhibitor in NF1-Related and Sporadic Malignant Peripheral Nerve Sheath TumorsAKT/mTOR and MAPK Pathways in MPNSTs. Clin. Cancer Res. 2013, 19, 450–461. [Google Scholar] [CrossRef]
- Widemann, B.C.; Lu, Y.; Reinke, D.; Okuno, S.H.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Hirbe, A.C.; Kim, A.; et al. Targeting Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST) in a Phase II Study of Everolimus in Combination with Bevacizumab (SARC016). Sarcoma 2019, 2019, 7656747. [Google Scholar] [CrossRef]
- Kim, A.; Lu, Y.; Okuno, S.H.; Reinke, D.; Maertens, O.; Perentesis, J.; Basu, M.; Wolters, P.L.; De Raedt, T.; Chawla, S.; et al. Targeting Refractory Sarcomas and Malignant Peripheral Nerve Sheath Tumors in a Phase I/II Study of Sirolimus in Combination with Ganetespib (SARC023). Sarcoma 2020, 2020, 5784876. [Google Scholar] [CrossRef]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V.; et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef]
- Peacock, J.D.; Pridgeon, M.G.; Tovar, E.A.; Essenburg, C.J.; Bowman, M.; Madaj, Z.; Koeman, J.; Boguslawski, E.A.; Grit, J.; Dodd, R.D.; et al. Genomic Status of MET Potentiates Sensitivity to MET and MEK Inhibition in NF1-Related Malignant Peripheral Nerve Sheath TumorsMET Promotes Sensitivity to MET and MEK Inhibition in MPNSTs. Cancer Res. 2018, 78, 3672–3687. [Google Scholar] [CrossRef]
- Jessen, W.J.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.O.; Dombi, E.; et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Investig. 2013, 123, 340–347. [Google Scholar] [CrossRef]
- Gross, A.M.; Wolters, P.; Baldwin, A.; Dombi, E.; Fisher, M.J.; Weiss, B.D.; Kim, A.; Blakeley, J.O.N.; Whitcomb, P.; Holmblad, M. SPRINT: Phase II Study of the MEK 1/2 Inhibitor Selumetinib (AZD6244, ARRY-142886) in Children with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN); American Society of Clinical Oncology: Alexandria, VA, USA, 2018. [Google Scholar]
- Vaassen, P.; Durr, N.; Rohrig, A.; Willing, R.; Rosenbaum, T. Trametinib Induces Neurofibroma Shrinkage and Enables Surgery. Neuropediatrics 2019, 50, 300–303. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Kaplan, H.G. Vemurafenib treatment of BRAF V600E-mutated malignant peripheral nerve sheath tumor. J. Natl. Compr. Cancer Netw. JNCCN 2013, 11, 1466–1470. [Google Scholar] [CrossRef]
- Valentin, T.; Le Cesne, A.; Ray-Coquard, I.; Italiano, A.; Decanter, G.; Bompas, E.; Isambert, N.; Thariat, J.; Linassier, C.; Bertucci, F.; et al. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur. J. Cancer 2016, 56, 77–84. [Google Scholar] [CrossRef]
- Watson, K.L.; Al Sannaa, G.A.; Kivlin, C.M.; Ingram, D.R.; Landers, S.M.; Roland, C.L.; Cormier, J.N.; Hunt, K.K.; Feig, B.W.; Guadagnolo, B.A.; et al. Patterns of recurrence and survival in sporadic, neurofibromatosis type 1–associated, and radiation-associated malignant peripheral nerve sheath tumors. Radiother. Oncol. 2017, 126, 319–329. [Google Scholar] [CrossRef]
- Martin, E.; Coert, J.H.; Flucke, U.E.; Slooff, W.M.; Ho, V.K.Y.; van der Graaf, W.T.; van Dalen, T.; van de Sande, M.A.J.; van Houdt, W.J.; Grunhagen, D.J.; et al. A nationwide cohort study on treatment and survival in patients with malignant peripheral nerve sheath tumours. Eur. J. Cancer 2020, 124, 77–87. [Google Scholar] [CrossRef]
- Woodruff, J.M. Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am. J. Med Genet. 1999, 89, 23–30. [Google Scholar] [CrossRef]
- Wakeman, K.M.; Zhang, Q.S.; Bandhlish, A.; Cranmer, L.D.; Ricciotti, R.W.; Mantilla, J.G. Fédération Nationale Des Centres de Lutte Contre Le Cancer (FNCLCC) Grading, Margin Status and Tumor Location Associate With Survival Outcomes in Malignant Peripheral Nerve Sheath Tumors. Am. J. Clin. Oncol. 2022, 45, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.G.; Vasudevan, H.N.; Chen, W.C.; Magill, S.T.; Braunstein, S.E.; Jacques, L.; Dahiya, S.; Rodriguez, F.J.; Horvai, A.E.; Perry, A.; et al. Histopathologic findings in malignant peripheral nerve sheath tumor predict response to radiotherapy and overall survival. Neuro-Oncol. Adv. 2020, 2, vdaa131. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.; Prasad, V.; Foster, L.; Dall, G.; Birch, R.; Grimer, R.J. Survival in malignant peripheral nerve sheath tumours: A comparison between sporadic and neurofibromatosis type 1-associated tumours. Sarcoma 2009, 2009, 756395. [Google Scholar] [CrossRef]
- Mohamad, T.; Plante, C.; Brosseau, J.P. Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development. Int. J. Mol. Sci. 2021, 22, 8620. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, C.; Zhou, H.; Dong, Y.; Alhaskawi, A.; Hasan Abdullah Ezzi, S.; Wang, Z.; Lai, J.; Goutham Kota, V.; Hasan Abdulla Hasan Abdulla, M.; Lu, H. Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management. Cancers 2023, 15, 1077. https://doi.org/10.3390/cancers15041077
Yao C, Zhou H, Dong Y, Alhaskawi A, Hasan Abdullah Ezzi S, Wang Z, Lai J, Goutham Kota V, Hasan Abdulla Hasan Abdulla M, Lu H. Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management. Cancers. 2023; 15(4):1077. https://doi.org/10.3390/cancers15041077
Chicago/Turabian StyleYao, Chengjun, Haiying Zhou, Yanzhao Dong, Ahmad Alhaskawi, Sohaib Hasan Abdullah Ezzi, Zewei Wang, Jingtian Lai, Vishnu Goutham Kota, Mohamed Hasan Abdulla Hasan Abdulla, and Hui Lu. 2023. "Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management" Cancers 15, no. 4: 1077. https://doi.org/10.3390/cancers15041077
APA StyleYao, C., Zhou, H., Dong, Y., Alhaskawi, A., Hasan Abdullah Ezzi, S., Wang, Z., Lai, J., Goutham Kota, V., Hasan Abdulla Hasan Abdulla, M., & Lu, H. (2023). Malignant Peripheral Nerve Sheath Tumors: Latest Concepts in Disease Pathogenesis and Clinical Management. Cancers, 15(4), 1077. https://doi.org/10.3390/cancers15041077