Anti-Leukaemic Activity of Rilpivirine Is Mediated by Aurora A Kinase Inhibition


 , and
, and 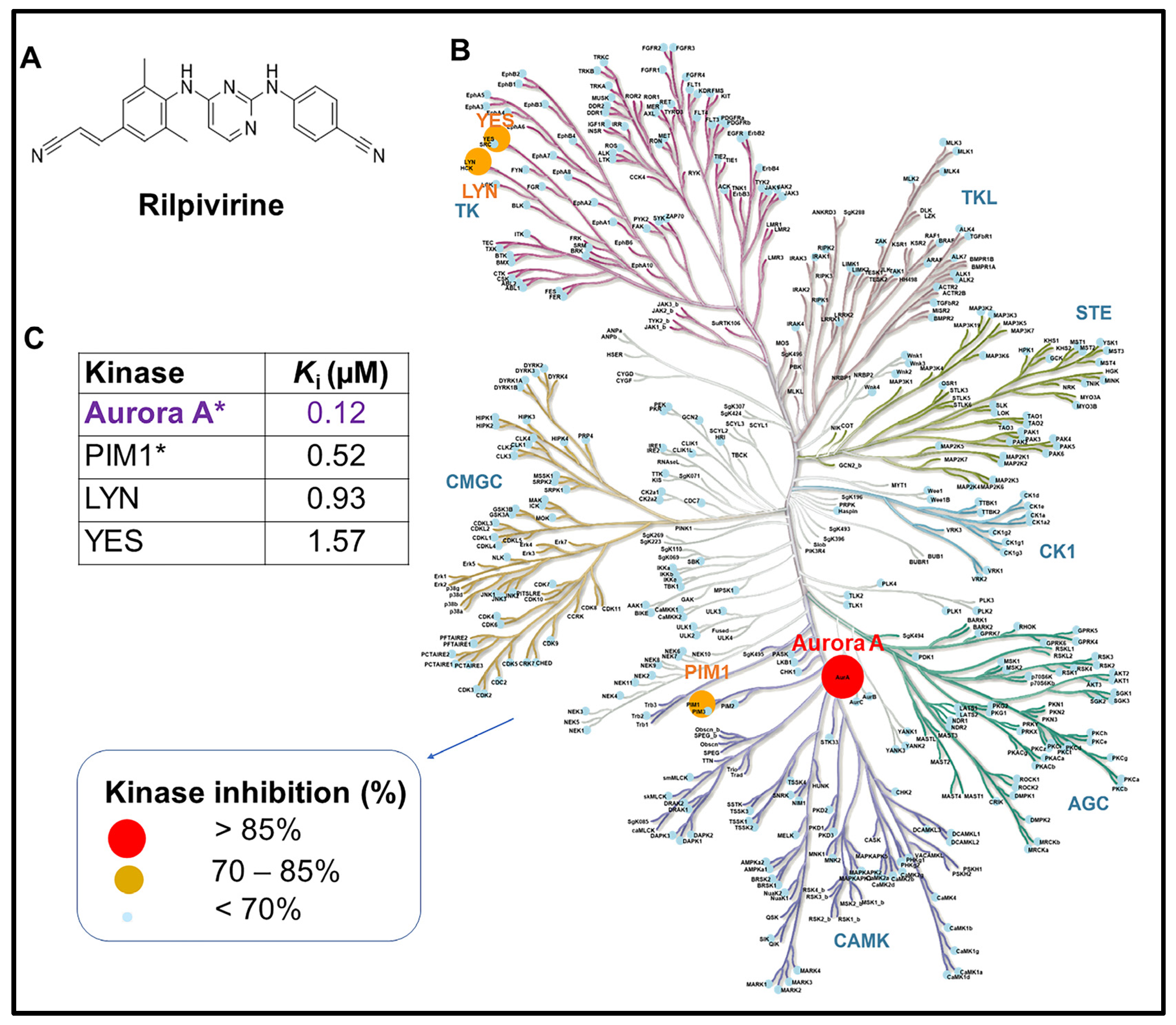{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Kinase Assay
2.3. Cell Culture
2.4. Cell Viability Assay
2.5. Colony Formation Assay
2.6. Cell Cycle Analysis
2.7. Apoptosis Analysis
2.8. Caspase-3/7 Activity Assay
2.9. Western Blotting
2.10. Animal Experiments
2.11. Pharmacokinetic Analysis
2.12. In Vivo Safety Profiles
2.13. In Vivo Anti-Cancer Efficacy Study
2.14. Statistical Analysis
3. Results
3.1. Rilpivirine Shows Significant Inhibitory Selectivity towards Aurora A Kinase
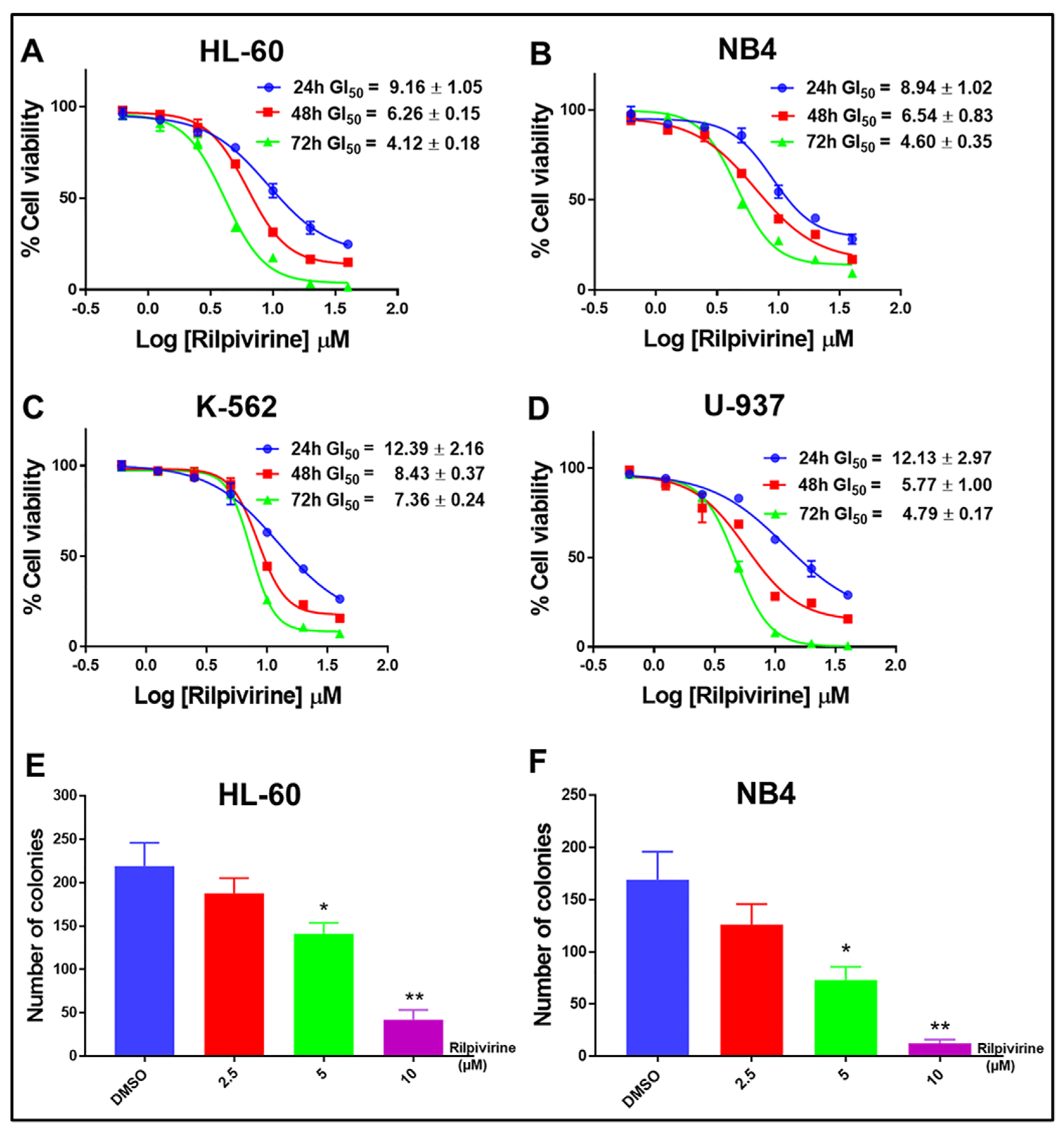
3.2. Rilpivirine Exhibits Anti-Proliferative Activities in Multiple Leukaemic Cell Lines
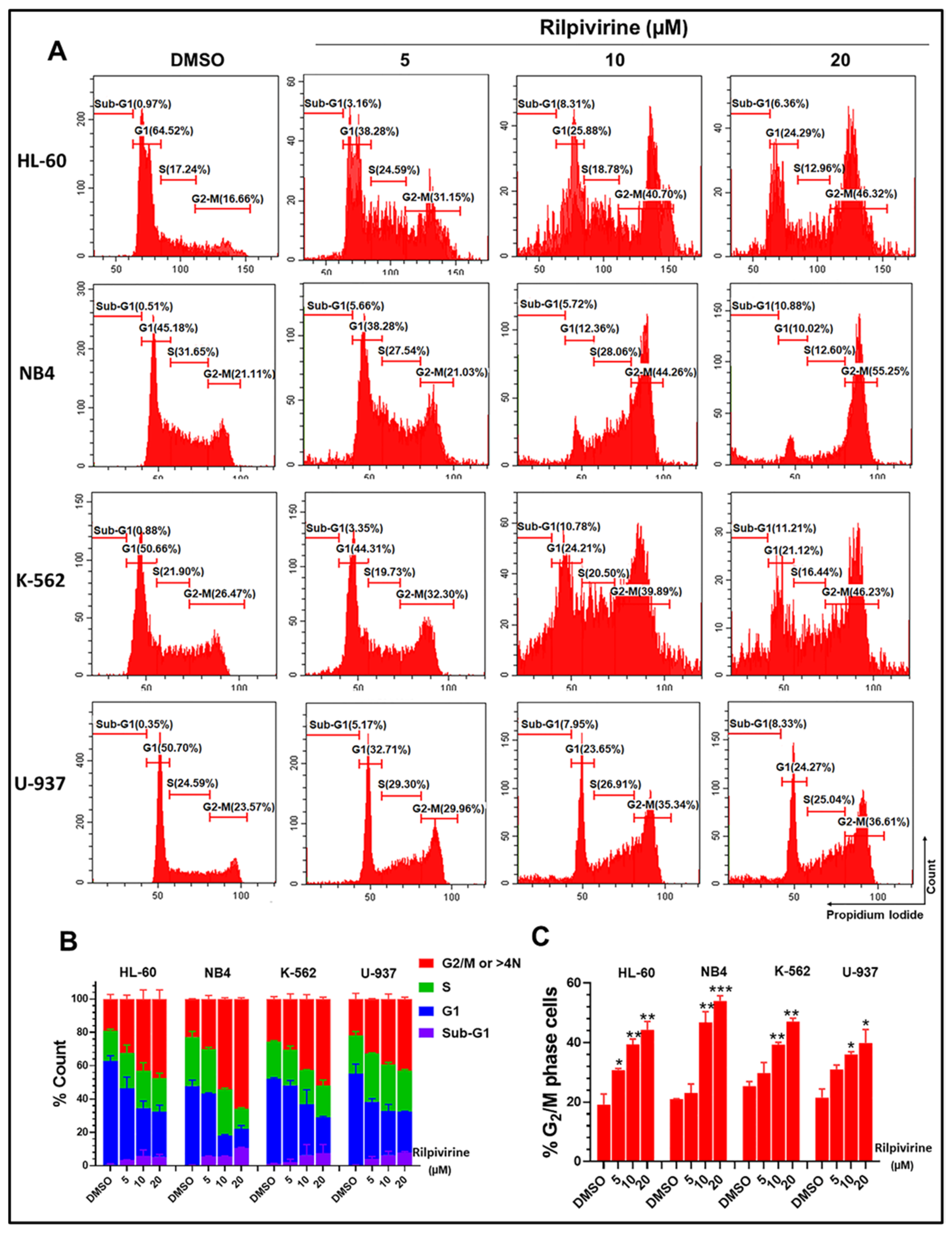
3.3. Rilpivirine Arrests Leukaemic Cells in the G2/M Phase of the Cell Cycle
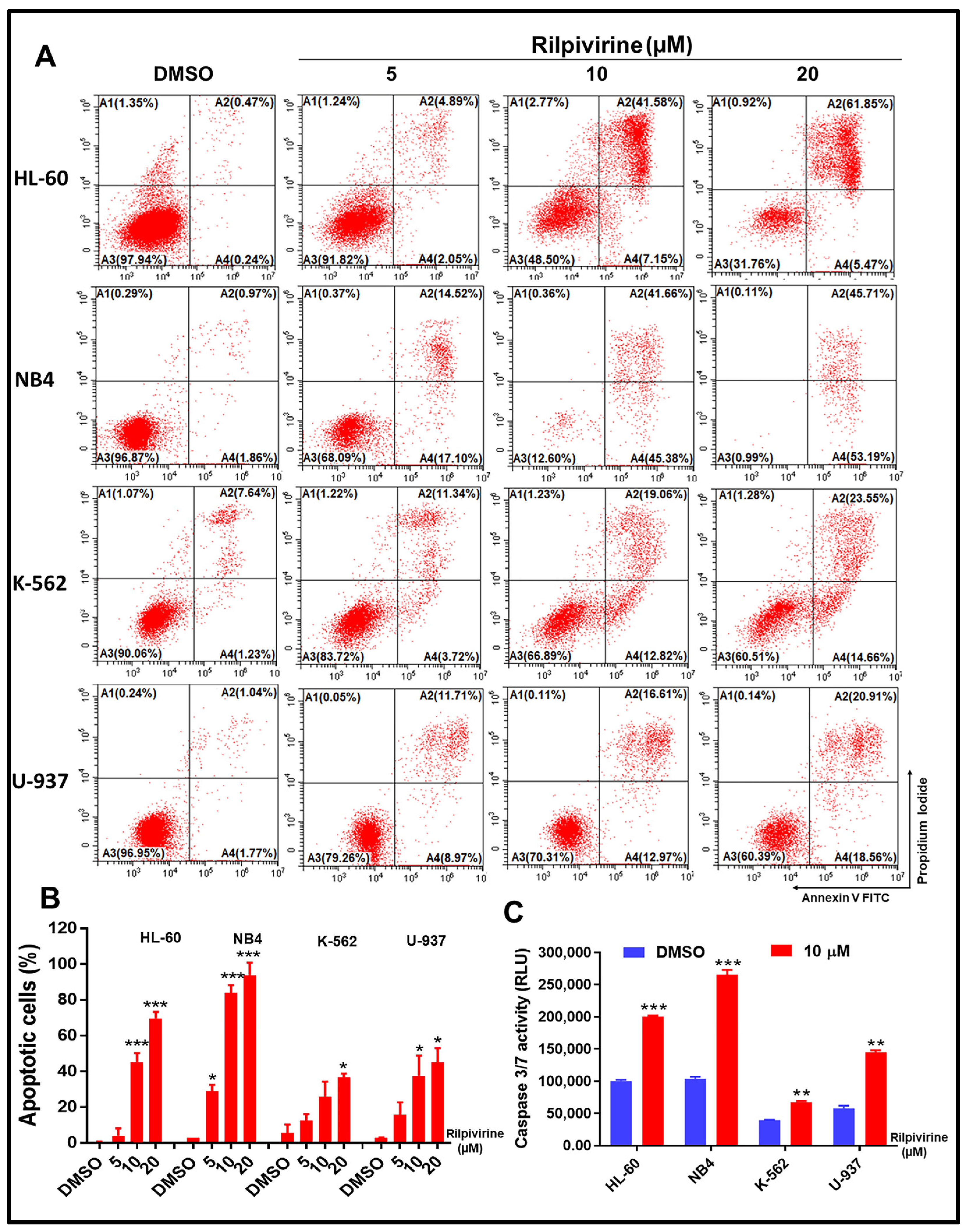
3.4. Rilpivirine Induces Apoptosis and Caspase 3/7 Activation in Leukaemic Cells
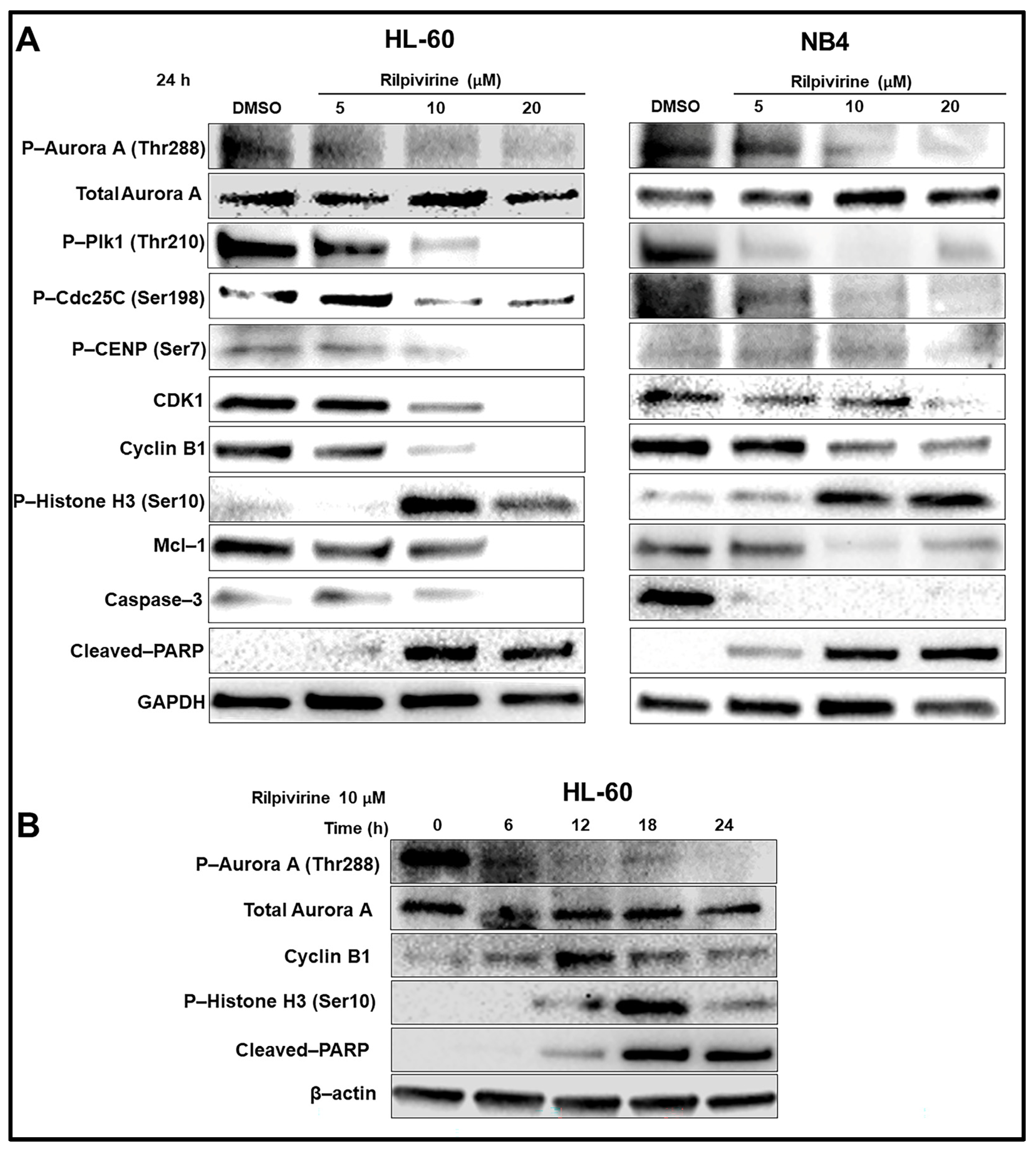
3.5. Rilpivirine Inhibits Aurora A Autophosphorylation and Modulates the Cell Cycle and Apoptosis Regulatory Proteins
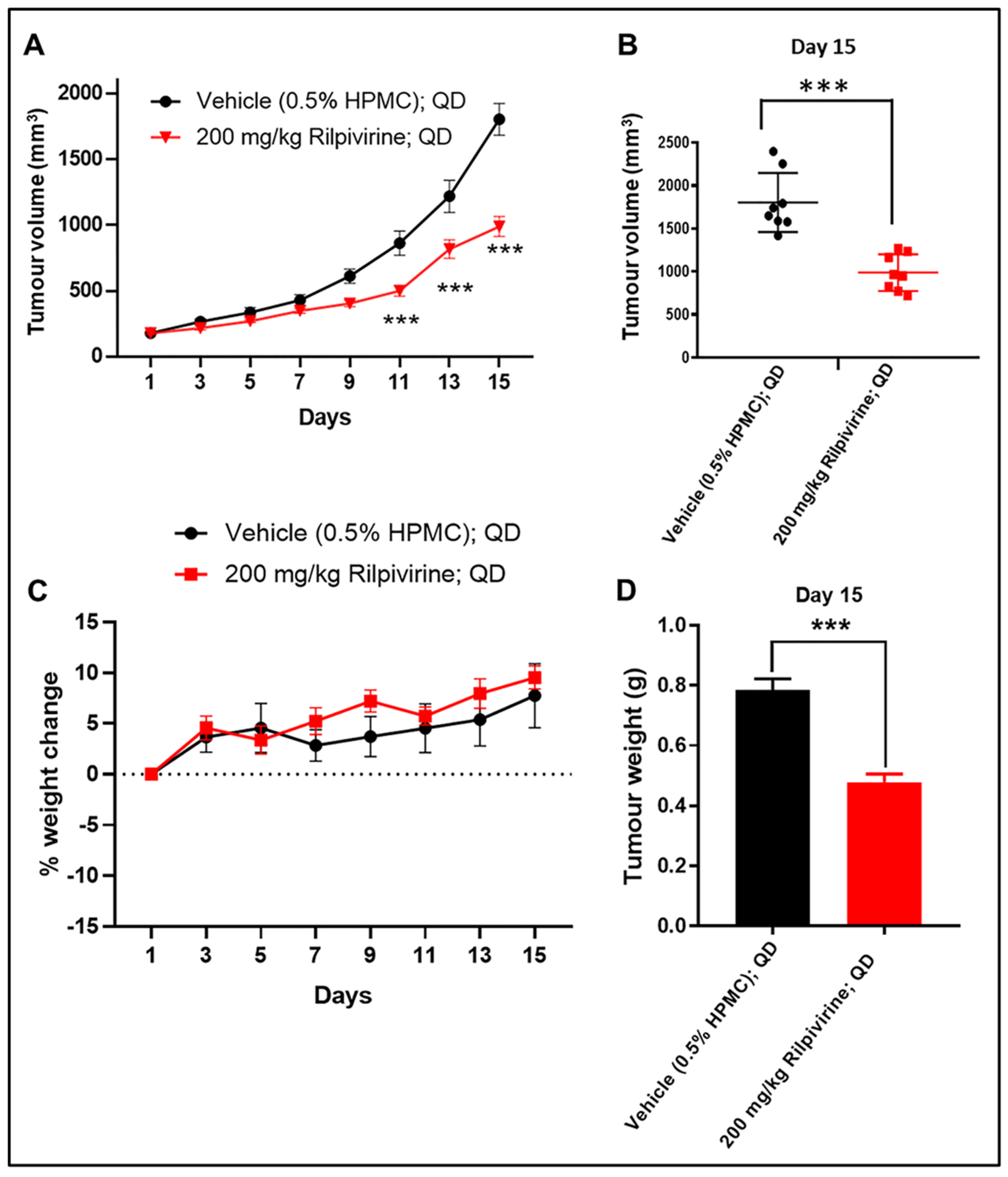
3.6. Rilpivirine Inhibits Leukaemic Tumour Growth In Vivo
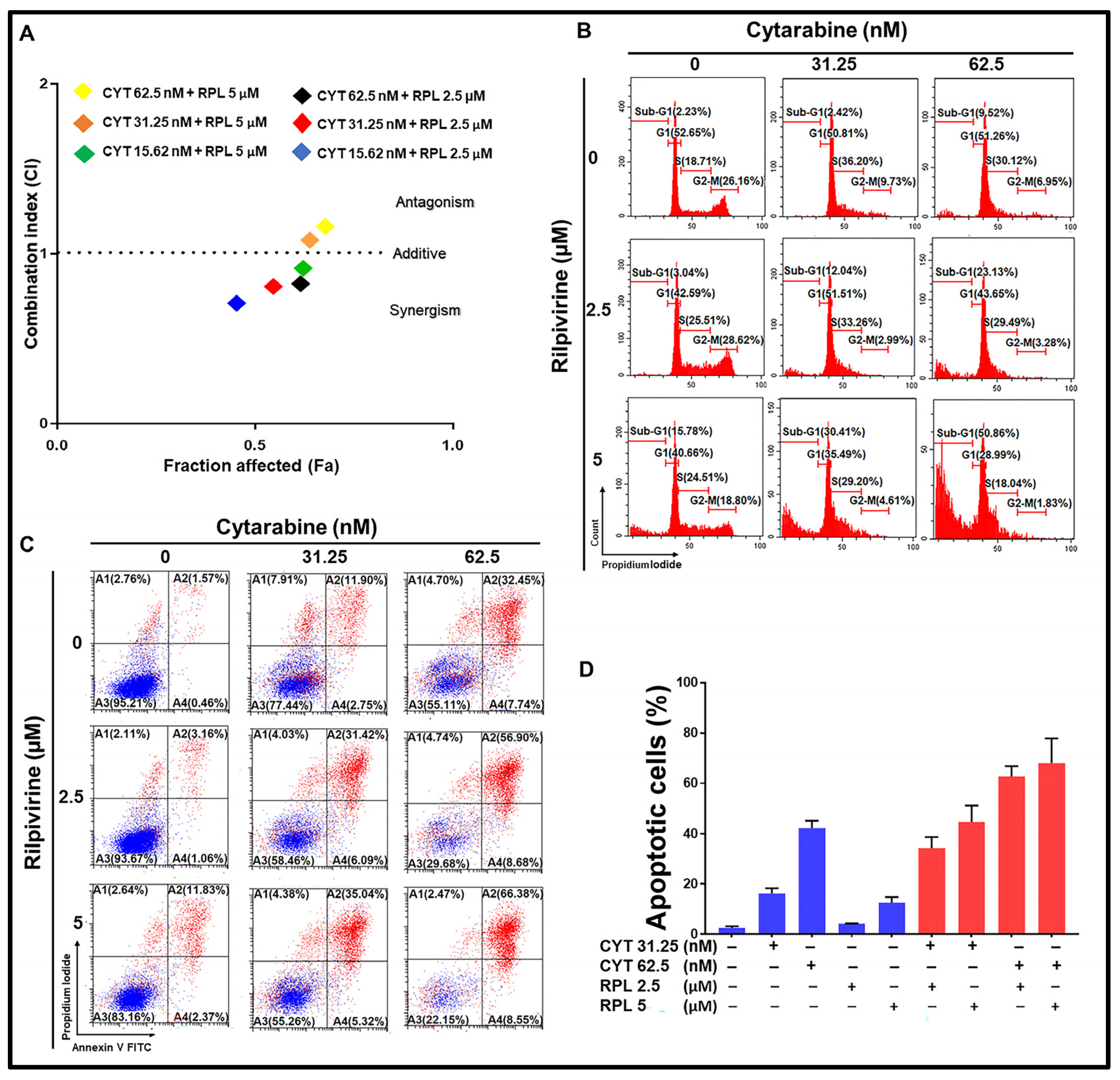
3.7. Rilpivirine Enhanced the Anti-Proliferative Effect of Cytarabine in Leukaemic Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J. Acute myeloid leukaemia. Nat. Rev. Dis. Prim. 2016, 2, 16010. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-F.; Luo, S.-K.; Xu, J.; Li, J.; Xu, D.-R.; Wang, L.-H.; Yan, M.; Wang, X.-R.; Wan, X.-B.; Zheng, F.-M. Aurora kinase inhibitory vx-680 increases bax/bcl-2 ratio and induces apoptosis in aurora-a-high acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2008, 111, 2854–2865. [Google Scholar] [CrossRef]
- Crossnohere, N.L.; Richardson, D.R.; Reinhart, C.; O’Donoghue, B.; Love, S.M.; Smith, B.D.; Bridges, J.F. Side effects from acute myeloid leukemia treatment: Results from a national survey. Curr. Med. Res. Opin. 2019, 35, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Nawrocki, S.T.; Espitia, C.M.; Zhang, M.; Yang, J.J.; Padmanabhan, S.; Ecsedy, J.; Giles, F.J.; Carew, J.S. Targeting aurora a kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a foxo-dependent mechanism. Int. J. Cancer 2012, 131, 2693–2703. [Google Scholar] [CrossRef]
- Perl, A.E. The role of targeted therapy in the management of patients with aml. Blood Adv. 2017, 1, 2281–2294. [Google Scholar] [CrossRef]
- Stegmeier, F.; Warmuth, M.; Sellers, W.; Dorsch, M. Targeted cancer therapies in the twenty-first century: Lessons from imatinib. Clin. Pharmacol. Ther. 2010, 87, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Wang, S.; Bowden, N.; Martin, J.; Head, R. Repurposing existing therapeutics, its importance in oncology drug development: Kinases as a potential target. Br. J. Clin. Pharmacol. 2022, 88, 64–74. [Google Scholar] [CrossRef]
- Knapp, S. New Opportunities for Kinase Drug Repurposing and Target Discovery; Nature Publishing Group: Berlin, Germany, 2018. [Google Scholar]
- Islam, S.; Teo, T.; Kumarasiri, M.; Slater, M.; Martin, J.H.; Wang, S.; Head, R. Combined in silico and in vitro evidence supporting an aurora a kinase inhibitory role of the anti-viral drug rilpivirine and an anti-proliferative influence on cancer cells. Pharmaceuticals 2022, 15, 1186. [Google Scholar] [CrossRef]
- Malumbres, M.; Perez de Castro, I. Aurora kinase a inhibitors: Promising agents in antitumoral therapy. Expert Opin. Ther. Targets 2014, 18, 1377–1393. [Google Scholar]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase a, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847. [Google Scholar] [CrossRef]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-a kinase: A potent oncogene and target for cancer therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Jang, J.E.; Cheong, J.-W.; Eom, J.-I.; Jeung, H.-K.; Kim, Y.; Hwang, D.Y.; Min, Y.H. Aurora a kinase expression is increased in leukemia stem cells, and a selective aurora a kinase inhibitor enhances ara-c-induced apoptosis in acute myeloid leukemia stem cells. Korean J. Hematol. 2012, 47, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Yang, J.; Nishioka, C.; Tasaka, T.; Taniguchi, A.; Kuwayama, Y.; Komatsu, N.; Bandobashi, K.; Togitani, K.; Koeffler, H.P. A novel treatment strategy targeting aurora kinases in acute myelogenous leukemia. Mol. Cancer Ther. 2007, 6, 1851–1857. [Google Scholar] [CrossRef]
- Fathi, A.T.; Wander, S.A.; Blonquist, T.M.; Brunner, A.M.; Amrein, P.C.; Supko, J.; Hermance, N.M.; Manning, A.L.; Sadrzadeh, H.; Ballen, K.K. Phase i study of the aurora a kinase inhibitor alisertib with induction chemotherapy in patients with acute myeloid leukemia. Haematologica 2017, 102, 719. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Blonquist, T.M.; DeAngelo, D.J.; McMasters, M.; Winer, E.S.; Hobbs, G.S.; Amrein, P.C.; Hock, H.; Steensma, D.P.; Garcia, J.S. Phase ii clinical trial of alisertib, an aurora a kinase inhibitor, in combination with induction chemotherapy in high-risk, untreated patients with acute myeloid leukemia. Blood 2018, 132, 766. [Google Scholar] [CrossRef]
- Farag, S.S. The potential role of aurora kinase inhibitors in haematological malignancies. Br. J. Haematol. 2011, 155, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C. Relationship between the inhibition constant (ki) and the concentration of inhibition, which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Diab, S.; Abdelaziz, A.M.; Li, P.; Teo, T.; Basnet, S.K.; Noll, B.; Rahaman, M.H.; Lu, J.; Hou, J.; Yu, M. Dual inhibition of mnk2 and flt3 for potential treatment of acute myeloid leukaemia. Eur. J. Med. Chem. 2017, 139, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.H.; Yu, Y.; Zhong, L.; Adams, J.; Lam, F.; Li, P.; Noll, B.; Milne, R.; Peng, J.; Wang, S. Cdki-73: An orally bioavailable and highly efficacious cdk9 inhibitor against acute myeloid leukemia. Investig. New Drugs 2019, 37, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Anshabo, A.T.; Bantie, L.; Diab, S.; Lenjisa, J.; Kebede, A.; Long, Y.; Heinemann, G.; Karanjia, J.; Noll, B.; Basnet, S.K. An orally bioavailable and highly efficacious inhibitor of cdk9/flt3 for the treatment of acute myeloid leukemia. Cancers 2022, 14, 1113. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.A.; Yourish, H.B.; Bunaciu, R.P.; Varner, J.D.; Yen, A. Induced myelomonocytic differentiation in leukemia cells is accompanied by noncanonical transcription factor expression. FEBS Open Biol. 2015, 5, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora a to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. Plk1 activation in late g2 sets up commitment to mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of cdc25c in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Shimomura, T.; Hasako, S.; Nakatsuru, Y.; Mita, T.; Ichikawa, K.; Kodera, T.; Sakai, T.; Nambu, T.; Miyamoto, M.; Takahashi, I. Mk-5108, a highly selective aurora-a kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol. Cancer Ther. 2010, 9, 157–166. [Google Scholar] [CrossRef]
- Kunitoku, N.; Sasayama, T.; Marumoto, T.; Zhang, D.; Honda, S.; Kobayashi, O.; Hatakeyama, K.; Ushio, Y.; Saya, H.; Hirota, T. Cenp-a phosphorylation by aurora-a in prophase is required for enrichment of aurora-b at inner centromeres and for kinetochore function. Dev. Cell 2003, 5, 853–864. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Wang, S.; Midgley, C.A.; Scaërou, F.; Grabarek, J.B.; Griffiths, G.; Jackson, W.; Kontopidis, G.; McClue, S.J.; McInnes, C.; Meades, C. Discovery of n-phenyl-4-(thiazol-5-yl) pyrimidin-2-amine aurora kinase inhibitors. J. Med. Chem. 2010, 53, 4367–4378. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Goldenson, B.; Silver, S.J.; Schenone, M.; Dancik, V.; Huang, Z.; Wang, L.-Z.; Lewis, T.A.; An, W.F.; Li, X. Identification of regulators of polyploidization presents therapeutic targets for treatment of amkl. Cell 2012, 150, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A. Azd1208, a potent and selective pan-pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2014, 123, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Xiong, Y.; Song, J.H.; Mahajan, S.; DuPont, R.; McEachern, K.; DeAngelo, D.J.; Cortes, J.E.; Minden, M.D.; Ebens, A. The pim-1 protein kinase is an important regulator of met receptor tyrosine kinase levels and signaling. Mol. Cell. Biol. 2014, 34, 2517–2532. [Google Scholar] [CrossRef]
- Alsubaie, M.; Matou-Nasri, S.; Aljedai, A.; Alaskar, A.; Al-Eidi, H.; Albabtain, S.A.; Aldilaijan, K.E.; Alsayegh, M.; Alabdulkareem, I.B. In vitro assessment of the efficiency of the pim-1 kinase pharmacological inhibitor as a potential treatment for burkitt’s lymphoma. Oncol. Lett. 2021, 22, 622. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, G.M.; Knegtel, R.M.; Coll, J.T.; Renwick, S.B.; Swenson, L.; Weber, P.; Lippke, J.A.; Austen, D.A. Crystal structure of aurora-2, an oncogenic serine/threonine kinase. J. Biol. Chem. 2002, 277, 42419–42422. [Google Scholar] [CrossRef]
- Garmendia, I.; Redin, E.; Montuenga, L.M.; Calvo, A. Yes1: A novel therapeutic target and biomarker in cancer. Mol. Cancer Ther. 2022, 21, 1371–1380. [Google Scholar] [CrossRef]
- Dos Santos, C.; Demur, C.; Bardet, V.; Prade-Houdellier, N.; Payrastre, B.; Récher, C. A critical role for lyn in acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2008, 111, 2269–2279. [Google Scholar] [CrossRef]
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of myeloid src-family kinases is associated with poor prognosis in aml and influences flt3-itd kinase inhibitor acquired resistance. PLoS ONE 2019, 14, e0225887. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F.; Zhou, Z.-W.; Xia, H.-C.; Wang, X.-Y.; Yang, Y.-X.; He, Z.-X.; Sun, T.; Zhou, S.-F. Alisertib induces g2/m arrest, apoptosis, and autophagy via pi3k/akt/mtor-and p38 mapk-mediated pathways in human glioblastoma cells. Am. J. Transl. Res. 2017, 9, 845. [Google Scholar] [PubMed]
- Jackman, M.; Lindon, C.; Nigg, E.A.; Pines, J. Active cyclin b1–cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 2003, 5, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P. Universal control mechanism regulating onset of m-phase. Nature 1990, 344, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Plk1 promotes nuclear translocation of human cdc25c during prophase. EMBO Rep. 2002, 3, 341–348. [Google Scholar] [CrossRef]
- Schmit, T.L.; Ahmad, N. Regulation of mitosis via mitotic kinases: New opportunities for cancer management. Mol. Cancer Ther. 2007, 6, 1920–1931. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; Tsao, T.; Nakakuma, H.; Andreeff, M. Concomitant inhibition of mdm2-p53 interaction and aurora kinases activates the p53-dependent postmitotic checkpoints and synergistically induces p53-mediated mitochondrial apoptosis along with reduced endoreduplication in acute myelogenous leukemia. Blood J. Am. Soc. Hematol. 2008, 112, 2886–2895. [Google Scholar] [CrossRef]
- Oussenko, I.A.; Holland, J.F.; Reddy, E.P.; Ohnuma, T. Effect of on 01910. Na, an anticancer mitotic inhibitor, on cell-cycle progression correlates with rangap1 hyperphosphorylation. Cancer Res. 2011, 71, 4968–4976. [Google Scholar] [CrossRef]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef]
- Oliver, F.J.; de la Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.C.; de Murcia, G.; Ménissier-de Murcia, J. Importance of poly (adp-ribose) polymerase and its cleavage in apoptosis lesson from an uncleavable mutant. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar] [CrossRef]
- Soldani, C.; Lazzè, M.C.; Bottone, M.G.; Tognon, G.; Biggiogera, M.; Pellicciari, C.E.; Scovassi, A.I. Poly (adp-ribose) polymerase cleavage during apoptosis: When and where? Exp. Cell Res. 2001, 269, 193–201. [Google Scholar] [CrossRef]
- Bavetsias, V.; Linardopoulos, S. Aurora kinase inhibitors: Current status and outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting aurka in cancer: Molecular mechanisms and opportunities for cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Hecht, M.; Erber, S.; Harrer, T.; Klinker, H.; Roth, T.; Parsch, H.; Fiebig, N.; Fietkau, R.; Distel, L.V. Efavirenz has the highest anti-proliferative effect of non-nucleoside reverse transcriptase inhibitors against pancreatic cancer cells. PLoS ONE 2015, 10, e0130277. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Malpicci, D.; Milite, C.; Castellano, S.; Sbardella, G.; Botti, G.; Ascierto, P.A.; Mancini, R.; Ciliberto, G. Reverse transcriptase inhibition potentiates target therapy in braf-mutant melanomas: Effects on cell proliferation, apoptosis, DNA-damage, ros induction and mitochondrial membrane depolarization. Cell Commun. Signal. 2020, 18, 150. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.; Lucariello, A.; Sellitto, C.; Agliata, I.; Carleo, M.A.; Sangiovanni, V.; Esposito, V.; Guerra, G.; Cobellis, L.; De Luca, A. Different cell cycle modulation in skov-3 ovarian cancer cell line by anti-hiv drugs. Oncol. Res. 2017, 25, 1617. [Google Scholar] [CrossRef]
- Chow, W.A.; Jiang, C.; Guan, M. Anti-hiv drugs for cancer therapeutics: Back to the future? Lancet Oncol. 2009, 10, 61–71. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T. Aids-related kaposi’s sarcoma: Current treatment options, future trends. Oncology 2000, 14, 867–878, discussion 878, 881. [Google Scholar] [PubMed]
- Xiao, B.; Si, H.; Cervini, A.; Gao, J.; Lu, J.; Upadhyay, S.; Verma, S.; Robertson, E. Kaposi’s sarcoma herpesvirus upregulates aurora a expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. 2012, 8, e1002566. [Google Scholar]
- Zhu, Q.; Ding, L.; Zi, Z.; Gao, S.; Wang, C.; Wang, Y.; Zhu, C.; Yuan, Z.; Wei, F.; Cai, Q. Viral-mediated aurkb cleavage promotes cell segregation and tumorigenesis. Cell Rep. 2019, 26, 3657–3671.e5. [Google Scholar] [CrossRef]
- Johnson, J.R.; Crosby, D.C.; Hultquist, J.F.; Kurland, A.P.; Adhikary, P.; Li, D.; Marlett, J.; Swann, J.; Hüttenhain, R.; Verschueren, E. Global post-translational modification profiling of hiv-1-infected cells reveals mechanisms of host cellular pathway remodeling. Cell Rep. 2022, 39, 110690. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology—Patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Carpinelli, P.; Moll, J. Aurora kinase inhibitors: Identification and preclinical validation of their biomarkers. Expert Opin. Ther. Targets 2008, 12, 69–80. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, S.; Rahaman, M.H.; Yu, M.; Noll, B.; Martin, J.H.; Wang, S.; Head, R. Anti-Leukaemic Activity of Rilpivirine Is Mediated by Aurora A Kinase Inhibition. Cancers 2023, 15, 1044. https://doi.org/10.3390/cancers15041044
Islam S, Rahaman MH, Yu M, Noll B, Martin JH, Wang S, Head R. Anti-Leukaemic Activity of Rilpivirine Is Mediated by Aurora A Kinase Inhibition. Cancers. 2023; 15(4):1044. https://doi.org/10.3390/cancers15041044
Chicago/Turabian StyleIslam, Saiful, Muhammed H. Rahaman, Mingfeng Yu, Benjamin Noll, Jennifer H. Martin, Shudong Wang, and Richard Head. 2023. "Anti-Leukaemic Activity of Rilpivirine Is Mediated by Aurora A Kinase Inhibition" Cancers 15, no. 4: 1044. https://doi.org/10.3390/cancers15041044
APA StyleIslam, S., Rahaman, M. H., Yu, M., Noll, B., Martin, J. H., Wang, S., & Head, R. (2023). Anti-Leukaemic Activity of Rilpivirine Is Mediated by Aurora A Kinase Inhibition. Cancers, 15(4), 1044. https://doi.org/10.3390/cancers15041044