
Systematic Review on CAR-T Cell Clinical Trials Up to 2022: Academic Center Input

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Study Selection
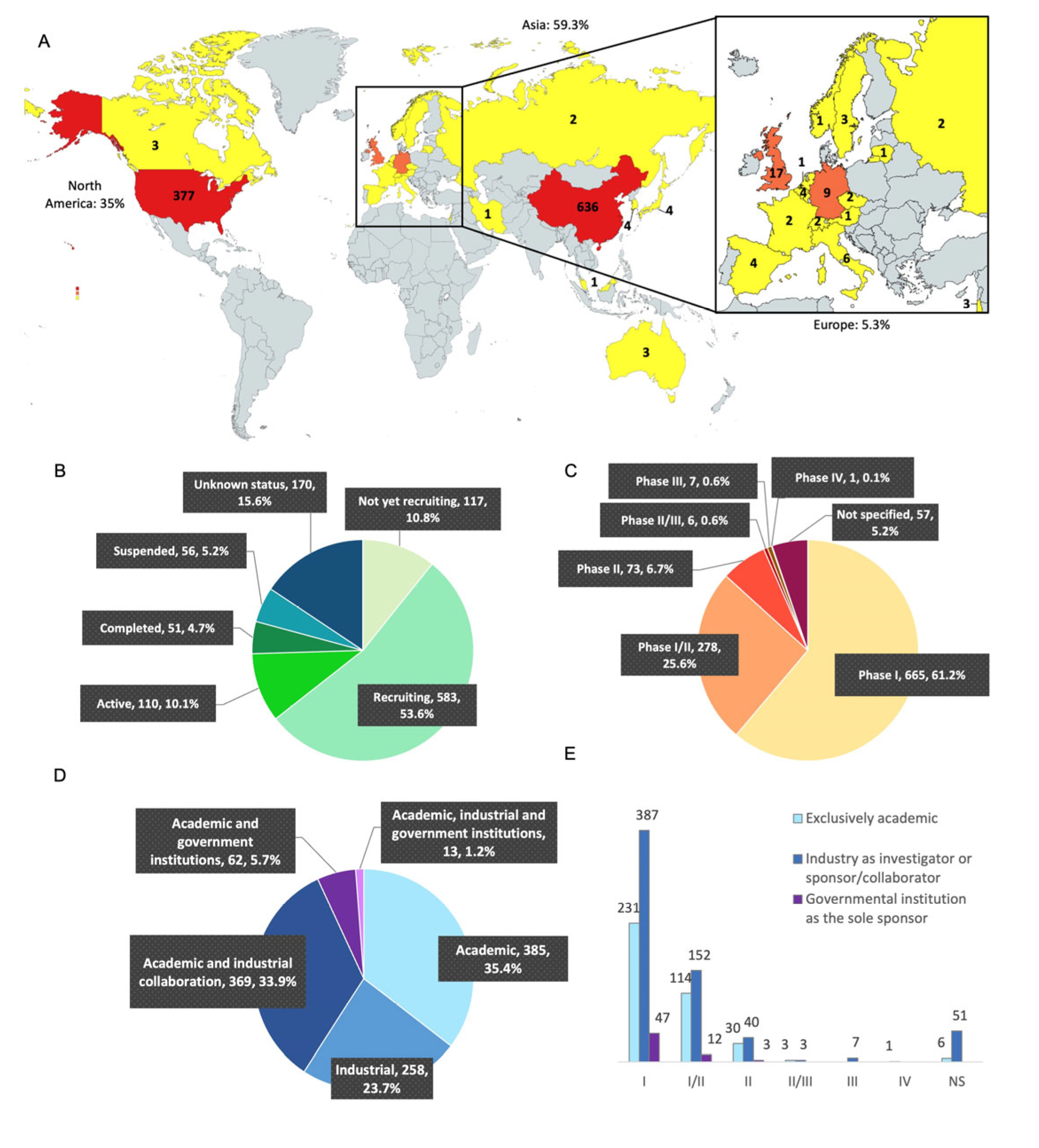
3.2. Geographic Situation, Clinical Trial Status, and Phases of the Clinical Trials
3.3. Analysis of the Clinical Trials According to Their Academic or Pharmaceutical Company Sponsorship
3.4. CAR-T Cell Clinical Trials in Europe Up to 2022
3.5. Academic Production of CAR-T Cells in European Clinical Trials
3.6. Two Different Examples of Academic Production in Europe
4. Discussion
4.1. Regulatory Differences
4.2. Financial Support
4.3. Academic Center for Autologous Production
4.4. Allogeneic CAR-T Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and Development of Therapies using Chimeric Antigen Receptor-Expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Haynes, N.M.; Trapani, J.A.; Teng, M.W.L.; Jackson, J.T.; Cerruti, L.; Jane, S.M.; Kershaw, M.H.; Smyth, M.J.; Darcy, P.K. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 2002, 100, 3155–3163. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef]
- Iyer, R.K.; Bowles, P.A.; Kim, H.; Dulgar-Tulloch, A. Industrializing Autologous Adoptive Immunotherapies: Manufacturing Advances and Challenges. Front. Med. 2018, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Yakoub-Agha, I.; Ferrand, C.; Chalandon, Y.; Ballot, C.; Castilla Llorente, C.; Deschamps, M.; Gauthier, J.; Labalette, M.; Larghero, J.; Maheux, C.; et al. Prérequis nécessaires pour la mise en place de protocoles de recherche clinique évaluant des thérapies cellulaires et géniques par lymphocytes T dotés de récepteur chimérique à l’antigène (CAR T-cells): Recommandations de la Société francophone de greffe de moelle et de thérapie cellulaire (SFGM-TC). Bull. Cancer 2017, 104, S43–S58. [Google Scholar]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells—Challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [PubMed]
- Blache, U.; Popp, G.; Dünkel, A.; Koehl, U.; Fricke, S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat. Commun. 2022, 13, 5225. [Google Scholar] [CrossRef]
- Abou-El-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable Manufacturing of CAR T cells for Cancer Immunotherapy. Blood Cancer Discov. 2021, 2, 408–422. [Google Scholar] [CrossRef]
- Mock, U.; Nickolay, L.; Philip, B.; Cheung, G.W.K.; Zhan, H.; Johnston, I.C.D.; Kaiser, A.D.; Peggs, K.; Pule, M.; Thrasher, A.J.; et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy 2016, 18, 1002–1011. [Google Scholar] [CrossRef]
- Vucinic, V.; Quaiser, A.; Lückemeier, P.; Fricke, S.; Platzbecker, U.; Koehl, U. Production and Application of CAR T Cells: Current and Future Role of Europe. Front. Med. 2021, 8, 713401. [Google Scholar] [CrossRef]
- Hort, S.; Herbst, L.; Bäckel, N.; Erkens, F.; Niessing, B.; Frye, M.; König, N.; Papantoniou, I.; Hudecek, M.; Jacobs, J.J.L.; et al. Toward Rapid, Widely Available Autologous CAR-T Cell Therapy—Artificial Intelligence and Automation Enabling the Smart Manufacturing Hospital. Front. Med. 2022, 9, 913–987. [Google Scholar]
- Castella, M.; Boronat, A.; Martín-Ibáñez, R.; Rodríguez, V.; Suñé, G.; Caballero, M.; Marzal, B.; Pérez-Amill, L.; Martín-Antonio, B.; Castaño, J.; et al. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol. Ther. Methods Clin. Dev. 2019, 12, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Bielorai, B.; Avigdor, A.; Itzhaki, O.; Hutt, D.; Nussboim, V.; Meir, A.; Kubi, A.; Levy, M.; Zikich, D.; et al. Locally produced CD19 CAR T cells leading to clinical remissions in medullary and extramedullary relapsed acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 1485–1492. [Google Scholar] [CrossRef]
- Itzhaki, O.; Jacoby, E.; Nissani, A.; Levi, M.; Nagler, A.; Kubi, A.; Brezinger, K.; Brayer, H.; Zeltzer, L.A.; Rozenbaum, M.; et al. Head-to-head comparison of in-house produced CD19 CAR-T cell in ALL and NHL patients. J. Immunother Cancer 2020, 8, e000148. [Google Scholar] [CrossRef]
- Ortíz-Maldonado, V.; Rives, S.; Castellà, M.; Alonso-Saladrigues, A.; Benítez-Ribas, D.; Caballero-Baños, M.; Baumann, T.; Cid, J.; Garcia-Rey, E.; Llanos, C.; et al. CART19-BE-01: A Multicenter Trial of ARI-0001 Cell Therapy in Patients with CD19+ Relapsed/Refractory Malignancies. Mol. Ther. 2021, 29, 636–644. [Google Scholar] [CrossRef]
- Ran, T.; Eichmüller, S.B.; Schmidt, P.; Schlander, M. Cost of decentralized CAR T-cell production in an academic nonprofit setting. Int. J. Cancer 2020, 147, 3438–3445. [Google Scholar] [CrossRef]
- Iglesias-Lopez, C.; Obach, M.; Vallano, A.; Agustí, A. Comparison of regulatory pathways for the approval of advanced therapies in the European Union and the United States. Cytotherapy 2021, 23, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Gao, J.; Li, G.; Hu, H.; Zhou, L.; Lu, S.; Chen, X. Gene and cell therapies in China: Booming landscape under dual-track regulation. J. Hematol. Oncol. 2022, 15, 139. [Google Scholar] [CrossRef] [PubMed]
- Pizevska, M.; Kaeda, J.; Fritsche, E.; Elazaly, H.; Reinke, P.; Amini, L. Advanced Therapy Medicinal Products’ Translation in Europe: A Developers’ Perspective. Front. Med. 2022, 9, 757647. [Google Scholar] [CrossRef]
- How China Is Making Progress in Cell and Gene Therapy EY—Global. Available online: https://www.ey.com/en_gl/life-sciences/how-china-is-making-progress-in-cell-and-gene-therapy (accessed on 10 December 2022).
- Braendstrup, P.; Levine, B.L.; Ruella, M. The long road to the first FDA-approved gene therapy: Chimeric antigen receptor T cells targeting CD19. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A first-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther. 2021, 28, 560–571. [Google Scholar] [CrossRef]
- SLAMF7-CAR T Cell Treatment of Multiple Myeloma Patients. Available online: https://www.caramba-cart.eu/ (accessed on 10 December 2022).
- Singh, H.; Moyes, J.S.E.; Huls, M.H.; Cooper, L.J.N. Manufacture of T cells using the Sleeping Beauty system to enforce expression of a CD19-specific chimeric antigen receptor. Cancer Gene Ther. 2015, 22, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Magnani, C.F.; Tettamanti, S.; Alberti, G.; Pisani, I.; Biondi, A.; Serafini, M.; Gaipa, G. Transposon-Based CAR T Cells in Acute Leukemias: Where are We Going? Cells 2020, 9, 1337. [Google Scholar] [CrossRef]
- Gee, A.P. GMP CAR-T cell production. Best Pract. Res. Clin. Haematol. 2018, 31, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, M.; Decot, V.; Giverne, C.; Pinturaud, M.; Vaissié, A.; Parquet, N.; Olivero, S.; Anne-Claire, M.; Bay, J.O.; Yakoub-Agha, I.; et al. Prérequis pour une production académique des cellules CART conforme aux bonnes pratiques pharmaceutiques (BPF). Recommandations de la Société francophone de greffe de moelle et de thérapie cellulaire (SFGM-TC). Bull. Cancer 2020, 107, S85–S93. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- AdvaBio. Available online: https://www.advabio.com/ (accessed on 10 December 2022).
- Wang, L.; Gong, W.; Wang, S.; Neuber, B.; Sellner, L.; Schubert, M.L.; Hückelhoven-Krauss, A.; Kunz, A.; Gern, U.; Michels, B.; et al. Improvement of in vitro potency assays by a resting step for clinical-grade chimeric antigen receptor engineered T cells. Cytotherapy 2019, 21, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Costariol, E.; Rotondi, M.C.; Amini, A.; Hewitt, C.J.; Nienow, A.W.; Heathman, T.R.J.; Rafiq, Q.A. Demonstrating the Manufacture of Human CAR-T Cells in an Automated Stirred-Tank Bioreactor. Biotechnol. J. 2020, 15, e2000177. [Google Scholar] [CrossRef]
- Mizukami, A.; Swiech, K. Platforms for Clinical-Grade CAR-T Cell Expansion. Methods Mol. Biol. 2020, 2086, 139–150. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, V.; Gauthier, M.; Decot, V.; Reppel, L.; Bensoussan, D. Systematic Review on CAR-T Cell Clinical Trials Up to 2022: Academic Center Input. Cancers 2023, 15, 1003. https://doi.org/10.3390/cancers15041003
Wang V, Gauthier M, Decot V, Reppel L, Bensoussan D. Systematic Review on CAR-T Cell Clinical Trials Up to 2022: Academic Center Input. Cancers. 2023; 15(4):1003. https://doi.org/10.3390/cancers15041003
Chicago/Turabian StyleWang, Valentine, Mélanie Gauthier, Véronique Decot, Loïc Reppel, and Danièle Bensoussan. 2023. "Systematic Review on CAR-T Cell Clinical Trials Up to 2022: Academic Center Input" Cancers 15, no. 4: 1003. https://doi.org/10.3390/cancers15041003
APA StyleWang, V., Gauthier, M., Decot, V., Reppel, L., & Bensoussan, D. (2023). Systematic Review on CAR-T Cell Clinical Trials Up to 2022: Academic Center Input. Cancers, 15(4), 1003. https://doi.org/10.3390/cancers15041003