
PARP Inhibitors and Proteins Interacting with SLX4

{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
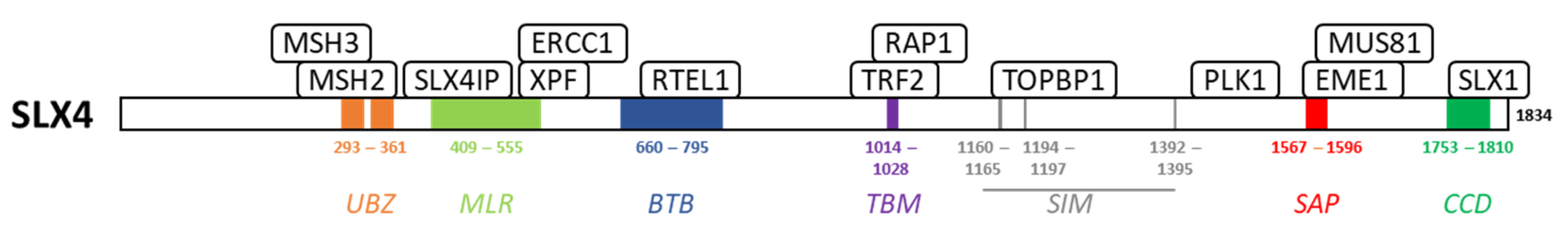
2. SLX4
3. MSH2/MSH3
4. ERCC1/XPF
5. TRF2/RAP1
6. PLK1/TOPBP1
7. MUS81/EME1
8. General Discussion and Perspectives
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Huang, D.; Kraus, W.L. The expanding universe of PARP1-mediated molecular and therapeutic mechanisms. Mol. Cell 2022, 82, 2315–2334. [Google Scholar] [CrossRef]
- Wang, Z.; Jia, R.; Wang, L.; Yang, Q.; Hu, X.; Fu, Q.; Zhang, X.; Li, W.; Ren, Y. The Emerging Roles of Rad51 in Cancer and Its Potential as a Therapeutic Target. Front. Oncol. 2022, 12, 935593. [Google Scholar] [CrossRef]
- Zhang, X.; Huo, X.; Guo, H.; Xue, L. Combined inhibition of PARP and EZH2 for cancer treatment: Current status, opportunities, and challenges. Front. Pharmacol. 2022, 13, 965244. [Google Scholar] [CrossRef]
- Guervilly, J.H.; Gaillard, P.H. SLX4: Multitasking to maintain genome stability. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 475–514. [Google Scholar] [CrossRef]
- Young, S.J.; West, S.C. Coordinated roles of SLX4 and MutSβ in DNA repair and the maintenance of genome stability. Crit. Rev. Biochem. Mol. Biol. 2021, 53, 157–177. [Google Scholar]
- Payliss, B.J.; Patel, A.; Sheppard, A.C.; Wyatt, H.D.M. Exploring the Structures and Functions of Macromolecular SLX4-Nuclease Complexes in Genome Stability. Front. Genet. 2021, 12, 784167. [Google Scholar] [CrossRef]
- Gonzalez-Prieto, R.; Cuijpers, S.A.; Luijsterburg, M.S.; van Attikum, H.; Vertegaal, A.C. SUMOylation and PARylation cooperate to recruit and stabilize SLX4 at DNA damage sites. EMBO Rep. 2015, 16, 512–519. [Google Scholar] [CrossRef]
- Kim, Y.; Spitz, G.S.; Veturi, U.; Lach, F.P.; Auerbach, A.D.; Smogorzewska, A. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 2013, 121, 54–63. [Google Scholar] [CrossRef]
- Liu, Q.; Underwood, T.S.A.; Kung, J.; Wang, M.; Lu, H.M.; Paganetti, H.; Held, K.D.; Hong, T.S.; Efstathiou, J.A.; Willers, H. Disruption of SLX4-MUS81 Function Increases the Relative Biological Effectiveness of Proton Radiation. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 78–85. [Google Scholar] [CrossRef]
- Shah, S.; Kim, Y.; Ostrovnaya, I.; Murali, R.; Schrader, K.A.; Lach, F.P.; Sarrel, K.; Rau-Murthy, R.; Hansen, N.; Zhang, L.; et al. Assessment of SLX4 Mutations in Hereditary Breast Cancers. PLoS ONE 2013, 8, e66961. [Google Scholar] [CrossRef]
- Guervilly, J.H.; Blin, M.; Laureti, L.; Baudelet, E.; Audebert, S.; Gaillard, P.H. SLX4 dampens MutSα-dependent mismatch repair. Nucleic Acids Res. 2022, 50, 2667–2680. [Google Scholar] [CrossRef]
- Elango, R.; Panday, A.; Lach, F.P.; Willis, N.A.; Nicholson, K.; Duffey, E.E.; Smogorzewska, A.; Scully, R. The structure-specific endonuclease complex SLX4-XPF regulates Tus-Ter-induced homologous recombination. Nat. Struct. Mol. Biol. 2022, 29, 801–812. [Google Scholar] [CrossRef]
- Takahashi, M.; Koi, M.; Balaguer, F.; Boland, C.R.; Goel, A. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J. Biol. Chem. 2011, 286, 12157–12165. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, W.; Cheng, C.T.; Ren, X.; Somlo, G.; Fong, M.Y.; Chin, A.R.; Li, H.; Yu, Y.; Xu, Y.; et al. TGFbeta induces “BRCAness” and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Mol. Cancer. Res. 2014, 12, 1597–1609. [Google Scholar] [CrossRef]
- Rahman, M.M.; Mohiuddin, M.; Shamima Keka, I.; Yamada, K.; Tsuda, M.; Sasanuma, H.; Andreani, J.; Guerois, R.; Borde, V.; Charbonnier, J.B.; et al. Genetic evidence for the involvement of mismatch repair proteins, PMS2 and MLH3, in a late step of homologous recombination. J. Biol. Chem. 2020, 295, 17460–17475. [Google Scholar] [CrossRef]
- Clark, C.C.; Weitzel, J.N.; O’Connor, T.R. Enhancement of synthetic lethality via combinations of ABT-888, a PARP inhibitor, and carboplatin in vitro and in vivo using BRCA1 and BRCA2 isogenic models. Mol. Cancer. Ther. 2012, 11, 1948–1958. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, Z.; Borczuk, A.; Powell, C.A.; Balajee, A.S.; Lieberman, H.B.; Halmos, B. PARP inhibition selectively increases sensitivity to cisplatin in ERCC1-low non-small cell lung cancer cells. Carcinogenesis 2013, 34, 739–749. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Bajrami, I.; Friboulet, L.; Elliott, R.; Fontebasso, Y.; Dorvault, N.; Olaussen, K.A.; Andre, F.; Soria, J.C.; Lord, C.J.; et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene 2013, 32, 5377–5387. [Google Scholar] [CrossRef] [PubMed]
- Boerner, J.L.; Nechiporchik, N.; Mueller, K.L.; Polin, L.; Heilbrun, L.; Boerner, S.A.; Zoratti, G.L.; Stark, K.; LoRusso, P.M.; Burger, A. Protein expression of DNA damage repair proteins dictates response to topoisomerase and PARP inhibitors in triple-negative breast cancer. PLoS ONE 2015, 10, e0119614. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Henon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Soria, J.C.; Lord, C.J.; Postel-Vinay, S. Beyond DNA repair: The novel immunological potential of PARP inhibitors. Mol. Cell Oncol. 2019, 6, 1585170. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Kim, J.W.; Min, A.; Im, S.A.; Jang, H.; Kim, Y.J.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Lee, K.W.; Oh, D.Y.; et al. TDP1 and TOP1 Modulation in Olaparib-Resistant Cancer Determines the Efficacy of Subsequent Chemotherapy. Cancers 2020, 12, 334. [Google Scholar] [CrossRef]
- Xie, K.; Ni, X.; Lv, S.; Zhou, G.; He, H. Synergistic effects of olaparib combined with ERCC1 on the sensitivity of cisplatin in non-small cell lung cancer. Oncol. Lett. 2021, 21, 365. [Google Scholar] [CrossRef]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef]
- Mesquita, K.A.; Alabdullah, M.; Griffin, M.; Toss, M.S.; Fatah, T.; Alblihy, A.; Moseley, P.; Chan, S.Y.T.; Rakha, E.A.; Madhusudan, S. ERCC1-XPF deficiency is a predictor of olaparib induced synthetic lethality and platinum sensitivity in epithelial ovarian cancers. Gynecol. Oncol. 2019, 153, 416–424. [Google Scholar] [CrossRef]
- Rajkumar-Calkins, A.S.; Szalat, R.; Dreze, M.; Khan, I.; Frazier, Z.; Reznichenkov, E.; Schnorenberg, M.R.; Tsai, Y.F.; Nguyen, H.; Kochupurakkal, B.; et al. Functional profiling of nucleotide Excision repair in breast cancer. DNA Repair 2019, 82, 102697. [Google Scholar] [CrossRef]
- Kong, X.; Cruz, G.M.S.; Trinh, S.L.; Zhu, X.D.; Berns, M.W.; Yokomori, K. Biphasic recruitment of TRF2 to DNA damage sites promotes non-sister chromatid homologous recombination repair. J. Cell. Sci. 2018, 131, jcs219311. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Shi, R.; Bian, J.; Li, Y.; Wang, P.; Wang, H.; Liao, J.; Zhu, W.G.; Xu, X. PARP1 and CHK1 coordinate PLK1 enzymatic activity during the DNA damage response to promote homologous recombination-mediated repair. Nucleic Acids Res. 2021, 49, 7554–7570. [Google Scholar] [CrossRef] [PubMed]
- Sourisseau, T.; Maniotis, D.; McCarthy, A.; Tang, C.; Lord, C.J.; Ashworth, A.; Linardopoulos, S. Aurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibition. EMBO Mol. Med. 2010, 2, 130–142. [Google Scholar] [CrossRef]
- Moudry, P.; Watanabe, K.; Wolanin, K.M.; Bartkova, J.; Wassing, I.E.; Watanabe, S.; Strauss, R.; Troelsgaard Pedersen, R.; Oestergaard, V.H.; Lisby, M.; et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell. Biol. 2016, 212, 281–288. [Google Scholar] [CrossRef]
- Li, J.; Wang, R.; Kong, Y.; Broman, M.M.; Carlock, C.; Chen, L.; Li, Z.; Farah, E.; Ratliff, T.L.; Liu, X. Targeting Plk1 to Enhance Efficacy of Olaparib in Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2017, 16, 469–479. [Google Scholar] [CrossRef]
- Lin, X.; Chen, D.; Zhang, C.; Zhang, X.; Li, Z.; Dong, B.; Gao, J.; Shen, L. Augmented antitumor activity by olaparib plus AZD1775 in gastric cancer through disrupting DNA damage repair pathways and DNA damage checkpoint. J. Exp. Clin. Cancer Res. 2018, 37, 129. [Google Scholar] [CrossRef]
- Carbajosa, S.; Pansa, M.F.; Paviolo, N.S.; Castellaro, A.M.; Andino, D.L.; Nigra, A.D.; Garcia, I.A.; Racca, A.C.; Rodriguez-Berdini, L.; Angiolini, V.; et al. Polo-like Kinase 1 Inhibition as a Therapeutic Approach to Selectively Target BRCA1-Deficient Cancer Cells by Synthetic Lethality Induction. Clin. Cancer Res. 2019, 25, 4049–4062. [Google Scholar] [CrossRef]
- Gasimli, K.; Raab, M.; Tahmasbi Rad, M.; Kurunci-Csacsko, E.; Becker, S.; Strebhardt, K.; Sanhaji, M. Sequential Targeting of PLK1 and PARP1 Reverses the Resistance to PARP Inhibitors and Enhances Platin-Based Chemotherapy in BRCA-Deficient High-Grade Serous Ovarian Cancer with KRAS Amplification. Int. J. Mol. Sci. 2022, 23, 10892. [Google Scholar] [CrossRef]
- Bailey, L.J.; Teague, R.; Kolesar, P.; Bainbridge, L.J.; Lindsay, H.D.; Doherty, A.J. PLK1 regulates the PrimPol damage tolerance pathway during the cell cycle. Sci. Adv. 2021, 7, eabh1004. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Kong, Y.; Yan, S.; Ahmad, N.; Liu, X. Plk1 Phosphorylation of Mre11 Antagonizes the DNA Damage Response. Cancer Res. 2017, 77, 3169–3180. [Google Scholar] [CrossRef]
- Koppensteiner, R.; Samartzis, E.P.; Noske, A.; von Teichman, A.; Dedes, I.; Gwerder, M.; Imesch, P.; Ikenberg, K.; Moch, H.; Fink, D.; et al. Effect of MRE11 loss on PARP-inhibitor sensitivity in endometrial cancer in vitro. PLoS ONE 2014, 9, e100041. [Google Scholar] [CrossRef] [PubMed]
- Vequaud, E.; Desplanques, G.; Jezequel, P.; Juin, P.; Barille-Nion, S. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res. Treat. 2016, 155, 53–63. [Google Scholar] [CrossRef]
- Zhong, A.; Zhang, H.; Xie, S.; Deng, M.; Zheng, H.; Wang, Y.; Chen, M.; Lu, R.; Guo, L. Inhibition of MUS81 improves the chemical sensitivity of olaparib by regulating MCM2 in epithelial ovarian cancer. Oncol. Rep. 2018, 39, 1747–1756. [Google Scholar] [CrossRef]
- Lu, R.; Xie, S.; Wang, Y.; Zheng, H.; Zhang, H.; Deng, M.; Shi, W.; Zhong, A.; Chen, M.; Zhang, M.; et al. MUS81 Participates in the Progression of Serous Ovarian Cancer Associated with Dysfunctional DNA Repair System. Front. Oncol. 2019, 9, 1189. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, P.; Li, C.; Liu, W.; Shen, Q.; Yang, L.; Xie, G.; Bai, J.; Li, R.; Tao, K.; et al. MUS81 Inhibition Enhances the Anticancer Efficacy of Talazoparib by Impairing ATR/CHK1 Signaling Pathway in Gastric Cancer. Front. Oncol. 2022, 12, 844135. [Google Scholar] [CrossRef]
- Gong, L.; Tang, Y.; Jiang, L.; Tang, W.; Luo, S. Expression of MUS81 Mediates the Sensitivity of Castration-Resistant Prostate Cancer to Olaparib. J. Immunol. Res. 2022, 2022, 4065580. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Huang, T.T.; Burkett, S.S.; Tandon, M.; Yamamoto, T.M.; Gupta, N.; Bitler, B.G.; Lee, J.M.; Nair, J.R. Distinct roles of treatment schemes and BRCA2 on the restoration of homologous recombination DNA repair and PARP inhibitor resistance in ovarian cancer. Oncogene 2022, 41, 5020–5031. [Google Scholar] [CrossRef]
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.Q.; Ruse, C.; Yates, J.R., 3rd; Russell, P.; et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef]
- Munoz, I.M.; Hain, K.; Declais, A.C.; Gardiner, M.; Toh, G.W.; Sanchez-Pulido, L.; Heuckmann, J.M.; Toth, R.; Macartney, T.; Eppink, B.; et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell. 2009, 35, 116–127. [Google Scholar] [CrossRef]
- Svendsen, J.M.; Smogorzewska, A.; Sowa, M.E.; O’Connell, B.C.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009, 138, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, M.; Gaur, V. Structure and mechanism of nucleases regulated by SLX4. Curr. Opin. Struct. Biol. 2016, 36, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lach, F.P.; Desetty, R.; Hanenberg, H.; Auerbach, A.D.; Smogorzewska, A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011, 43, 142–146. [Google Scholar] [CrossRef]
- Stoepker, C.; Hain, K.; Schuster, B.; Hilhorst-Hofstee, Y.; Rooimans, M.A.; Steltenpool, J.; Oostra, A.B.; Eirich, K.; Korthof, E.T.; Nieuwint, A.W.; et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011, 43, 138–141. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordheim, L.P. PARP Inhibitors and Proteins Interacting with SLX4. Cancers 2023, 15, 997. https://doi.org/10.3390/cancers15030997
Jordheim LP. PARP Inhibitors and Proteins Interacting with SLX4. Cancers. 2023; 15(3):997. https://doi.org/10.3390/cancers15030997
Chicago/Turabian StyleJordheim, Lars Petter. 2023. "PARP Inhibitors and Proteins Interacting with SLX4" Cancers 15, no. 3: 997. https://doi.org/10.3390/cancers15030997
APA StyleJordheim, L. P. (2023). PARP Inhibitors and Proteins Interacting with SLX4. Cancers, 15(3), 997. https://doi.org/10.3390/cancers15030997