
CDK4/6 Inhibitors in Pancreatobiliary Cancers: Opportunities and Challenges

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
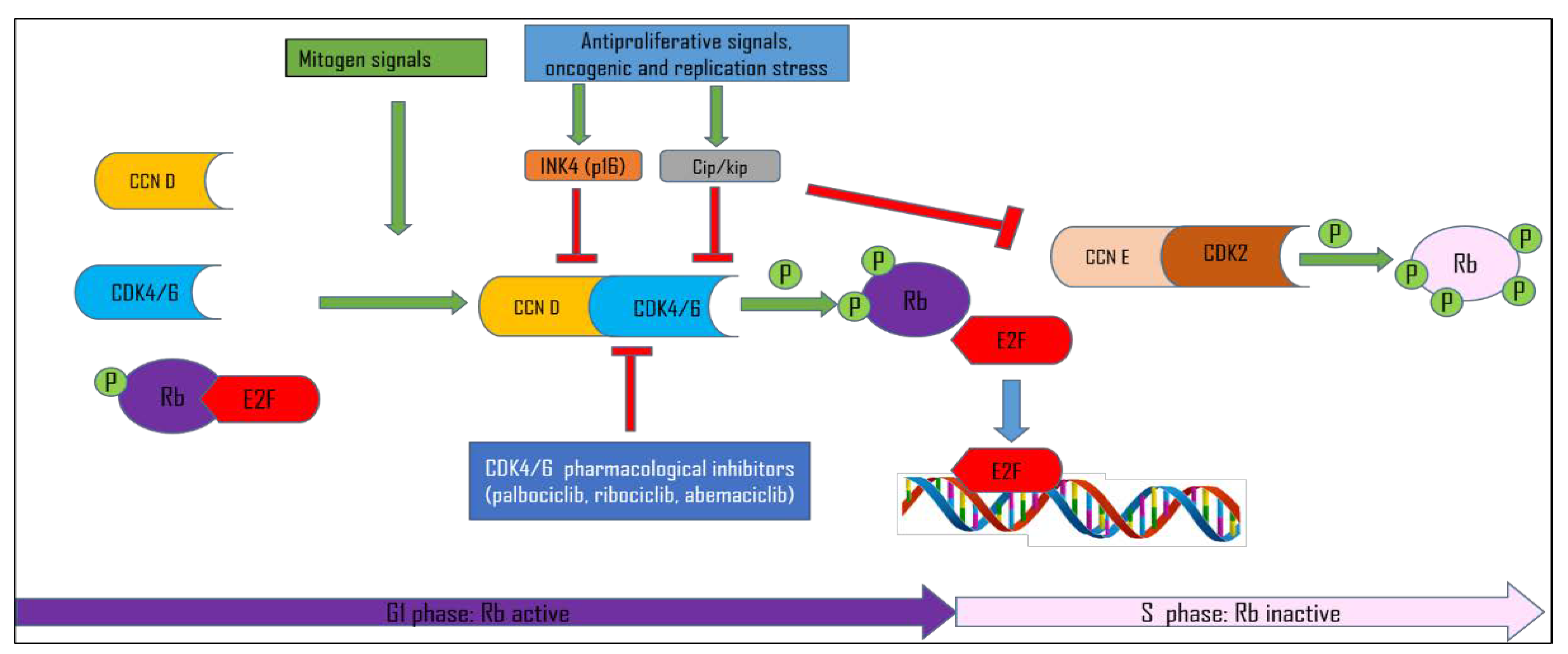
1.1. Cell Cycle Control
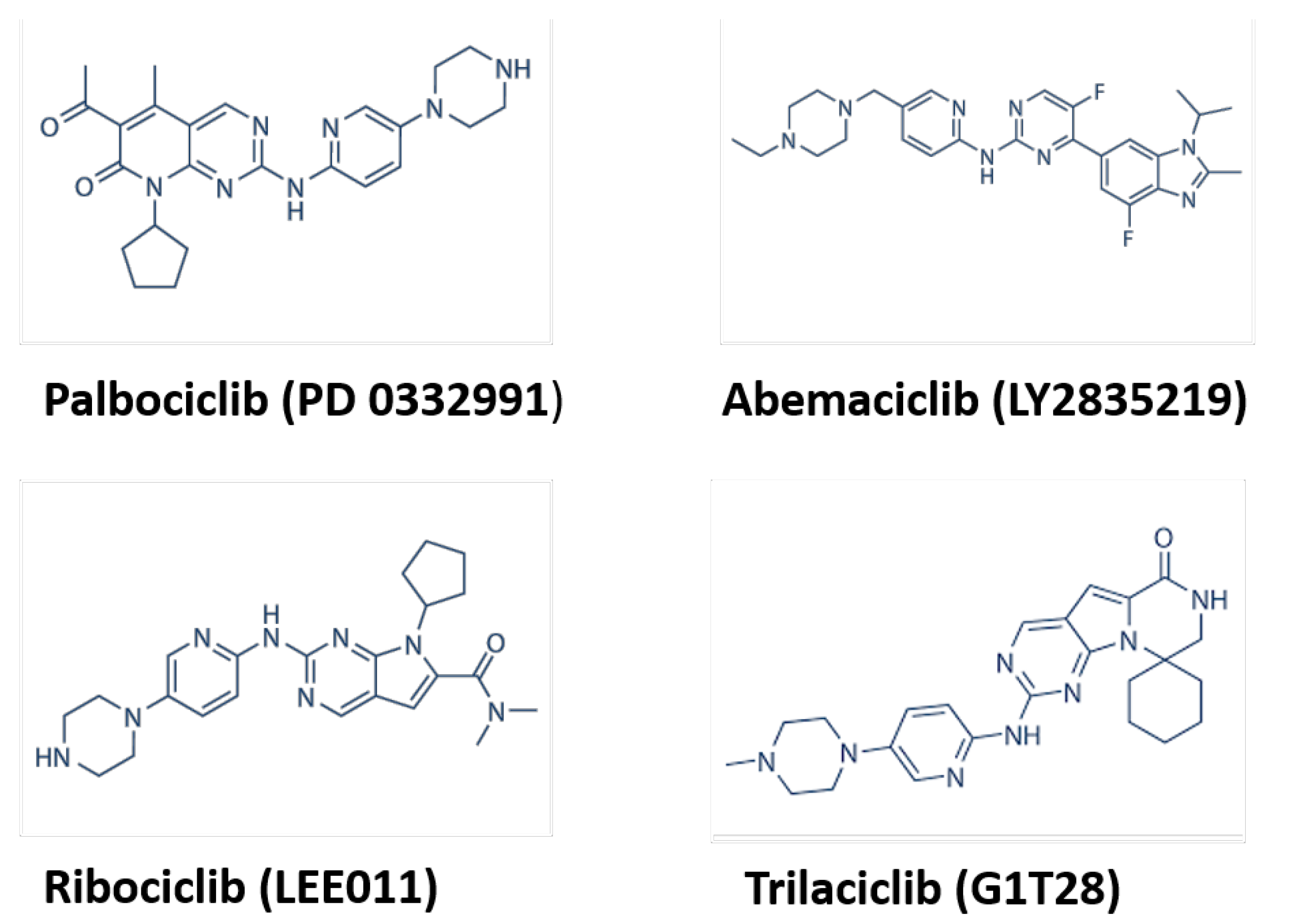
1.2. CDK Inhibitors
1.3. Pancreatobiliary Cancers
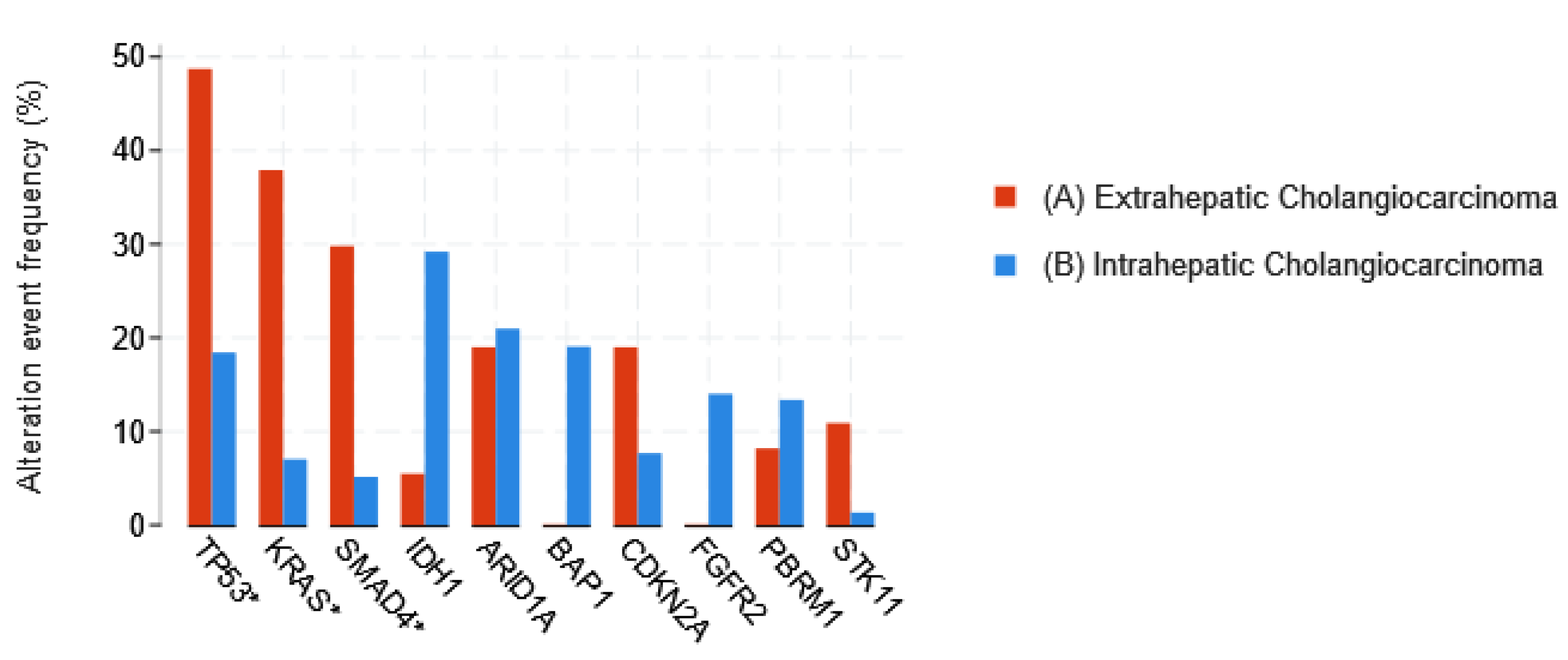
1.3.1. Cholangiocarcinoma
1.3.2. Pancreatic Ductal Adenocarcinoma
2. CDK4/6 Inhibitors in Preclinical Models of CCA and PDAC
2.1. CDK4/6 Inhibitor Monotherapy in Preclinical Models of CCA and PDAC
2.2. The Effects of CDK4/6 Inhibitors in Monotherapy: Quiescence, Senescence, or Resistance?
2.3. Combination Therapies to Overcome CDK4/6 Inhibitor Resistance
2.4. CDK4/6 Inhibitors in Combination with Chemotherapy
2.5. CDK4/6is Impact on Immunogenic Mechanisms
3. Challenges in the Clinical Use of CDK4/6is in Pancreatobiliary Cancers
4. Potential Biomarkers for Personalizing CDK4/6 Target Therapy
4.1. RB Protein
4.2. Cyclin D1
4.3. Cyclin E
4.4. Phosphorylation Status of CDK4
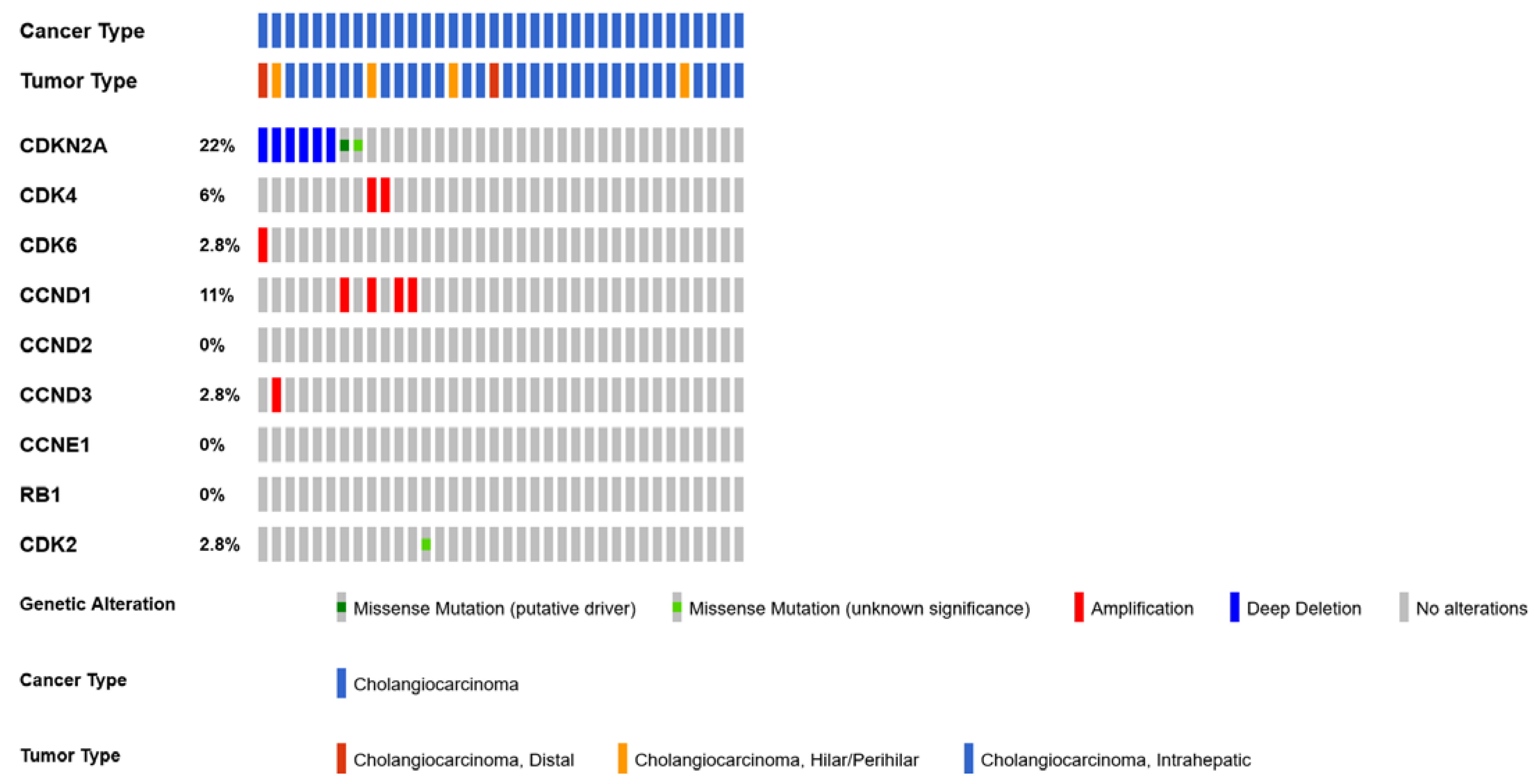
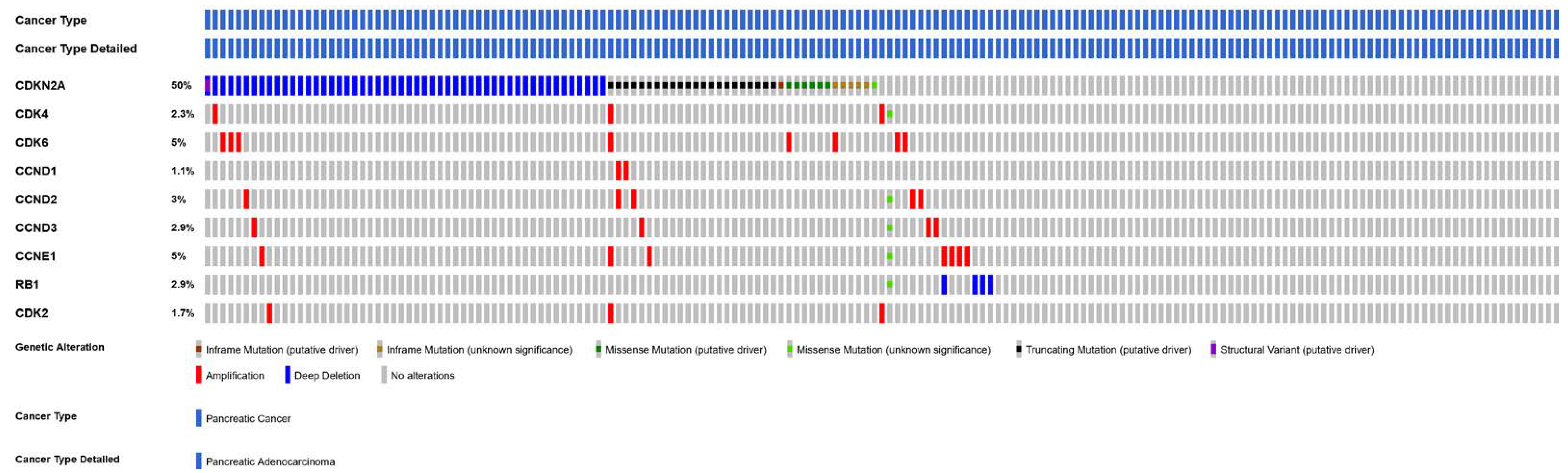
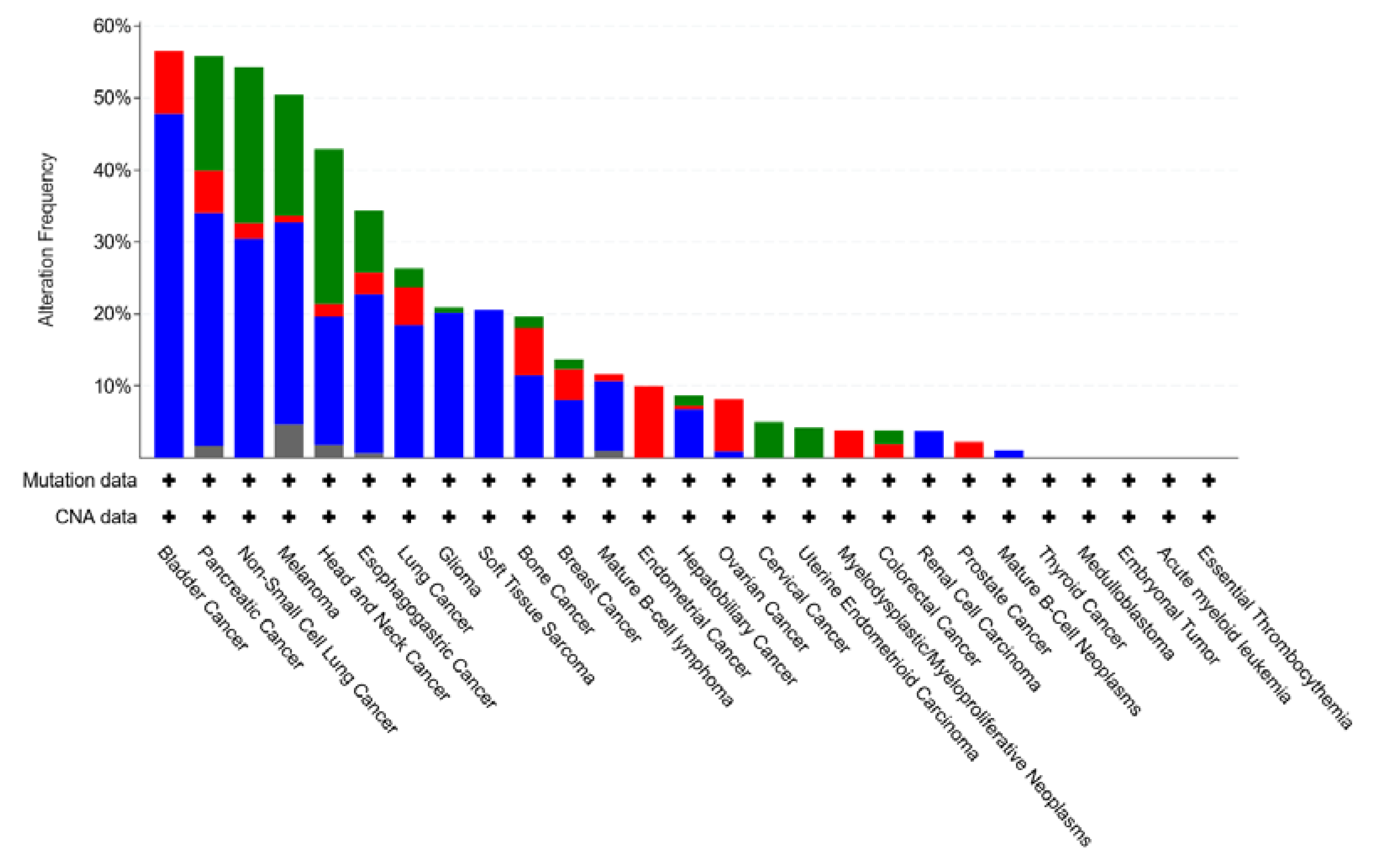
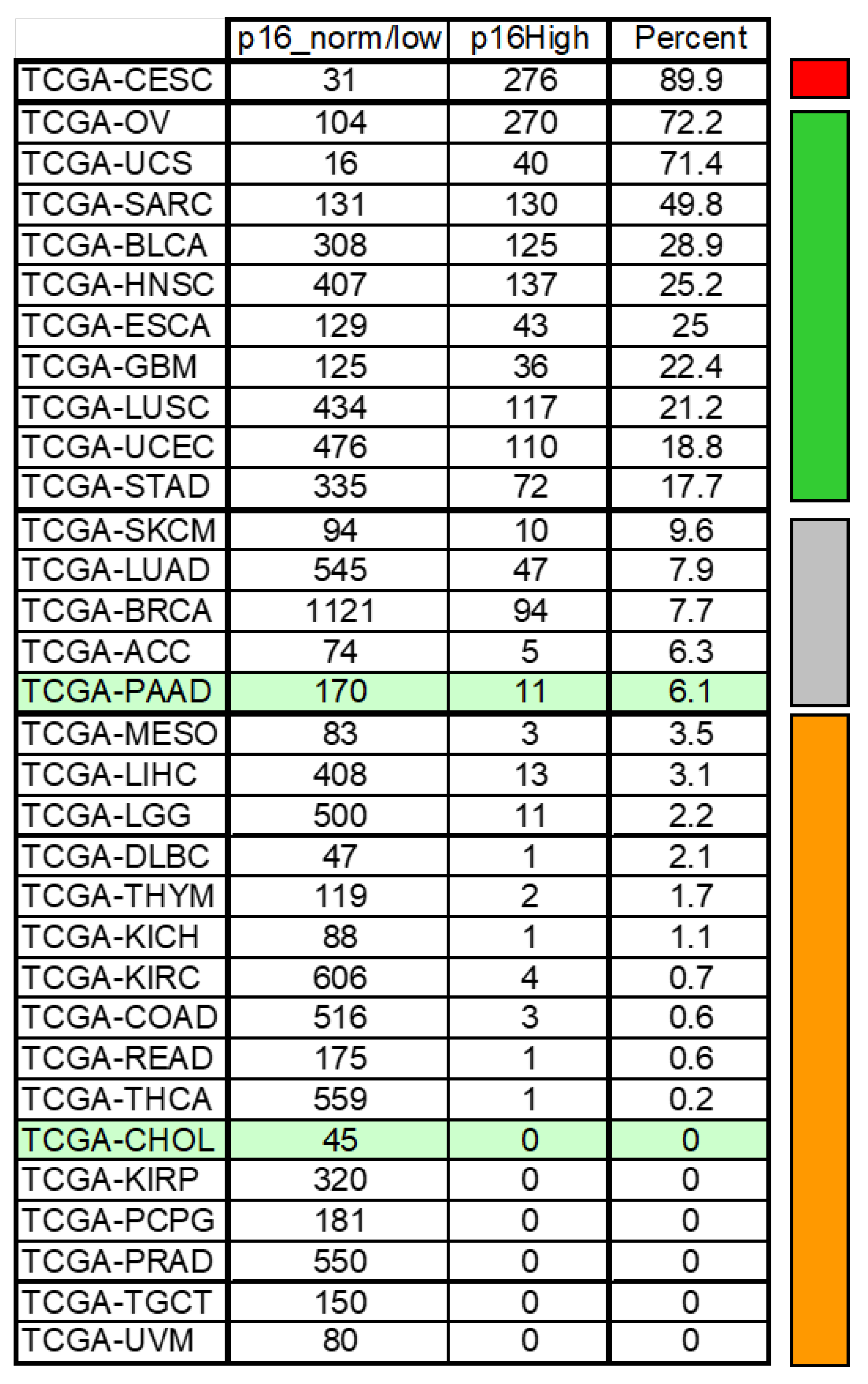
4.5. CDKN2A
4.6. CDK6 and CDK4 Gene Amplification
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Glossary
| AKT | protein kinase B |
| AP1 | activator protein 1 (transcription factor) |
| BrdU | 5-bromo-2-deoxyuridine |
| CCA | cholangiocarcinoma |
| CCND | cyclin D |
| CCNE1 | cyclin E1 |
| CDK2 | cyclin dependent kinase 2 |
| CDKN2A/B | cyclin-dependent kinase inhibitor 2A/B gene |
| CDK4/6 | cyclin-dependent kinase 4/6 |
| CDK4/6i | cyclin-dependent kinase 4/6 inhibitor |
| CDK7 | cyclin dependent kinase 7 |
| eCAA | extrahepatic cholangiocarcinoma |
| ECM | extracellular matrix |
| EdU | 5-ethynyl-2′-deoxyuridine |
| E2F | E2F transcription factor |
| FACS | Fluorescence-activated Cell Sorting |
| FAK | focal adhesion kinase |
| FFPE | formalin-fixed, paraffin-embedded |
| FOLFOX | chemotherapeutic regimen composed of folinic acid, fluorouracil, and oxaliplatin |
| FOLFIRINOX | chemotherapeutic regimen composed of irinotecan, oxaliplatin and leucovirin-modulated fluorouracil |
| HPV | Human papillomavirus |
| HuR | human antigen R |
| iCC | intrahepatic cholangiocarcinoma |
| IGF1 | insulin-like growth factor 1 |
| KRAS | kirsten rat sarcoma virus |
| mTOR | mammalian target of rapamycin |
| MEK | mitogen-activated protein kinase |
| MEKi | mitogen-activated protein kinase inhibitor |
| PDAC | pancreatic ductal adenocarcinoma |
| PDTX | patient-derived tumor xenographt |
| PD-1 | programmed cell death protein 1 |
| PI3K | phosphatidylinositol 3-kinase |
| p21 | cyclin-dependent kinase (Cdk) inhibitor p21 |
| p27 | cyclin-dependent kinase (Cdk) inhibitor p27 |
| PROTAC | proteolysis targeting chimera |
| RB | retinoblastoma protein |
| SASP | senescence-associated secretory phenotype |
| SMAD | suppressor of mothers against decapentaplegic |
| α-SMA | alpha-smooth muscle actin |
| TCGA | The Cancer Genome Atlas Program |
| TGF-β | transforming growth factor β |
| TβRI | type-I transforming growth factor-β receptor |
| YAP1 | yes1-associated transcriptional regulator |
References
- Sherr, C.J. D-type cyclins. Trends Biochem. Sci. 1995, 20, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Bockstaele, L.; Coulonval, K.; Kooken, H.; Paternot, S.; Roger, P.P. Regulation of CDK4. Cell Div. 2006, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Alonso, D.; Malumbres, M. Mammalian cell cycle cyclins. Semin. Cell Dev. Biol. 2020, 107, 28–35. [Google Scholar] [CrossRef]
- Kato, J.Y.; Matsuoka, M.; Strom, D.K.; Sherr, C.J. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol. Cell. Biol. 1994, 14, 2713–2721. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Bartkova, J.; Lukas, J. The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol. 1996, 8, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Bockstaele, L.; Kooken, H.; Libert, F.; Paternot, S.; Dumont, J.E.; De Launoit, Y.; Roger, P.P.; Coulonval, K. Regulated activating Thr172 phosphorylation of cyclin-dependent kinase 4 (CDK4): Its relationship with cyclins and CDK “inhibitors. ” Mol. Cell. Biol. 2006, 26, 5070–5085. [Google Scholar] [CrossRef] [PubMed]
- Paternot, S.; Bockstaele, L.; Bisteau, X.; Kooken, H.; Coulonval, K.; Roger, P. Rb inactivation in cell cycle and cancer: The puzzle of highly regulated activating phosphorylation of CDK4 versus constitutively active CDK-activating kinase. Cell Cycle 2010, 9, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Bisteau, X.; Paternot, S.; Colleoni, B.; Ecker, K.; Coulonval, K.; De Groote, P.; Declercq, W.; Hengst, L.; Roger, P.P. CDK4 T172 phosphorylation is central in a CDK7-dependent bidirectional CDK4/CDK2 interplay mediated by p21 phosphorylation at the restriction point. PLoS Genet. 2013, 9, e1003546. [Google Scholar] [CrossRef]
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 1998, 18, 753–761. [Google Scholar] [CrossRef]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Zerjatke, T.; Gak, I.A.; Kirova, D.; Fuhrmann, M.; Daniel, K.; Gonciarz, M.; Müller, D.; Glauche, I.; Mansfeld, J. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Rep. 2017, 19, 1953–1966. [Google Scholar] [CrossRef]
- Chung, M.; Liu, C.; Yang, H.W.; Köberlin, M.S.; Cappell, S.D.; Meyer, T. Transient hysteresis in CDK4/6 activity underlies passage of the restriction point in G1. Mol. Cell 2019, 76, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.; Miller, I.; Carter, D.; Spencer, S.L. Control of the Restriction Point by Rb and p21. Proc. Natl. Acad. Sci. USA 2018, 115, E8219–E8227. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Lynce, F.; Shajahan-Haq, A.N.; Swain, S.M. CDK4/6 inhibitors in breast cancer therapy: Current practice and future opportunities. Pharmacol. Ther. 2018, 191, 65–73. [Google Scholar] [CrossRef]
- Coulonval, K.; Bockstaele, L.; Paternot, S.; Dumont, J.E.; Roger, P.P. The cyclin D3-CDK4-p27kip1 holoenzyme in thyroid epithelial cells: Activation by TSH, inhibition by TGFβ, and phosphorylations of its subunits demonstrated by two-dimensional gel electrophoresis. Exp. Cell Res. 2003, 291, 135–149. [Google Scholar] [CrossRef]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef]
- Colleoni, B.; Paternot, S.; Pita, J.M.; Bisteau, X.; Coulonval, K.; Davis, R.J.; Raspé, E.; Roger, P.P. JNKs function as CDK4-activating kinases by phosphorylating CDK4 and p21. Oncogene 2017, 36, 4349–4361. [Google Scholar] [CrossRef] [PubMed]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; James, M.K.; Larochelle, S.; Fisher, R.P.; Blain, S.W. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol. Cell. Biol. 2009, 29, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Coulonval, K.; Vercruysse, V.; Paternot, S.; Pita, J.M.; Corman, R.; Raspé, E.; Roger, P.P. Monoclonal antibodies to activated CDK4: Use to investigate normal and cancerous cell cycle regulation and involvement of phosphorylations of p21 and p27. Cell Cycle 2022, 21, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Herrera, R.E.; Lam, F.; Weinberg, R.A. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc. Natl. Acad. Sci. USA 1995, 92, 6289–6293. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Wang, J.Y.J. Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. 2010, 16, 1094–1099. [Google Scholar] [CrossRef]
- Roskoski, R. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Losiewicz, M.D.; Carlson, B.A.; Kaur, G.; Sausville, E.A.; Worland, P.J. Potent inhibition of Cdc2 kinase activity by the flavonoid L86-8275. Biochem. Biophys. Res. Commun. 1994, 201, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.J.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.-G.; Moulinoux, J.-P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Jorda, R.; Hendrychová, D.; Voller, J.; Řezníčková, E.; Gucký, T.; Kryštof, V. How Selective Are Pharmacological Inhibitors of Cell-Cycle-Regulating Cyclin-Dependent Kinases? J. Med. Chem. 2018, 61, 9105–9120. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Siemeister, G.; Lücking, U.; Wengner, A.M.; Lienau, P.; Steinke, W.; Schatz, C.; Mumberg, D.; Ziegelbauer, K. BAY 1000394, a Novel Cyclin-Dependent Kinase Inhibitor, with Potent Antitumor Activity in Mono- and in Combination Treatment upon Oral Application. Mol. Cancer Ther. 2012, 11, 2265–2273. [Google Scholar] [CrossRef]
- Lücking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 2017, 12, 1776–1793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gan, Y.; Li, H.; Yin, J.; He, X.; Lin, L.; Xu, S.; Fang, Z.; Kim, B.; Gao, L.; et al. Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat. Commun. 2022, 13, 2835. [Google Scholar] [CrossRef]
- Olson, C.M.; Liang, Y.; Leggett, A.; Park, W.D.; Li, L.; Mills, C.E.; Elsarrag, S.Z.; Ficarro, S.B.; Zhang, T.; Düster, R.; et al. Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem. Biol. 2019, 26, 792–803.e10. [Google Scholar] [CrossRef]
- Bisi, J.E.; Sorrentino, J.A.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Mol. Cancer Ther. 2016, 15, 783–793. [Google Scholar] [CrossRef]
- Ferrarotto, R.; Anderson, I.; Medgyasszay, B.; García-Campelo, M.R.; Edenfield, W.; Feinstein, T.M.; Johnson, J.M.; Kalmadi, S.; Lammers, P.E.; Sanchez-Hernandez, A.; et al. Trilaciclib prior to chemotherapy reduces the usage of supportive care interventions for chemotherapy-induced myelosuppression in patients with small cell lung cancer: Pooled analysis of three randomized phase 2 trials. Cancer Med. 2021, 10, 5748–5756. [Google Scholar] [CrossRef]
- O’leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Cousins, E.M.; Goldfarb, D.; Yan, F.; Roques, J.; Darr, D.; Johnson, G.L.; Major, M.B. Competitive Kinase Enrichment Proteomics Reveals that Abemaciclib Inhibits GSK3β and Activates WNT Signaling. Mol. Cancer Res. 2018, 16, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.I.; Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Ingold, M.R.; Pickett, J.E.; Drewry, D.H.; Pugh, K.M.; Schwinn, M.K.; et al. Quantifying CDK inhibitor selectivity in live cells. Nat. Commun. 2020, 11, 2743. [Google Scholar] [CrossRef] [PubMed]
- Sumi, N.J.; Kuenzi, B.M.; Knezevic, C.E.; Rix, L.L.R.; Rix, U. Chemoproteomics Reveals Novel Protein and Lipid Kinase Targets of Clinical CDK4/6 Inhibitors in Lung Cancer. ACS Chem. Biol. 2015, 10, 2680–2686. [Google Scholar] [CrossRef]
- Kim, S.; Tiedt, R.; Loo, A.; Horn, T.; Delach, S.; Kovats, S.; Haas, K.; Schacher Engstler, B.; Cao, A.; Pinzon-Ortiz, M.; et al. The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 2018, 9, 35226. [Google Scholar] [CrossRef]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef]
- Sangar, M.L.C.; Genovesi, L.A.; Nakamoto, M.W.; Davis, M.J.; Knobluagh, S.E.; Ji, P.; Millar, A.; Wainwright, B.J.; Olson, J.M. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res. 2017, 23, 5802–5813. [Google Scholar] [CrossRef]
- Iriyama, N.; Hino, H.; Moriya, S.; Hiramoto, M.; Hatta, Y.; Takei, M.; Miyazawa, K. The cyclin-dependent kinase 4/6 inhibitor, abemaciclib, exerts dose-dependent cytostatic and cytocidal effects and induces autophagy in multiple myeloma cells. Leuk. Lymphoma 2018, 59, 1439–1450. [Google Scholar] [CrossRef]
- Dickson, M.A.; Schwartz, G.K.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Chi, P.; Antonescu, C.R.; Landa, J.; Qin, L.-X.; Crago, A.M.; et al. Progression-Free Survival Among Patients with Well-Differentiated or Dedifferentiated Liposarcoma Treated with CDK4 Inhibitor Palbociclib: A Phase 2 Clinical Trial. JAMA Oncol. 2016, 2, 937–940. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non–small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Johnson, S.M.; Torrice, C.D.; Bell, J.F.; Monahan, K.B.; Jiang, Q.; Wang, Y.; Ramsey, M.R.; Jin, J.; Wong, K.-K.; Su, L. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J. Clin. Investig. 2010, 120, 2528–2536. [Google Scholar] [CrossRef]
- Thill, M.; Schmidt, M. Management of adverse events during cyclin-dependent kinase 4/6 (CDK4/6) inhibitor-based treatment in breast cancer. Ther. Adv. Med. Oncol. 2018, 10, 1758835918793326. [Google Scholar] [CrossRef]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39, 7–18. [Google Scholar] [CrossRef]
- Allen, R.; Halpern, N.; Algaze, S.; Golan, T.; El-Khoueiry, A.B.; Shroff, R.T. Moving Beyond Chemotherapy for Pancreaticobiliary Tumors: Targeted and Immunotherapy Strategies. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e333–e343. [Google Scholar] [CrossRef]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Horgan, A.M.; Amir, E.; Walter, T.; Knox, J.J. Adjuvant Therapy in the Treatment of Biliary Tract Cancer: A Systematic Review and Meta-Analysis. J. Clin. Oncol. 2012, 30, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Nakagohri, T.; Kinoshita, T.; Konishi, M.; Takahashi, S.; Gotohda, N. Surgical outcome and prognostic factors in intrahepatic cholangiocarcinoma. World J. Surg. 2008, 32, 2675–2680. [Google Scholar] [CrossRef]
- Primrose, J.N.; Fox, R.P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): A randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019, 20, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Fletcher, P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Long-Term Outcomes and Exploratory Analyses of the Randomized Phase III BILCAP Study. J. Clin. Oncol. 2022, 40, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Palmer, D.; Wasan, H.; Ryder, W.D.; Davies, L.; Flight, H.; Rogan, J.; Hubner, R.; Bridgewater, J.A.; Valle, J.W. 748TiP—Abc-06: A Randomised Phase Iii, Multi-Centre, Open-Label Study of Active Symptom Control (Asc) Alone or Asc with Oxaliplatin/5-Fu Chemotherapy for Patients with Locally Advanced/Metastatic Biliary Tract Cancers (Abc) Previously Treated with C. Ann. Oncol. 2014, 25, iv252. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): A phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef]
- Boerner, T.; Drill, E.; Pak, L.M.; Nguyen, B.; Sigel, C.S.; Doussot, A.; Shin, P.; Goldman, D.A.; Gonen, M.; Allen, P.J.; et al. Genetic Determinants of Outcome in Intrahepatic Cholangiocarcinoma. Hepatology 2021, 74, 1429–1444. [Google Scholar] [CrossRef]
- Tannapfel, A.; Sommerer, F.; Benicke, M.; Weinans, L.; Katalinic, A.; Geißler, F.; Uhlmann, D.; Hauss, J.; Wittekind, C. Genetic and epigenetic alterations of the INK4a–ARF pathway in cholangiocarcinoma. J. Pathol. 2002, 197, 624–631. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, J.-L. Induction chemotherapy in borderline (non-)resectable pancreatic cancer: A unique window of opportunity for understanding pancreatic cancer. Eur. J. Cancer 2019, 106, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Bouchart, C.; Engelholm, J.-L.; Closset, J.; Navez, J.; Loi, P.; Gökburun, Y.; De Grez, T.; Mans, L.; Hendlisz, A.; Bali, M.A.; et al. Isotoxic high-dose stereotactic body radiotherapy integrated in a total multimodal neoadjuvant strategy for the treatment of localized pancreatic ductal adenocarcinoma. Ther. Adv. Med. Oncol. 2021, 13, 17588359211045860. [Google Scholar] [CrossRef] [PubMed]
- Bouchart, C.; Navez, J.; Closset, J.; Hendlisz, A.; Van Gestel, D.; Moretti, L.; Van Laethem, J.-L. Novel strategies using modern radiotherapy to improve pancreatic cancer outcomes: Toward a new standard? Ther. Adv. Med. Oncol. 2020, 12, 1758835920936093. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.; Mair, F. Tackling cancers of unmet need: The pancreatic cancer pathway. Lancet Gastroenterol. Hepatol. 2016, 1, 266–267. [Google Scholar] [CrossRef]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up †. Ann. Oncol. 2015, 26, v56–v68. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Van Laethem, J.-L.; Bali, M.A.; Borbath, I.; Verset, G.; Demols, A.; Puleo, F.; Peeters, M.; Annet, L.; Ceratti, A.; Ghilain, A.; et al. Preoperative gemcitabine-nab-paclitaxel (G-NP) for (borderline) resectable (BLR) or locally advanced (LA) pancreatic ductal adenocarcinoma (PDAC): Feasibility results and early response monitoring by Diffusion-Weighted (DW) MR. J. Clin. Oncol. 2016, 34, 4116. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef]
- Cowan, R.W.; Maitra, A. Genetic progression of pancreatic cancer. Cancer J. 2014, 20, 80–84. [Google Scholar] [CrossRef]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yang, P.; Liu, B.; Tang, Y. Is there a CDKN2A-centric network in pancreatic ductal adenocarcinoma? Onco. Targets. Ther. 2020, 13, 2551–2562. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef]
- Sittithumcharee, G.; Suppramote, O.; Vaeteewoottacharn, K.; Sirisuksakun, C.; Jamnongsong, S.; Laphanuwat, P.; Suntiparpluacha, M.; Matha, A.; Chusorn, P.; Buraphat, P.; et al. Dependency of Cholangiocarcinoma on Cyclin D–Dependent Kinase Activity. Hepatology 2019, 70, 1614–1630. [Google Scholar] [CrossRef] [PubMed]
- Saqub, H.; Proetsch-Gugerbauer, H.; Bezrookove, V.; Nosrati, M.; Vaquero, E.M.; de Semir, D.; Ice, R.J.; McAllister, S.; Soroceanu, L.; Kashani-Sabet, M.; et al. Dinaciclib, a cyclin-dependent kinase inhibitor, suppresses cholangiocarcinoma growth by targeting CDK2/5/9. Sci. Rep. 2020, 10, 18489. [Google Scholar] [CrossRef]
- Raspé, E.; Coulonval, K.; Pita, J.M.; Paternot, S.; Rothé, F.; Twyffels, L.; Brohée, S.; Craciun, L.; Larsimont, D.; Kruys, V.; et al. CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol. Med. 2017, 9, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Froio, D.; Nagrial, A.M.; Parkin, A.; Murphy, K.J.; Chin, V.T.; Wohl, D.; Steinmann, A.; Stark, R.; Drury, A.; et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut 2018, 67, 2142–2155. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Borja, N.A.; Franco, J.; Brody, J.R.; Yeo, C.J.; Mansour, J.; Choti, M.A.; McCue, P.; Knudsen, E.S. Selective impact of CDK4/6 suppression on patient-derived models of pancreatic cancer. Oncotarget 2015, 6, 15788–15801. [Google Scholar] [CrossRef]
- Franco, J.; Witkiewicz, A.K.; Knudsen, E.S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014, 5, 6512–6525. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Kumarasamy, V.; Ruiz, A.; Sivinski, J.; Chung, S.; Grant, A.; Vail, P.; Chauhan, S.S.; Jie, T.; Riall, T.S.; et al. Cell cycle plasticity driven by MTOR signaling: Integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene 2019, 38, 3355–3370. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, A.M.; Perera, R.M.; Ecker, V.; Nicolay, B.N.; Bardeesy, N.; Benes, C.H.; Dyson, N.J. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res. 2014, 74, 3947–3958. [Google Scholar] [CrossRef]
- Liu, F.; Korc, M. Cdk4/6 inhibition induces epithelial–mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol. Cancer Ther. 2012, 11, 2138–2148. [Google Scholar] [CrossRef]
- Rencuzogulları, O.; Yerlikaya, P.O.; Gürkan, A.Ç.; Arısan, E.D.; Telci, D. Palbociclib, a selective CDK4/6 inhibitor, restricts cell survival and epithelial-mesenchymal transition in Panc-1 and MiaPaCa-2 pancreatic cancer cells. J. Cell. Biochem. 2020, 121, 508–523. [Google Scholar] [CrossRef]
- Song, X.; Liu, X.; Wang, H.; Wang, J.; Qiao, Y.; Cigliano, A.; Utpatel, K.; Ribback, S.; Pilo, M.G.; Serra, M.; et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2019, 25, 403–413. [Google Scholar] [CrossRef]
- Song, X.; Xu, H.; Wang, P.; Wang, J.; Affo, S.; Wang, H.; Xu, M.; Liang, B.; Che, L.; Qiu, W.; et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J. Hepatol. 2021, 75, 888–899. [Google Scholar] [CrossRef]
- Willobee, B.A.; Gaidarski, A.A.; Dosch, A.R.; Castellanos, J.A.; Dai, X.; Mehra, S.; Messaggio, F.; Srinivasan, S.; VanSaun, M.N.; Nagathihalli, N.S.; et al. Combined Blockade of MEK and CDK4/6 Pathways Induces Senescence to Improve Survival in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2021, 20, 1246–1256. [Google Scholar] [CrossRef]
- Le Duff, M.; Gouju, J.; Jonchère, B.; Guillon, J.; Toutain, B.; Boissard, A.; Henry, C.; Guette, C.; Lelièvre, E.; Coqueret, O. Regulation of senescence escape by the cdk4–EZH2–AP2M1 pathway in response to chemotherapy. Cell Death Dis. 2018, 9, 199. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Witkiewicz, A.K. The strange case of CDK4/6 inhibitors: Mechanisms, resistance, and combination strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Demidenko, Z.N.; Blagosklonny, M. V MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013, 20, 1241–1249. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Watt, A.C.; Cejas, P.; DeCristo, M.J.; Metzger-Filho, O.; Lam, E.Y.N.; Qiu, X.; BrinJones, H.; Kesten, N.; Coulson, R.; Font-Tello, A.; et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nat. Cancer 2021, 2, 34–48. [Google Scholar] [CrossRef]
- Dhir, T.; Schultz, C.W.; Jain, A.; Brown, S.Z.; Haber, A.; Goetz, A.; Xi, C.; Su, G.H.; Xu, L.; Posey, J., 3rd; et al. Abemaciclib Is Effective Against Pancreatic Cancer Cells and Synergizes with HuR and YAP1 Inhibition. Mol. Cancer Res. 2019, 17, 2029–2041. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Kumarasamy, V.; Ruiz, A.; Nambiar, R.; Witkiewicz, A.K.; Knudsen, E.S. Chemotherapy impacts on the cellular response to CDK4/6 inhibition: Distinct mechanisms of interaction and efficacy in models of pancreatic cancer. Oncogene 2020, 39, 1831–1845. [Google Scholar] [CrossRef]
- Suppramote, O.; Prasopporn, S.; Aroonpruksakul, S.; Ponvilawan, B.; Makjaroen, J.; Suntiparpluacha, M.; Korphaisarn, K.; Charngkaew, K.; Chanwat, R.; Pisitkun, T.; et al. The Acquired Vulnerability Caused by CDK4/6 Inhibition Promotes Drug Synergism Between Oxaliplatin and Palbociclib in Cholangiocarcinoma. Front. Oncol. 2022, 12, 877194. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Hsieh, F.-S.; Wang, C.-Y.; Chen, L.-J.; Chang, S.-S.; Tsai, M.-H.; Hung, M.-H.; Kuo, C.-W.; Shih, C.-T.; Chao, T.-I.; et al. Palbociclib enhances radiosensitivity of hepatocellular carcinoma and cholangiocarcinoma via inhibiting ataxia telangiectasia–mutated kinase–mediated DNA damage response. Eur. J. Cancer 2018, 102, 10–22. [Google Scholar] [CrossRef]
- Salvador-Barbero, B.; Álvarez-Fernández, M.; Zapatero-Solana, E.; El Bakkali, A.; del Camino Menéndez, M.; López-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353.e6. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-dependent kinases in DNA damage response. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188716. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular senescence: A translational perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Johnson, C.A.; Prasad, N.; Yang, J.; Chen, S.-C.; Ayers, G.D.; Pawlikowski, J.S.; Raman, D.; Sosman, J.A.; Kelley, M. Connecting the dots: Therapy-induced senescence and a tumor-suppressive immune microenvironment. JNCI J. Natl. Cancer Inst. 2016, 108, djv406. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Kumarasamy, V.; Chung, S.; Ruiz, A.; Vail, P.; Tzetzo, S.; Wu, J.; Nambiar, R.; Sivinski, J.; Chauhan, S.S.; et al. Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut 2021, 70, 127–138. [Google Scholar] [CrossRef]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e21. [Google Scholar] [CrossRef]
- Hidalgo, M.; Carbonero, R.G.; Lim, K.-H.; Messersmith, W.; Garrido-Laguna, I.; Borazanci, E.; Lowy, A.; Rodriguez, L.M.; Laheru, D.; Huang, X.; et al. Abstract CT135: A phase 1b study of palbociclib + nab-paclitaxel in patients (pts) with metastatic adenocarcinoma of the pancreas (PDAC). Cancer Res. 2020, 80, CT135. [Google Scholar] [CrossRef]
- Azaro, A.; Massard, C.; Tap, W.D.; Cassier, P.A.; Merchan, J.; Italiano, A.; Anderson, B.; Yuen, E.; Yu, D.; Oakley, G.; et al. A phase 1b study of the Notch inhibitor crenigacestat (LY3039478) in combination with other anticancer target agents (taladegib, LY3023414, or abemaciclib) in patients with advanced or metastatic solid tumors. Investig. N. Drugs 2021, 39, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Van Sciver, R.; Lee, M.; Lee, C.; Lafever, A.; Svyatova, E.; Kanda, K.; Collier, A.; Siewertsz van Reesema, L.; Tang-Tan, A.; Zheleva, V. A new strategy to control and eradicate “undruggable” oncogenic k-ras-driven pancreatic cancer: Molecular insights and core principles learned from developmental and evolutionary biology. Cancers 2018, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The Role of Stroma in Cholangiocarcinoma: The Intriguing Interplay between Fibroblastic Component, Immune Cell Subsets and Tumor Epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef]
- Konstantinidou, M.; Li, J.; Zhang, B.; Wang, Z.; Shaabani, S.; Ter Brake, F.; Essa, K.; Dömling, A. PROTACs– a game-changing technology. Expert Opin. Drug Discov. 2019, 14, 1255–1268. [Google Scholar] [CrossRef]
- Adon, T.; Shanmugarajan, D.; Kumar, H.Y. CDK4/6 inhibitors: A brief overview and prospective research directions. RSC Adv. 2021, 11, 29227–29246. [Google Scholar] [CrossRef]
- Wu, X.; Yang, X.; Xiong, Y.; Li, R.; Ito, T.; Ahmed, T.A.; Karoulia, Z.; Adamopoulos, C.; Wang, H.; Wang, L.; et al. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat. Cancer 2021, 2, 429–443. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, B.; Guo, J.; Shao, H.; Del Priore, I.S.; Chang, Q.; Kudo, R.; Li, Z.; Razavi, P.; Liu, B.; et al. INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors. Cancer Discov. 2022, 12, 356–371. [Google Scholar] [CrossRef]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C.; et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 2018, 4, 38. [Google Scholar] [CrossRef]
- Finn, R.S.; Liu, Y.; Zhu, Z.; Martin, M.; Rugo, H.S.; Diéras, V.; Im, S.-A.; Gelmon, K.A.; Harbeck, N.; Lu, D.R. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naïve Metastatic Breast CancerBiomarker Analyses of Palbociclib in Breast Cancer. Clin. Cancer Res. 2020, 26, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor–positive metastatic breast cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Pogacar, Z.; Johnson, J.L.; Krenning, L.; De Conti, G.; Jochems, F.; Lieftink, C.; Velds, A.; Wardak, L.; Groot, K.; Schepers, A.; et al. Indisulam synergizes with palbociclib to induce senescence through inhibition of CDK2 kinase activity. PLoS ONE 2022, 17, e0273182. [Google Scholar] [CrossRef]
- Palafox, M.; Monserrat, L.; Bellet, M.; Villacampa, G.; Gonzalez-Perez, A.; Oliveira, M.; Brasó-Maristany, F.; Ibrahimi, N.; Kannan, S.; Mina, L.; et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER+ breast cancer. Nat. Commun. 2022, 13, 5258. [Google Scholar] [CrossRef]
- Paternot, S.; Raspé, E.; Meiller, C.; Tarabichi, M.; Assié, J.-B.; Libert, F.; Remmelink, M.; Bisteau, X.; Pauwels, P.; Blum, Y.; et al. Preclinical evaluation of CDK4 phosphorylation predicts high sensitivity of pleural mesotheliomas to CDK4/6 inhibition. Mol. Oncol. 2022. [CrossRef]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.N.; El Messaoudi-Aubert, S.; Racek, T.; Stock, J.K.; Nicholls, J.; Rodriguez-Niedenführ, M.; Gil, J.; Peters, G. Several Distinct Polycomb Complexes Regulate and Co-Localize on the INK4a Tumor Suppressor Locus. PLoS ONE 2009, 4, e6380. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Peters, G. Regulation of the INK4b–ARF–INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16ink4a expression in tumors. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef]
- Gopalan, P.K.; Pinder, M.C.; Chiappori, A.; Ivey, A.M.; Gordillo Villegas, A.; Kaye, F.J. A phase II clinical trial of the CDK 4/6 inhibitor palbociclib (PD 0332991) in previously treated, advanced non-small cell lung cancer (NSCLC) patients with inactivated CDKN2A. J. Clin. Oncol. 2014, 32, 8077. [Google Scholar] [CrossRef]
- Al Baghdadi, T.; Halabi, S.; Garrett-Mayer, E.; Mangat, P.K.; Ahn, E.R.; Sahai, V.; Alvarez, R.H.; Kim, E.S.; Yost, K.J.; Rygiel, A.L.; et al. Palbociclib in Patients with Pancreatic and Biliary Cancer with CDKN2A Alterations: Results From the Targeted Agent and Profiling Utilization Registry Study. JCO Precis. Oncol. 2019, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, V.; Vail, P.; Nambiar, R.; Witkiewicz, A.K.; Knudsen, E.S. Functional Determinants of Cell Cycle Plasticity and Sensitivity to CDK4/6 Inhibition. Cancer Res. 2021, 81, 1347–1360. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT Number | Title | Status | Interventions | Conditions | N |
|---|---|---|---|---|---|
| NCT03454035 | Ulixertinib/Palbociclib in Patients with Advanced Pancreatic and Other Solid Tumours | Active, not recruiting |
|
| 45 |
| NCT03339843 | Multiorgan Metabolic Imaging Response Assessment of Abemaciclib | Active, not recruiting |
|
| 85 |
| NCT03065062 | Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination with the PI3K/mTOR Inhibitor Gedatolisib (PF-05212384) for Patients with Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumours | Recruiting |
|
| 96 |
| NCT02981342 | A Study of Abemaciclib (LY2835219) Alone or in Combination with Other Agents in Participants with Previously Treated Pancreatic Ductal Adenocarcinoma | Completed |
|
| 106 |
| NCT02784795 | Study of LY3039478 in Participants with Advanced or Metastatic Solid Tumours | Completed |
|
| 94 |
| NCT02501902 | Dose-Escalation Study of Palbociclib + Nab-Paclitaxel In mPDAC | Completed |
|
| 76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arsenijevic, T.; Coulonval, K.; Raspé, E.; Demols, A.; Roger, P.P.; Van Laethem, J.-L. CDK4/6 Inhibitors in Pancreatobiliary Cancers: Opportunities and Challenges. Cancers 2023, 15, 968. https://doi.org/10.3390/cancers15030968
Arsenijevic T, Coulonval K, Raspé E, Demols A, Roger PP, Van Laethem J-L. CDK4/6 Inhibitors in Pancreatobiliary Cancers: Opportunities and Challenges. Cancers. 2023; 15(3):968. https://doi.org/10.3390/cancers15030968
Chicago/Turabian StyleArsenijevic, Tatjana, Katia Coulonval, Eric Raspé, Anne Demols, Pierre P. Roger, and Jean-Luc Van Laethem. 2023. "CDK4/6 Inhibitors in Pancreatobiliary Cancers: Opportunities and Challenges" Cancers 15, no. 3: 968. https://doi.org/10.3390/cancers15030968
APA StyleArsenijevic, T., Coulonval, K., Raspé, E., Demols, A., Roger, P. P., & Van Laethem, J.-L. (2023). CDK4/6 Inhibitors in Pancreatobiliary Cancers: Opportunities and Challenges. Cancers, 15(3), 968. https://doi.org/10.3390/cancers15030968