
Vemurafenib and Dabrafenib Downregulates RIPK4 Level

 , , ,
, , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General Chemicals and Reagents
2.2. Clinical Samples
2.3. Detection of BRAF Mutations from Paraffin-Embedded Tissues
2.4. Immunohistochemistry for RIPK4 and Section Assessment
2.5. Cell Culture
2.6. Transfection with Small Interfering RNA (siRNA)
2.7. Overexpression of RIPK4
2.8. Western Blotting
2.9. Viability and Apoptosis Assay
2.10. Quantitative Real-Time PCR
2.11. Ki67 Analysis Using Immunofluorescence Staining
2.12. Sequence Similarity-Blast
2.13. Structural Similarity and Molecular Docking
2.14. Statistical Analysis
3. Results
3.1. RIPK4 Shows Sequence and Structural Similarity to the BRAF Protein
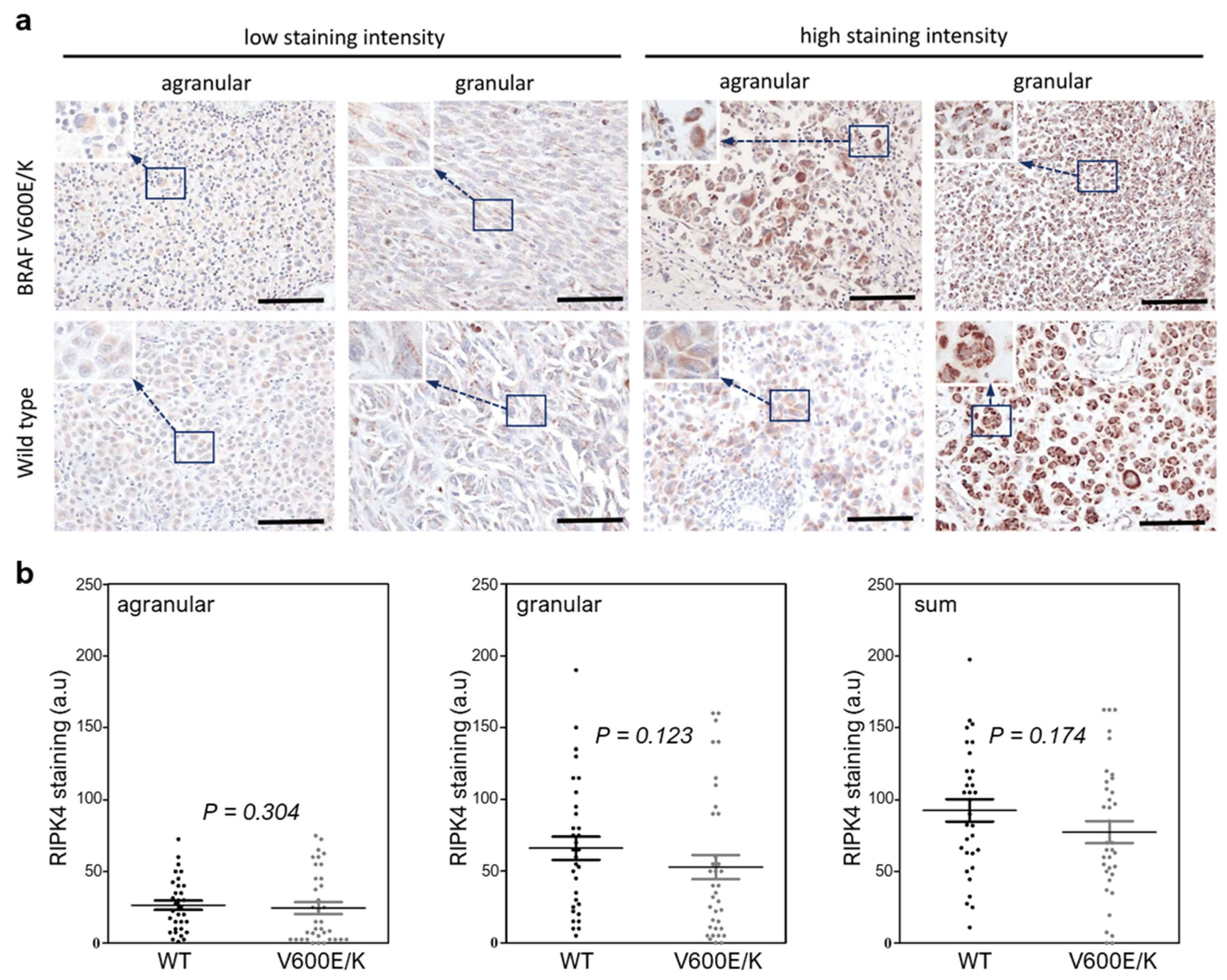
3.2. The BRAF Mutation Does Not Affect RIPK4 Expression in Metastatic Melanoma
3.3. BRAF Inhibitors Decrease the Level of the RIPK4 Protein in Melanoma Cells
3.4. Neither the Decrease in RIPK4 Using siRNA nor Crisp/Cas9-Mediated Overexpression of RIPK4 Affects PEBP1 or BRAF/ERK/MEK Signaling Pathway in Melanoma Cell Lines
3.5. Silencing RIPK4 Altered the CDK4/CDK6/RB and FAK/AKT Pathways in WM266.4, but Not in A375
3.6. RIPK4 Downregulation Sensitized WM266.4 but Not A375 Melanoma Cells to BRAFi Treatment
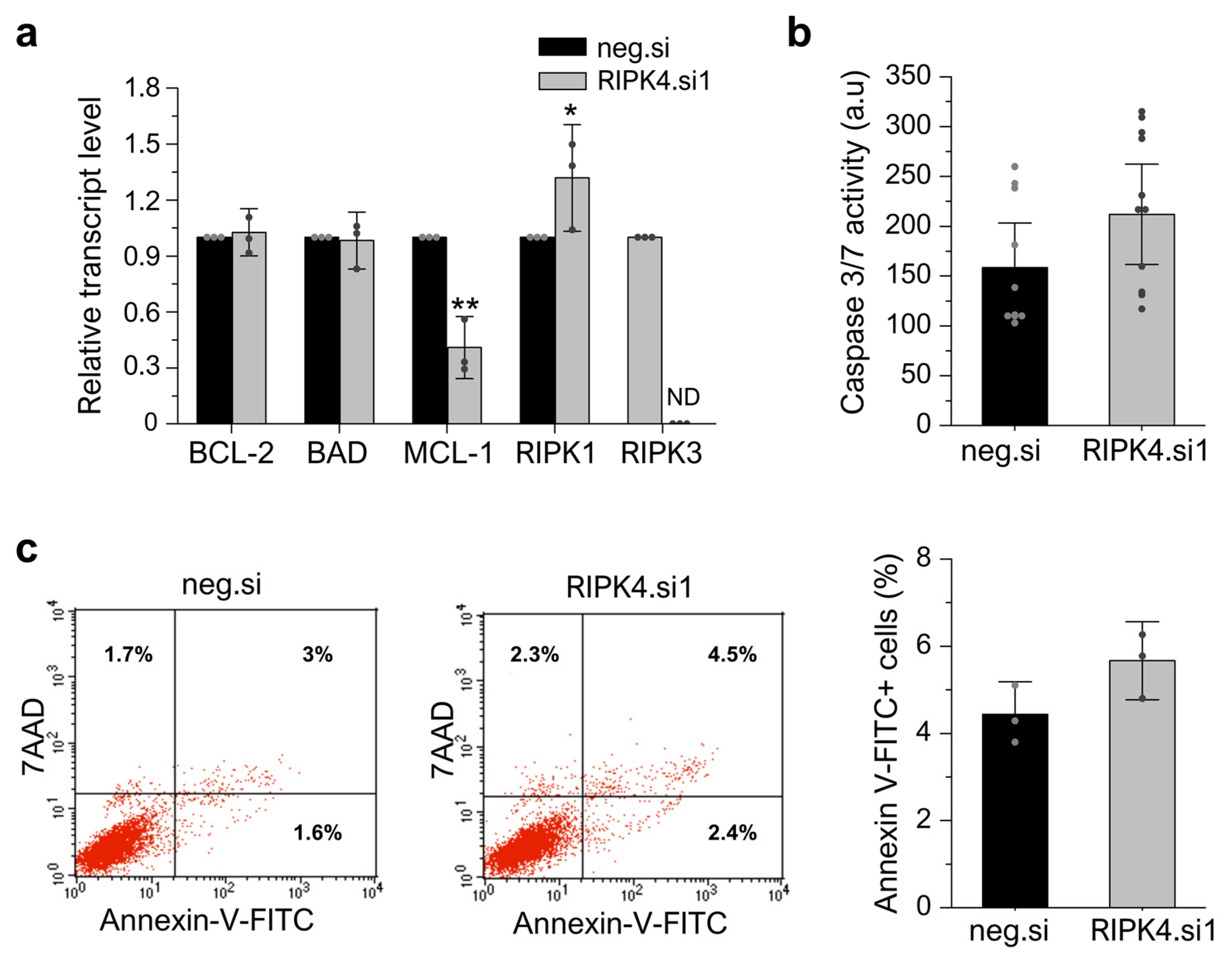
3.7. RIPK4 Silencing Using siRNA Has No Impact on Apoptosis or Necroptosis in WM266.4 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MacKenzie, A.; Boycott, K. MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 1364–1365. [Google Scholar] [CrossRef]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; Maclennan, G.T.; Montironi, R. Molecular Testing for BRAF Mutations to Inform Melanoma Treatment Decisions: A Move toward Precision Medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of Mutations in BRAF, NRAS, and KIT in Different Populations and Histological Subtypes of Melanoma: A Systemic Review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef]
- Spathis, A.; Katoulis, A.; Damaskou, V.; Liakou, A.; Kottaridi, C.; Leventakou, D.; Sgouros, D.; Mamantopoulos, A.; Rigopoulos, D.; Karakitsos, P.; et al. BRAF Mutation Status in Primary, Recurrent, and Metastatic Malignant Melanoma and Its Relation to Histopathological Parameters. Dermatol. Pract. Concept. 2019, 9, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Manfredini, M.; Greco, S.; Pellacani, G.; Depenni, R.; Tomasi, A.; Maccaferri, M.; Cascinu, S. BRAF, NRAS and C-KIT Advanced Melanoma: Clinico-Pathological Features, Targeted-Therapy Strategies and Survival. Anticancer Res. 2017, 37, 7043–7048. [Google Scholar] [CrossRef]
- Viros, A.; Fridlyand, J.; Bauer, J.; Lasithiotakis, K.; Garbe, C.; Pinkel, D.; Bastian, B.C. Improving Melanoma Classification by Integrating Genetic and Morphologic Features. PLoS Med. 2008, 5, 0941–0952. [Google Scholar] [CrossRef]
- Garnett, M.J.; Marais, R. Guilty as Charged: B-RAF Is a Human Oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Al Hashmi, M.; Sastry, K.S.; Silcock, L.; Chouchane, L.; Mattei, V.; James, N.; Mathew, R.; Bedognetti, D.; De Giorgi, V.; Murtas, D.; et al. Differential Responsiveness to BRAF Inhibitors of Melanoma Cell Lines BRAF V600E-Mutated. J. Transl. Med. 2020, 18, 192. [Google Scholar] [CrossRef]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-Mutant Melanoma: Treatment Approaches, Resistance Mechanisms, and Diagnostic Strategies. OncoTargets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Gupta, R.; Janostiak, R.; Wajapeyee, N. Transcriptional Regulators and Alterations That Drive Melanoma Initiation and Progression. Oncogene 2020, 39, 7093–7105. [Google Scholar] [CrossRef]
- Haugh, A.M.; Salama, A.K.S.; Johnson, D.B. Advanced Melanoma: Resistance Mechanisms to Current Therapies. Hematol. Oncol. Clin. N. Am. 2021, 35, 111–128. [Google Scholar] [CrossRef]
- Rossi, A.; Roberto, M.; Panebianco, M.; Botticelli, A.; Mazzuca, F.; Marchetti, P. Drug Resistance of BRAF-Mutant Melanoma: Review of up-to-Date Mechanisms of Action and Promising Targeted Agents. Eur. J. Pharmacol. 2019, 862, 172621. [Google Scholar] [CrossRef]
- Huang, C.S.; Oberbeck, N.; Hsiao, Y.C.; Liu, P.; Johnson, A.R.; Dixit, V.M.; Hymowitz, S.G. Crystal Structure of Ripk4 Reveals Dimerization-Dependent Kinase Activity. Structure 2018, 26, 767–777.e5. [Google Scholar] [CrossRef]
- Cuny, G.D.; Degterev, A. RIPK Protein Kinase Family: Atypical Lives of Typical Kinases. Semin. Cell Dev. Biol. 2021, 109, 96–105. [Google Scholar] [CrossRef]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP Kinases: Key Decision Makers in Cell Death and Innate Immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef]
- Xu, J.; Wei, Q.; He, Z. Insight Into the Function of RIPK4 in Keratinocyte Differentiation and Carcinogenesis. Front. Oncol. 2020, 10, 1562. [Google Scholar] [CrossRef] [PubMed]
- Azizmohammadi, S.; Azizmohammadi, S.; Safari, A.; Kaghazian, M.; Sadrkhanlo, M.; Behnod, V.; Seifoleslami, M. High-Level Expression of RIPK4 and EZH2 Contributes to Lymph Node Metastasis and Predicts Favorable Prognosis in Patients with Cervical Cancer. Oncol. Res. 2017, 25, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Q.; Li, F.F.; Zhang, J.B.; Zhou, T.J.; Xue, W.Q.; Zheng, X.H.; Chen, Y.B.; Liao, X.Y.; Zhang, L.; Zhang, S.D.; et al. Increased RIPK4 Expression Is Associated with Progression and Poor Prognosis in Cervical Squamous Cell Carcinoma Patients. Sci. Rep. 2015, 5, 11955. [Google Scholar] [CrossRef]
- Qi, Z.H.; Xu, H.X.; Zhang, S.R.; Xu, J.Z.; Li, S.; Gao, H.L.; Jin, W.; Wang, W.Q.; Wu, C.T.; Ni, Q.X.; et al. RIPK4/PEBP1 Axis Promotes Pancreatic Cancer Cell Migration and Invasion by Activating RAF1/MEK/ERK Signaling. Int. J. Oncol. 2018, 52, 1105–1116. [Google Scholar] [CrossRef]
- Heim, D.; Cornils, K.; Schulze, K.; Fehse, B.; Lohse, A.W.; Brümmendorf, T.H.; Wege, H. Retroviral Insertional Mutagenesis in Telomerase-Immortalized Hepatocytes Identifies RIPK4 as Novel Tumor Suppressor in Human Hepatocarcinogenesis. Oncogene 2015, 34, 385–393. [Google Scholar] [CrossRef]
- Kopparam, J.; Chiffelle, J.; Angelino, P.; Piersigilli, A.; Zangger, N.; Delorenzi, M.; Meylan, E. RIP4 Inhibits STAT3 Signaling to Sustain Lung Adenocarcinoma Differentiation. Cell Death Differ. 2017, 24, 1761–1771. [Google Scholar] [CrossRef]
- Madej, E.; Ryszawy, D.; Brożyna, A.A.; Czyz, M.; Czyz, J.; Wolnicka-Glubisz, A. Deciphering the Functional Role of RIPK4 in Melanoma. Int. J. Mol. Sci. 2021, 22, 11504. [Google Scholar] [CrossRef] [PubMed]
- Alla, J.A.; Quitterer, U. The RAF Kinase Inhibitor Protein (RKIP): Good as Tumour Suppressor, Bad for the Heart. Cells 2022, 11, 654. [Google Scholar] [CrossRef]
- Granovsky, A.E.; Rosner, M.R. Raf Kinase Inhibitory Protein: A Signal Transduction Modulator and Metastasis Suppressor. Cell Res. 2008, 18, 452–457. [Google Scholar] [CrossRef]
- Cardile, V.; Malaponte, G.; Loreto, C.; Libra, M.; Caggia, S.; Trovato, F.M.; Musumeci, G. Raf Kinase Inhibitor Protein (RKIP) and Phospho-RKIP Expression in Melanomas. Acta Histochem. 2013, 115, 795–802. [Google Scholar] [CrossRef]
- Park, S.; Yeung, M.L.; Beach, S.; Shields, J.M.; Yeung, K.C. RKIP Downregulates B-Raf Kinase Activity in Melanoma Cancer Cells. Oncogene 2005, 24, 3535–3540. [Google Scholar] [CrossRef]
- Huang, X.; Mcgann, J.C.; Liu, B.Y.; Hannoush, R.N.; Jennie, R.; Hymowitz, G.; Hongo, J.; Wynshaw-boris, A.; Polakis, P.; Richard, M. RIPK4 Phosphorylates Dishevelled Proteins to Regulate Canonical Wnt Signaling. Science 2013, 339, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Munz, B. RIP4 Is a Target of Multiple Signal Transduction Pathways in Keratinocytes: Implications for Epidermal Differentiation and Cutaneous Wound Repair. Exp. Cell Res. 2010, 316, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Madonna, G.; Ullman, C.D.; Gentilcore, G.; Palmieri, G.; Ascierto, P.A. NF-ΚB as Potential Target in the Treatment of Melanoma. J. Transl. Med. 2012, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Sinnberg, T.; Levesque, M.P.; Krochmann, J.; Cheng, P.F.; Ikenberg, K.; Meraz-Torres, F.; Niessner, H.; Garbe, C.; Busch, C. Wnt-Signaling Enhances Neural Crest Migration of Melanoma Cells and Induces an Invasive Phenotype. Mol. Cancer 2018, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Skalniak, L.; Smejda, M.; Cierniak, A.; Adamczyk, A.; Konieczny, P.; Madej, E.; Wolnicka-Glubisz, A. P38 but Not P53 Is Responsible for UVA-Induced MCPIP1 Expression. Mech. Ageing Dev. 2017, 172, 96–106. [Google Scholar] [CrossRef]
- Wolnicka-Glubisz, A.; Pawlak, A.; Insinska-Rak, M.; Zadlo, A. Analysis of Photoreactivity and Phototoxicity of Riboflavin’s Analogue 3MeTARF. J. Photochem. Photobiol. B 2020, 205, 111820. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Muto, A.; Ruland, J.; McAllister-Lucas, L.M.; Lucas, P.C.; Yamaoka, S.; Chen, F.F.; Lin, A.; Mak, T.W.; Núñez, G.; Inohara, N. Protein Kinase C-Associated Kinase (PKK) Mediates Bcl10-Independent NF-ΚB Activation Induced by Phorbol Ester. J. Biol. Chem. 2002, 277, 31871–31876. [Google Scholar] [CrossRef]
- Tham, M.; Stark, H.J.; Jauch, A.; Harwood, C.; Pavez Lorie, E.; Boukamp, P. Adverse Effects of Vemurafenib on Skin Integrity: Hyperkeratosis and Skin Cancer Initiation Due to Altered MEK/ERK-Signaling and MMP Activity. Front. Oncol. 2022, 12, 827985. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; O’Sullivan, J.; Missero, C.; Blair, E.; Richardson, R.; Anderson, B.; Antonini, D.; Murray, J.C.; Shanske, A.L.; Schutte, B.C.; et al. Exome Sequence Identifies RIPK4 as the Bartsocas-Papas Syndrome Locus. Am. J. Hum. Genet. 2012, 90, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Garcia, L.F.; Mannella, V.; Gammon, L.; Borg, T.M.; Maffucci, T.; Scatolini, M.; Chiorino, G.; Vergani, E.; Rodolfo, M.; et al. Targeting P63 Upregulation Abrogates Resistance to MAPK Inhibitors in Melanoma. Cancer Res. 2020, 80, 2676–2688. [Google Scholar] [CrossRef]
- Kalay, E.; Sezgin, O.; Chellappa, V.; Mutlu, M.; Morsy, H.; Kayserili, H.; Kreiger, E.; Cansu, A.; Toraman, B.; Abdalla, E.M.; et al. Mutations in RIPK4 Cause the Autosomal-Recessive Form of Popliteal Pterygium Syndrome. Am. J. Hum. Genet. 2012, 90, 76–85. [Google Scholar] [CrossRef]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-RafV600E Inhibitor Dabrafenib Selectively Inhibits RIP3 and Alleviates Acetaminophen-Induced Liver Injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef]
- Xu, J.; Wu, D.; Zhang, B.; Pan, C.; Guo, Y.; Wei, Q. Depletion of RIPK4 Parallels Higher Malignancy Potential in Cutaneous Squamous Cell Carcinoma. PeerJ 2022, 10, e12932. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. A New Cell-Cycle Target in Cancer—Inhibiting Cyclin D–Dependent Kinases 4 and 6. N. Engl. J. Med. 2016, 375, 1920–1923. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, P.; Zhang, Q.; Wu, H. CDK14/Β-catenin/TCF4/MiR-26b Positive Feedback Regulation Modulating Pancreatic Cancer Cell Phenotypes in Vitro and Tumor Growth in Mice Model in Vivo. J. Gene Med. 2022, 24, e3343. [Google Scholar] [CrossRef]
- Shang, Y.; Jiang, Y.-L.; Ye, L.-J.; Chen, L.-N.; Ke, Y. Resveratrol Acts via Melanoma-Associated Antigen A12 (MAGEA12)/Protein Kinase B (Akt) Signaling to Inhibit the Proliferation of Oral Squamous Cell Carcinoma Cells. Bioengineered 2021, 12, 2253–2262. [Google Scholar] [CrossRef]
- Paul, R.; Luo, M.; Mo, X.; Lu, J.; Yeo, S.K.; Guan, J.L. FAK Activates AKT-MTOR Signaling to Promote the Growth and Progression of MMTV-Wnt1-Driven Basal-like Mammary Tumors. Breast Cancer Res. 2020, 22, 59. [Google Scholar] [CrossRef]
- Byron, S.A.; Loch, D.C.; Wellens, C.L.; Wortmann, A.; Wu, J.; Wang, J.; Nomoto, K.; Pollock, P.M. Sensitivity to the MEK Inhibitor E6201 in Melanoma Cells Is Associated with Mutant BRAF and Wildtype PTEN Status. Mol. Cancer 2012, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Geserick, P.; Wang, J.; Schilling, R.; Horn, S.; Harris, P.A.; Bertin, J.; Gough, P.J.; Feoktistova, M.; Leverkus, M. Absence of RIPK3 Predicts Necroptosis Resistance in Malignant Melanoma. Cell Death Dis. 2015, 6, e1884. [Google Scholar] [CrossRef]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M.; Arpino, G.; De Laurentiis, M.; Puglisi, F.; De Placido, S.; et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front. Oncol. 2018, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in Cancer: Challenges and Opportunities for Therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Gera, R.; Mokbel, L.; Jiang, W.G.; Mokbel, K. MRNA Expression of CDK2AP1 in Human Breast Cancer: Correlation with Clinical and Pathological Parameters. Cancer Genom.-Proteom. 2018, 15, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Eng, V.V.; Wemyss, M.A.; Pearson, J.S. The Diverse Roles of RIP Kinases in Host-Pathogen Interactions. Semin. Cell Dev. Biol. 2021, 109, 125–143. [Google Scholar] [CrossRef]
- He, S.; Wang, X. RIP Kinases as Modulators of Inflammation and Immunity. Nat. Immunol. 2018, 19, 912–922. [Google Scholar] [CrossRef]
- Pawlikowska, M.; Jadrzejewski, T.; Broayna, A.A.; Wrotek, S. Protein-Bound Polysaccharides from Coriolus Versicolor Induce RIPK1/RIPK3/MLKL-Mediated Necroptosis in ER-Positive Breast Cancer and Amelanotic Melanoma Cells. Cell. Physiol. Biochem. 2020, 54, 591–604. [Google Scholar] [CrossRef]
- Torre, L.D.; Nebbioso, A.; Stunnenberg, H.G.; Martens, J.H.A.; Carafa, V.; Altucci, L. The Role of Necroptosis: Biological Relevance and Its Involvement in Cancer. Cancers 2021, 13, 684. [Google Scholar] [CrossRef]
- Speir, M.; Djajawi, T.M.; Conos, S.A.; Tye, H.; Lawlor, K.E. Targeting Rip Kinases in Chronic Inflammatory Disease. Biomolecules 2021, 11, 646. [Google Scholar] [CrossRef]
- Meylan, E.; Martinon, F.; Thome, M.; Gschwendt, M.; Tschopp, J. RIP4 (DIK/PKK), a Novel Member of the RIP Kinase Family, Activates NF-ΚB and Is Processed during Apoptosis. EMBO Rep. 2002, 3, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Poligone, B.; Gilmore, E.S.; Alexander, C.V.; Oleksyn, D.; Gillespie, K.; Zhao, J.; Ibrahim, S.F.; Pentland, A.P.; Brown, M.D.; Chen, L. PKK Suppresses Tumor Growth and Is Decreased in Squamous Cell Carcinoma of the Skin. J. Investig. Dermatol. 2015, 135, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, W.; Zhou, Y.; Xu, W.; Wang, H. RIPK4 Is Downregulated in Poorly Differentiated Tongue Cancer and Is Associated with Migration/Invasion and Cisplatin-Induced Apoptosis. Int. J. Biol. Markers 2014, 29, 150–159. [Google Scholar] [CrossRef]
- Lee, E.F.; Harris, T.J.; Tran, S.; Evangelista, M.; Arulananda, S.; John, T.; Ramnac, C.; Hobbs, C.; Zhu, H.; Gunasingh, G.; et al. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death Dis. 2019, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Categorization | Analyzable n (%) |
|---|---|---|
| Age | <60 years | 14 (20.6) |
| >60 years | 54 (79.4) | |
| Gender | Female | 42 (61.8) |
| Male | 36 (52.9) | |
| Melanoma site | Primary melanoma | 6 (8.8) |
| Lymph node | 41 (60.3) | |
| Recurrence | 19 (27.9) | |
| Others | 2 (2.9) | |
| Treatment | Surgery | 23 (35.3) |
| Radiotherapy | 4 (5.9) | |
| Chemotherapy/immunotherapy | 30 (44.1) | |
| Radiotherapy + chemotherapy | 10 (14.7) | |
| Surgery + radiotherapy | 1 (1.5) | |
| BRAF status | V600E | 27 (39.7) |
| V600K | 5 (7.4) | |
| Wild-type | 36 (52.9) | |
| Mitotic index | ≤5 mitoses/1 mm2 | 22 (32.3) |
| >5 mitoses/1 mm2 | 46 (67.7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madej, E.; Brożyna, A.A.; Adamczyk, A.; Wronski, N.; Harazin-Lechowska, A.; Muzyk, A.; Makuch, K.; Markiewicz, M.; Rys, J.; Wolnicka-Glubisz, A. Vemurafenib and Dabrafenib Downregulates RIPK4 Level. Cancers 2023, 15, 918. https://doi.org/10.3390/cancers15030918
Madej E, Brożyna AA, Adamczyk A, Wronski N, Harazin-Lechowska A, Muzyk A, Makuch K, Markiewicz M, Rys J, Wolnicka-Glubisz A. Vemurafenib and Dabrafenib Downregulates RIPK4 Level. Cancers. 2023; 15(3):918. https://doi.org/10.3390/cancers15030918
Chicago/Turabian StyleMadej, Ewelina, Anna A. Brożyna, Agnieszka Adamczyk, Norbert Wronski, Agnieszka Harazin-Lechowska, Anna Muzyk, Krzysztof Makuch, Michal Markiewicz, Janusz Rys, and Agnieszka Wolnicka-Glubisz. 2023. "Vemurafenib and Dabrafenib Downregulates RIPK4 Level" Cancers 15, no. 3: 918. https://doi.org/10.3390/cancers15030918
APA StyleMadej, E., Brożyna, A. A., Adamczyk, A., Wronski, N., Harazin-Lechowska, A., Muzyk, A., Makuch, K., Markiewicz, M., Rys, J., & Wolnicka-Glubisz, A. (2023). Vemurafenib and Dabrafenib Downregulates RIPK4 Level. Cancers, 15(3), 918. https://doi.org/10.3390/cancers15030918