

P38 Mediates Tumor Suppression through Reduced Autophagy and Actin Cytoskeleton Changes in NRAS-Mutant Melanoma

 , , ,
, , ,  ,
,
Simple Summary
Abstract
1. Introduction
2. Results
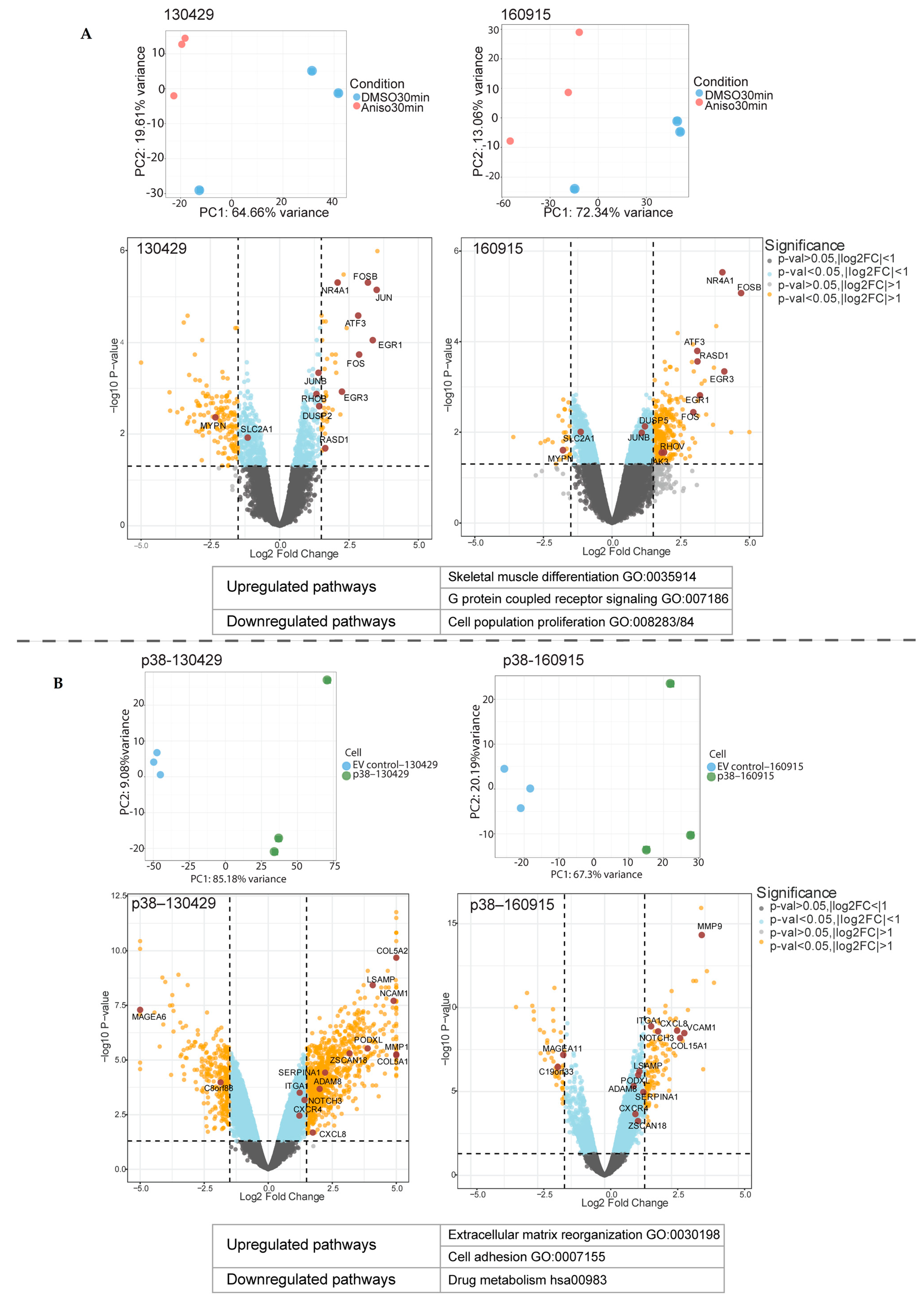
2.1. Short- and Long-Term p38 Activation by Anisomycin and Stable Transduction Leads to a Distinct Transcriptional Signature in Patient-Derived NRAS Melanoma Cell Lines
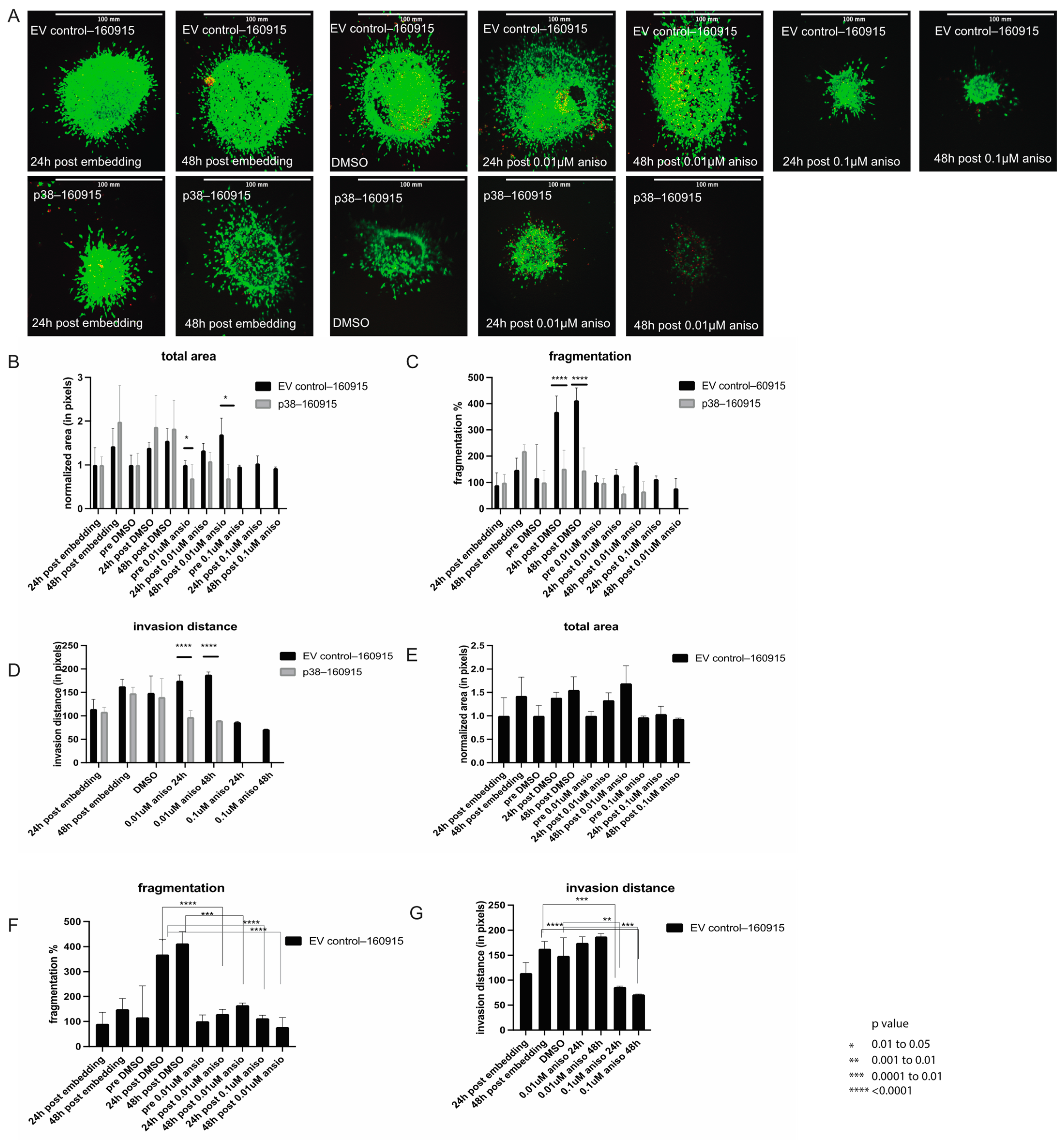
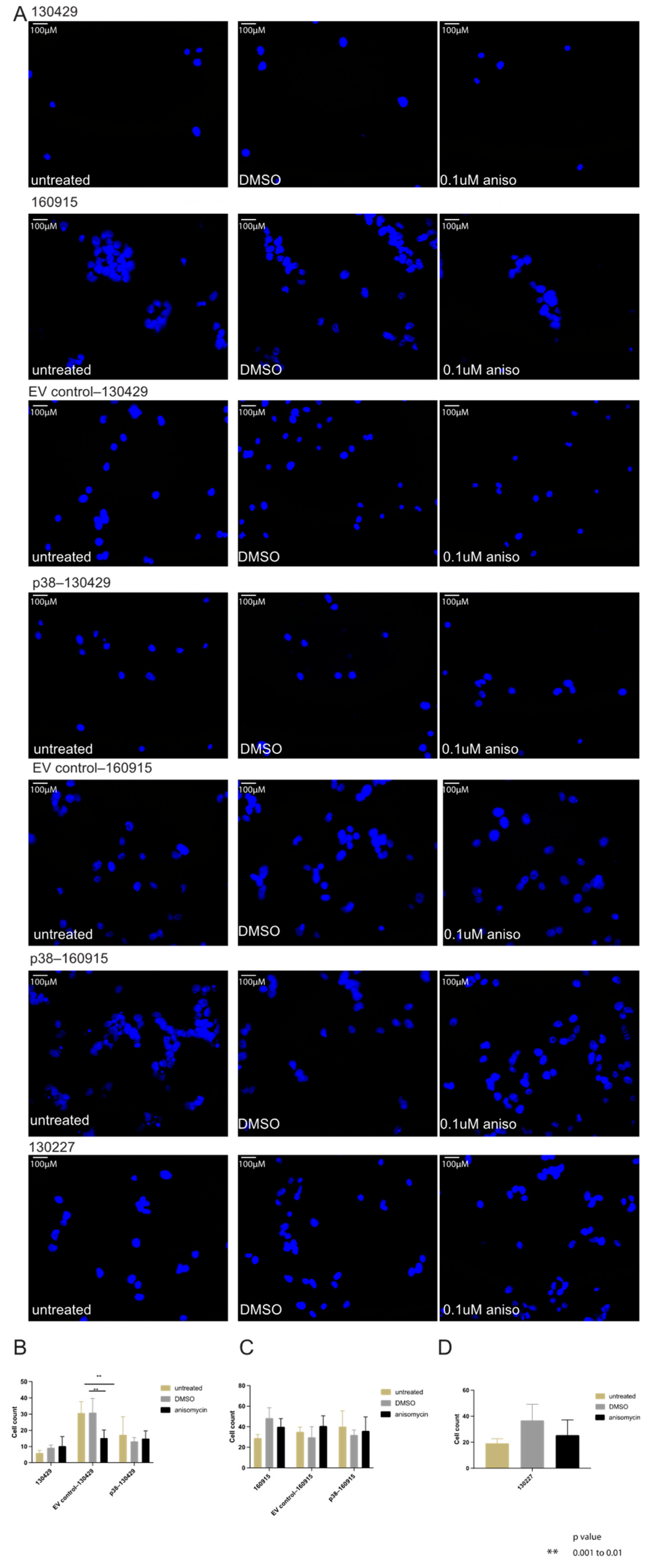
2.2. Activation of p38 in 160915 Spheroids Does Not Increase Invasion
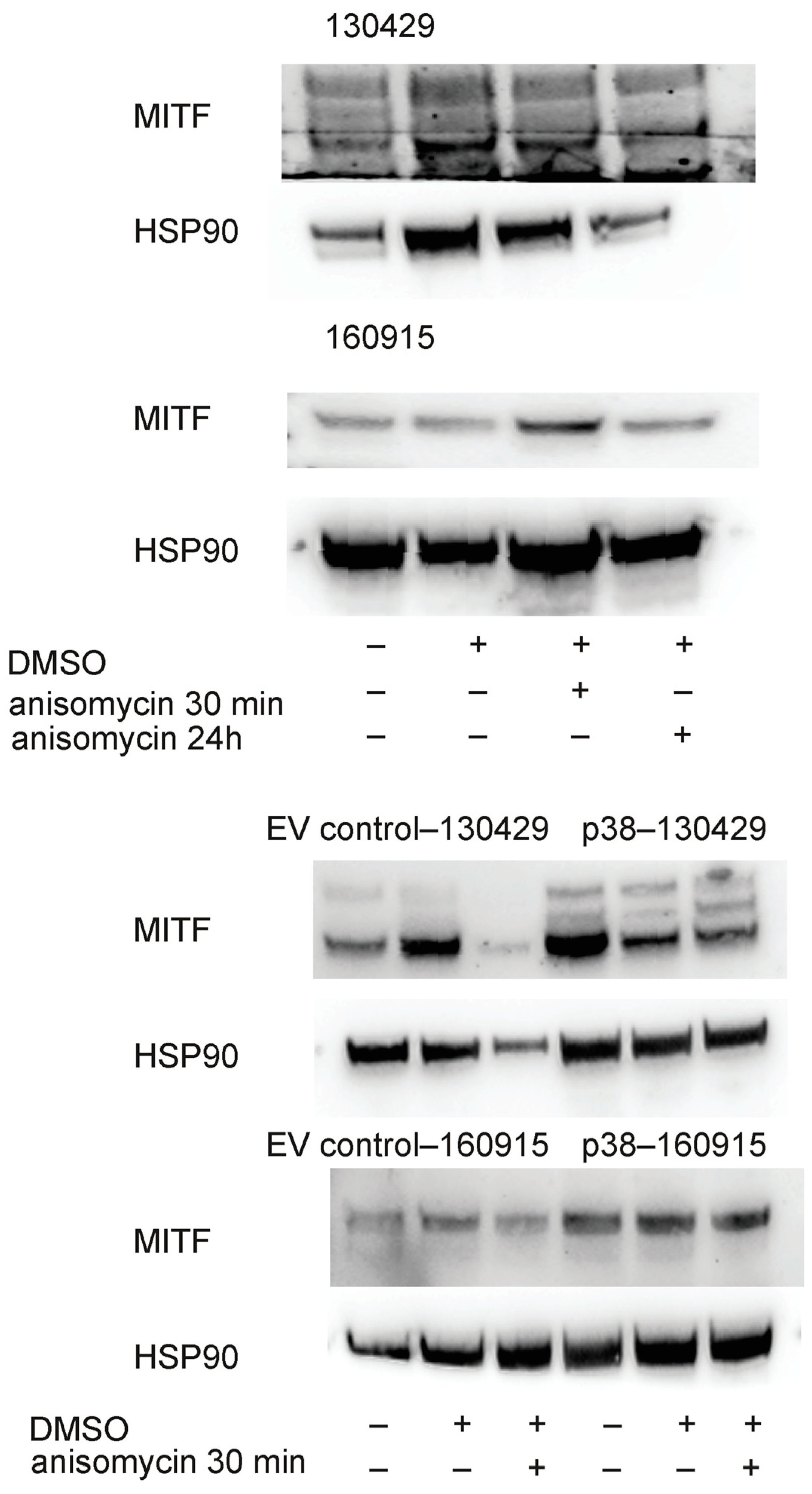
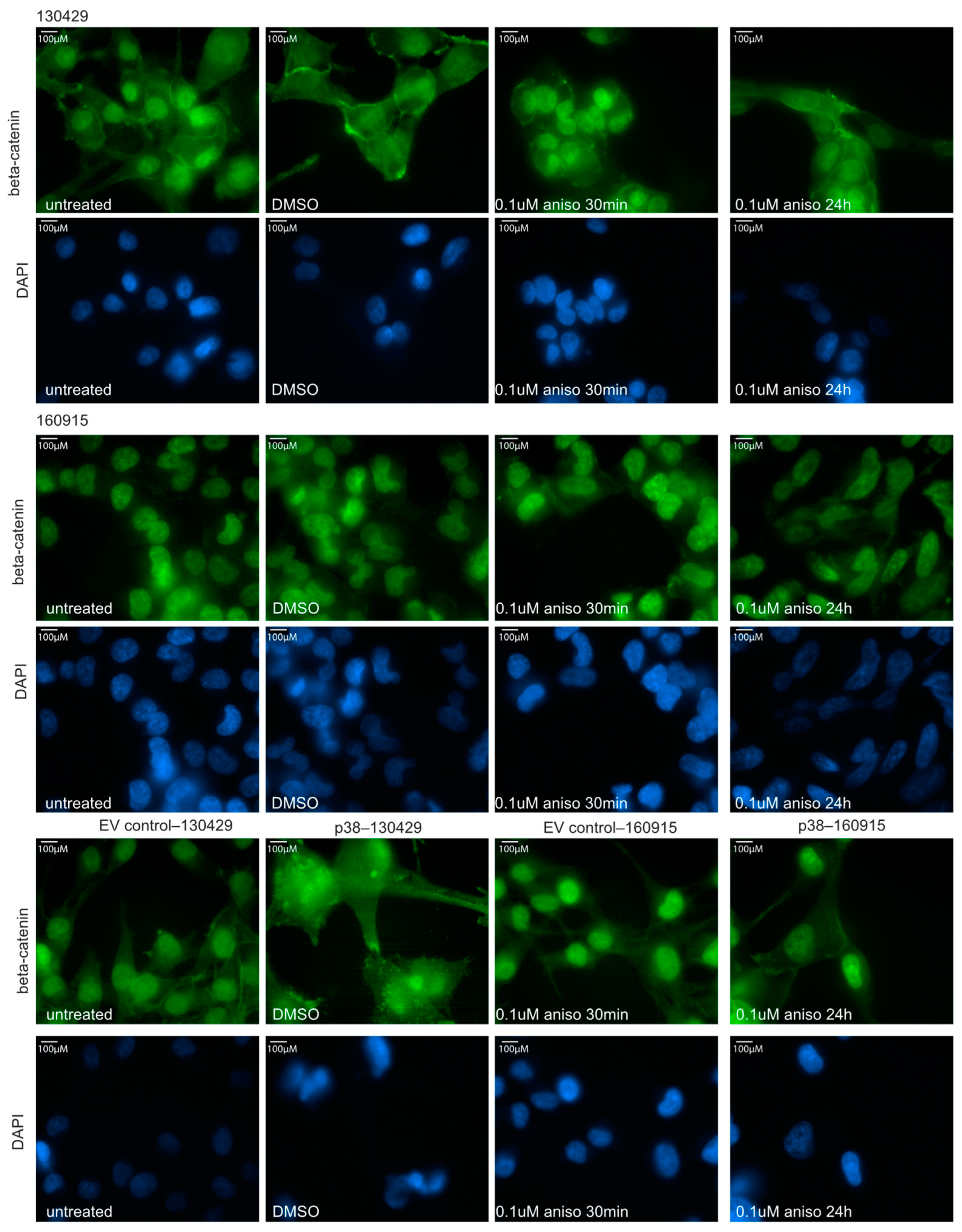
2.3. Activation of p38 Does Not Affect Beta-Catenin Localization or Cell Motility
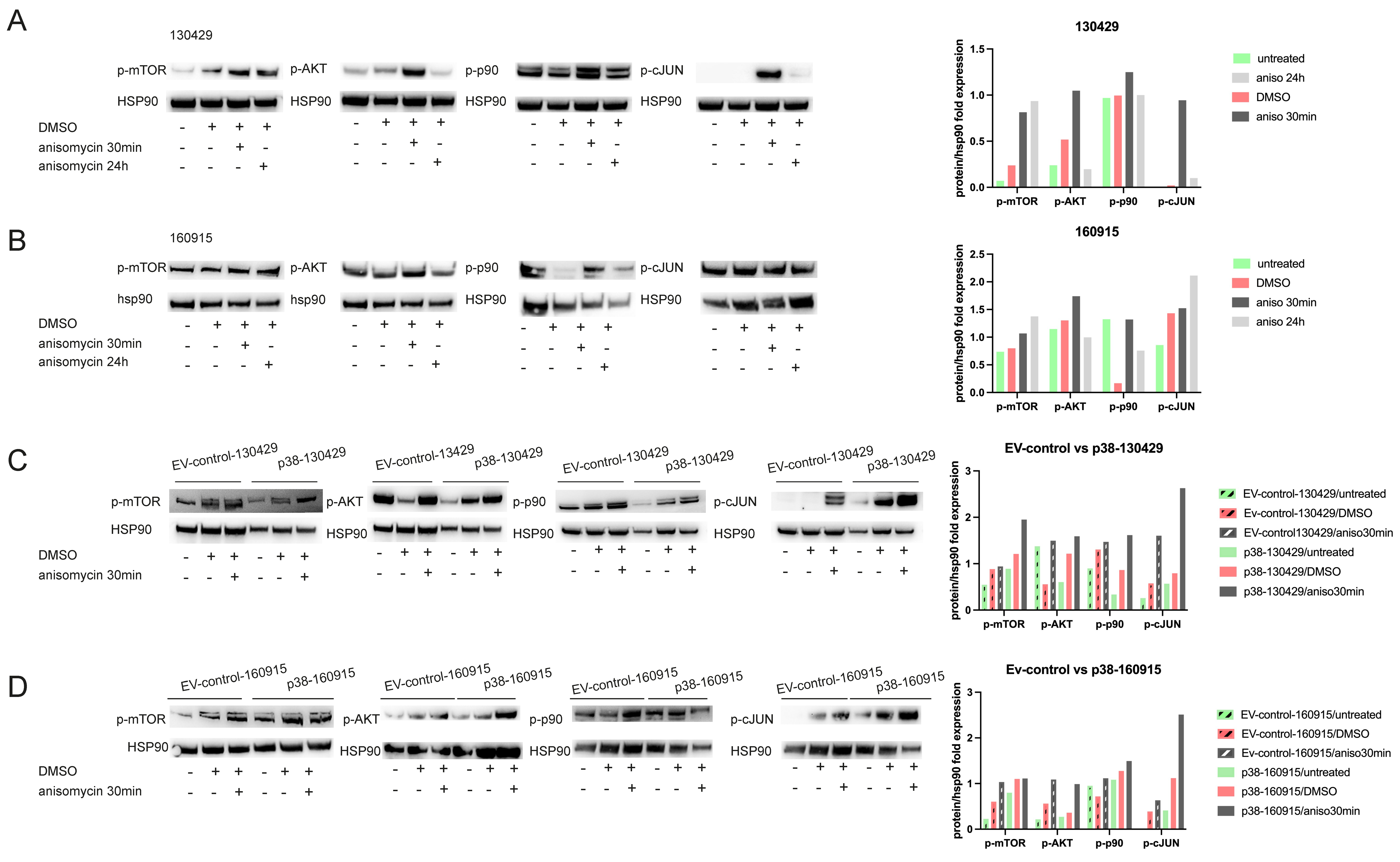
2.4. p38 Activation Results in Transient Activation of Phospho-p90 and AKT and Stable Activation of Phospho-mTOR
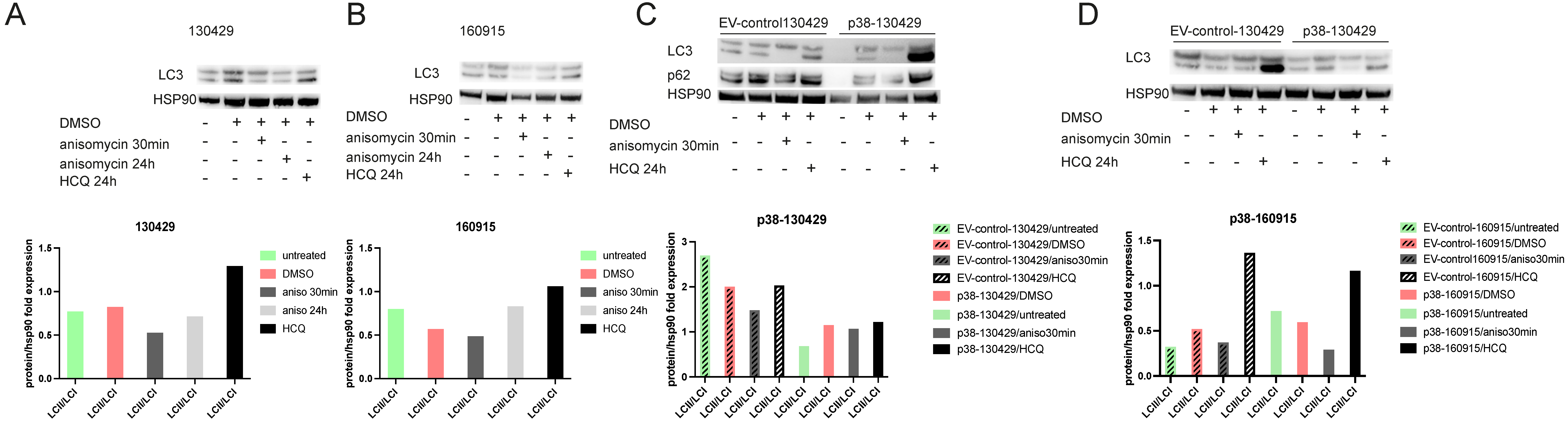
2.5. Phosphorylation of mTOR Leads to Reduced Autophagy
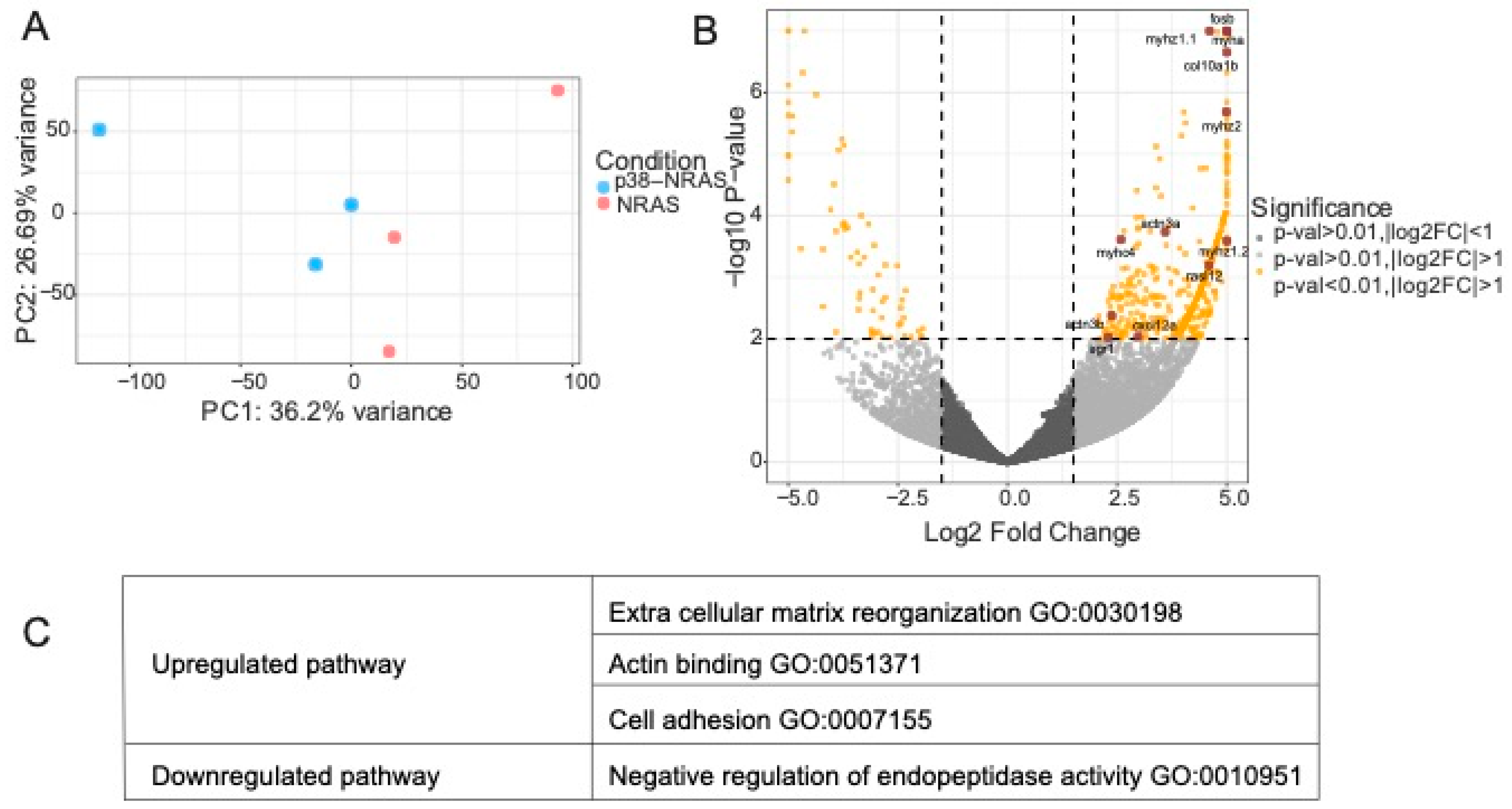
2.6. RNA Sequencing of Transgenic Zebrafish Overexpressing p38 and Actin Staining In Vitro Suggests Actin Remodeling with p38 Overexpression
3. Discussion
4. Methods
4.1. Cell Lines and Culture Conditions
4.2. Collagen-Invasion Assay in Spheroids
4.3. Motility Assay
4.4. RNA Extraction
4.5. Laser-Capture Microdissection (LCM)
4.6. Library Preparation
4.7. RNA Seq Analysis
4.8. Phospho Kinase Assay
4.9. Immunofluorescence Assay
4.10. Immunoblotting
4.11. Statistical Analysis and Blinding Approach
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guy, G.P., Jr.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982–2030. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 591–596. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Brito, C.; Barral, D.C.; Pojo, M. Subversion of Ras Small GTPases in Cutaneous Melanoma Aggressiveness. Front. Cell Dev. Biol. 2020, 8, 575223. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Kirchberger, M.C.; Ugurel, S.; Mangana, J.; Heppt, M.V.; Eigentler, T.K.; Berking, C.; Schadendorf, D.; Schuler, G.; Dummer, R.; Heinzerling, L. MEK inhibition may increase survival of NRAS-mutated melanoma patients treated with checkpoint blockade: Results of a retrospective multicentre analysis of 364 patients. Eur. J. Cancer 2018, 98, 10–16. [Google Scholar] [CrossRef]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco Targets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef]
- Hacker, E.; Nagore, E.; Cerroni, L.; Woods, S.L.; Hayward, N.K.; Chapman, B.; Montgomery, G.W.; Soyer, H.P.; Whiteman, D.C. NRAS and BRAF mutations in cutaneous melanoma and the association with MC1R genotype: Findings from Spanish and Austrian populations. J. Investig. Dermatol. 2013, 133, 1027–1033. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, J.W.; Kim, Y.S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef]
- Banik, I.; Cheng, P.F.; Dooley, C.M.; Travnickova, J.; Merteroglu, M.; Dummer, R.; Patton, E.E.; Busch-Nentwich, E.M.; Levesque, M.P. NRAS(Q61K) melanoma tumor formation is reduced by p38–MAPK14 activation in zebrafish models and NRAS-mutated human melanoma cells. Pigment. Cell Melanoma Res. 2020, 34, 150–162. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef]
- Bonney, E.A. Mapping out p38MAPK. Am. J. Reprod. Immunol 2017, 77, e12652. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Sanz-Ezquerro, J.J. p38γ and p38δ: From Spectators to Key Physiological Players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef]
- Chen, G.; Hitomi, M.; Han, J.; Stacey, D.W. The p38 pathway provides negative feedback for Ras proliferative signaling. J. Biol. Chem. 2000, 275, 38973–38980. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Imoto, I.; Inoue, J.; Onda, M.; Emi, M.; Inazawa, J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene 2007, 26, 1178–1187. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Peng, Y.; Austin, R.J.; van Eyndhoven, W.G.; Nguyen, K.C.; Gabriele, T.; McCurrach, M.E.; Marks, J.R.; Hoey, T.; et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat. Genet. 2002, 31, 133–134. [Google Scholar] [CrossRef]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFβRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef]
- Trempolec, N.; Dave-Coll, N.; Nebreda, A.R. SnapShot: p38 MAPK substrates. Cell 2013, 152, 924–924.e1. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Sugden, P.H.; Clerk, A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ. Res. 1998, 83, 345–352. [Google Scholar] [CrossRef]
- Han, Q.; Leng, J.; Bian, D.; Mahanivong, C.; Carpenter, K.A.; Pan, Z.K.; Han, J.; Huang, S. Rac1-MKK3-p38–MAPKAPK2 pathway promotes urokinase plasminogen activator mRNA stability in invasive breast cancer cells. J. Biol. Chem. 2002, 277, 48379–48385. [Google Scholar] [CrossRef]
- Huang, S.; New, L.; Pan, Z.; Han, J.; Nemerow, G.R. Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38alpha mitogen-activated protein kinase activity. J. Biol. Chem. 2000, 275, 12266–12272. [Google Scholar] [CrossRef]
- Kim, M.-S.; Lee, E.-J.; Kim, H.-R.C.; Moon, A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003, 63, 5454–5461. [Google Scholar]
- Puujalka, E.; Heinz, M.; Hoesel, B.; Friedl, P.; Schweighofer, B.; Wenzina, J.; Pirker, C.; Schmid, J.A.; Loewe, R.; Wagner, E.F.; et al. Opposing Roles of JNK and p38 in Lymphangiogenesis in Melanoma. J. Investig. Dermatol. 2016, 136, 967–977. [Google Scholar] [CrossRef]
- Wenzina, J.; Holzner, S.; Puujalka, E.; Cheng, P.F.; Forsthuber, A.; Neumüller, K.; Schossleitner, K.; Lichtenberger, B.M.; Levesque, M.P.; Petzelbauer, P. Inhibition of p38/MK2 Signaling Prevents Vascular Invasion of Melanoma. J. Investig. Dermatol. 2020, 140, 878–890.e5. [Google Scholar] [CrossRef]
- Naffa, R.; Vogel, L.; Hegedűs, L.; Pászty, K.; Tóth, S.; Kelemen, K.; Singh, N.; Reményi, A.; Kállay, E.; Cserepes, M.; et al. P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b. Cells 2020, 9, 1209. [Google Scholar] [CrossRef]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; Larue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Webster, M.R.; Weeraratna, A.T. A Wnt-er migration: The confusing role of β-catenin in melanoma metastasis. Sci. Signal. 2013, 6, pe11. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Wang, Q.J.; Ding, Y.; Kohtz, D.S.; Mizushima, N.; Cristea, I.M.; Rout, M.P.; Chait, B.T.; Zhong, Y.; Heintz, N.; Yue, Z. Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 2006, 26, 8057–8068. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, X.Y.; Jiang, H.; Yang, M.H.; Yuan, G.H.; Tang, Z.; Wang, H. Hydroxychloroquine facilitates autophagosome formation but not degradation to suppress the proliferation of cervical cancer SiHa cells. Oncol. Lett. 2014, 7, 1057–1062. [Google Scholar] [CrossRef]
- Maninová, M.; Vomastek, T. Dorsal stress fibers, transverse actin arcs, and perinuclear actin fibers form an interconnected network that induces nuclear movement in polarizing fibroblasts. FEBS J. 2016, 283, 3676–3693. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef]
- Larue, L.; Delmas, V. The WNT/Beta-catenin pathway in melanoma. Front. Biosci. A J. Virtual Libr. 2006, 11, 733–742. [Google Scholar] [CrossRef]
- Sinnberg, T.; Menzel, M.; Ewerth, D.; Sauer, B.; Schwarz, M.; Schaller, M.; Garbe, C.; Schittek, B. β-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS ONE 2011, 6, e23429. [Google Scholar] [CrossRef]
- Sinnberg, T.; Levesque, M.P.; Krochmann, J.; Cheng, P.F.; Ikenberg, K.; Meraz-Torres, F.; Niessner, H.; Garbe, C.; Busch, C. Wnt-signaling enhances neural crest migration of melanoma cells and induces an invasive phenotype. Mol. Cancer 2018, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Bischof, H.; Gilby, D.; Belloni, B.; Dummer, R.; Wellbrock, C. In melanoma, beta-catenin is a suppressor of invasion. Oncogene 2011, 30, 4531–4543. [Google Scholar] [CrossRef]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Kageshita, T.; Hamby, C.V.; Ishihara, T.; Matsumoto, K.; Saida, T.; Ono, T. Loss of beta-catenin expression associated with disease progression in malignant melanoma. Br. J. Dermatol. 2001, 145, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Maelandsmo, G.M.; Holm, R.; Nesland, J.M.; Fodstad, Ø.; Flørenes, V.A. Reduced beta-catenin expression in the cytoplasm of advanced-stage superficial spreading malignant melanoma. Clin. Cancer Res. 2003, 9, 3383–3388. [Google Scholar] [PubMed]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.-L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd ed). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12 (Suppl. S2), 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Kunz, J.; Hall, M.N. TOR2 is required for organization of the actin cytoskeleton in yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 13780–13785. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef]
- Han, Y.; Kim, S.J. Simvastatin-dependent actin cytoskeleton rearrangement regulates differentiation via the extracellular signal-regulated kinase-1/2 and p38 kinase pathways in rabbit articular chondrocytes. Eur. J. Pharm. 2018, 834, 197–205. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.H.; Lee, J.K.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef]
- De Wever, O.; Hendrix, A.; De Boeck, A.; Westbroek, W.; Braems, G.; Emami, S.; Sabbah, M.; Gespach, C.; Bracke, M. Modeling and quantification of cancer cell invasion through collagen type I matrices. Int. J. Dev. Biol. 2010, 54, 887–896. [Google Scholar] [CrossRef]
- Hendrix, A.; Maynard, D.; Pauwels, P.; Braems, G.; Denys, H.; Van den Broecke, R.; Lambert, J.; Van Belle, S.; Cocquyt, V.; Gespach, C.; et al. Effect of the secretory small GTPase Rab27B on breast cancer growth, invasion, and metastasis. J. Natl. Cancer Inst. 2010, 102, 866–880. [Google Scholar] [CrossRef]
- Beebe, E.; Motamed, Z.; Opitz, L.; Cheng, P.F.; Levesque, M.P.; Markkanen, E.; Feldmeyer, L. Defining the molecular landscape of cancer-associated stroma in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2022, 142, 3304–3312. [Google Scholar] [CrossRef]
- Amini, P.; Ettlin, J.; Opitz, L.; Clementi, E.; Malbon, A.; Markkanen, E. An optimised protocol for isolation of RNA from small sections of laser-capture microdissected FFPE tissue amenable for next-generation sequencing. BMC Mol. Biol. 2017, 18, 22. [Google Scholar] [CrossRef]
- Amini, P.; Nassiri, S.; Ettlin, J.; Malbon, A.; Markkanen, E. Next-generation RNA sequencing of FFPE subsections reveals highly conserved stromal reprogramming between canine and human mammary carcinoma. Dis. Model Mech. 2019, 12, dmm040444. [Google Scholar] [CrossRef]
- Ettlin, J.; Clementi, E.; Amini, P.; Malbon, A.; Markkanen, E. Analysis of Gene Expression Signatures in Cancer-Associated Stroma from Canine Mammary Tumours Reveals Molecular Homology to Human Breast Carcinomas. Int. J. Mol. Sci. 2017, 18, 1101. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Dilution | Manufacturer | Primary Antibody | Dilution | Manufacturer |
|---|---|---|---|---|---|
| Phospho cJUN 2361 | 1:1000 | Cell Signaling | Phospho-p90 9341 | 1:1000 | Cell Signaling |
| Total cJUN 9165 | 1:1000 | Cell Signaling | Total p90 9355 | 1:1000 | Cell Signaling |
| Phospho-mTORS2481 2974 | 1:1000 | Cell Signaling | Phospho-pRAS40 13175 | 1:1000 | Cell Signaling |
| Phospho-mTORS2448 5536 | 1:1000 | Cell Signaling | Total pRAS40 2610 | 1:1000 | Cell Signaling |
| Total mTOR 2983 | 1:1000 | Cell Signaling | Phospho-PDK1 3061 | 1:1000 | Cell Signaling |
| Phospho-AKTS437 4060 | 1:1000 | Cell Signaling | Total PDK1 3062 | 1:1000 | Cell Signaling |
| Phospho-AKTT803 13038 | 1:1000 | Cell Signaling | HSP90 4877 | 1:2000 | Cell Signaling |
| Pan AKT 4691 | 1:2000 | Cell Signaling | P62 sc28359 | 1:250 | Santa Cruz |
| Phospho-p70 9234 | 1:1000 | Cell Signaling | BECLIN 3738 | 1:1000 | Cell Signaling |
| Total p70 9202 | 1:1000 | Cell Signaling | ATG4 5299 | 1:1000 | Cell Signaling |
| Phospho GSK-ß 9322 | 1:1000 | Cell Signaling | LC3 ab51520 | 1:3000 | Abcam |
| Total GSK-ß 9315 | 1:1000 | Cell Signaling | MITF ab12039 | 1:1000 | Abcam |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banik, I.; Ghosh, A.; Beebe, E.; Burja, B.; Frank Bertoncelj, M.; Dooley, C.M.; Markkanen, E.; Dummer, R.; Busch-Nentwich, E.M.; Levesque, M.P. P38 Mediates Tumor Suppression through Reduced Autophagy and Actin Cytoskeleton Changes in NRAS-Mutant Melanoma. Cancers 2023, 15, 877. https://doi.org/10.3390/cancers15030877
Banik I, Ghosh A, Beebe E, Burja B, Frank Bertoncelj M, Dooley CM, Markkanen E, Dummer R, Busch-Nentwich EM, Levesque MP. P38 Mediates Tumor Suppression through Reduced Autophagy and Actin Cytoskeleton Changes in NRAS-Mutant Melanoma. Cancers. 2023; 15(3):877. https://doi.org/10.3390/cancers15030877
Chicago/Turabian StyleBanik, Ishani, Adhideb Ghosh, Erin Beebe, Blaž Burja, Mojca Frank Bertoncelj, Christopher M. Dooley, Enni Markkanen, Reinhard Dummer, Elisabeth M. Busch-Nentwich, and Mitchell P. Levesque. 2023. "P38 Mediates Tumor Suppression through Reduced Autophagy and Actin Cytoskeleton Changes in NRAS-Mutant Melanoma" Cancers 15, no. 3: 877. https://doi.org/10.3390/cancers15030877
APA StyleBanik, I., Ghosh, A., Beebe, E., Burja, B., Frank Bertoncelj, M., Dooley, C. M., Markkanen, E., Dummer, R., Busch-Nentwich, E. M., & Levesque, M. P. (2023). P38 Mediates Tumor Suppression through Reduced Autophagy and Actin Cytoskeleton Changes in NRAS-Mutant Melanoma. Cancers, 15(3), 877. https://doi.org/10.3390/cancers15030877