

Causal Link of Human Papillomavirus in Barrett Esophagus and Adenocarcinoma: Are We There Yet?

Abstract
Simple Summary
Abstract
1. Introduction
2. Literature Search and Study Selection
3. HPV as a Common Denominator in Esophageal Adenocarcinoma and Cervical Cancer
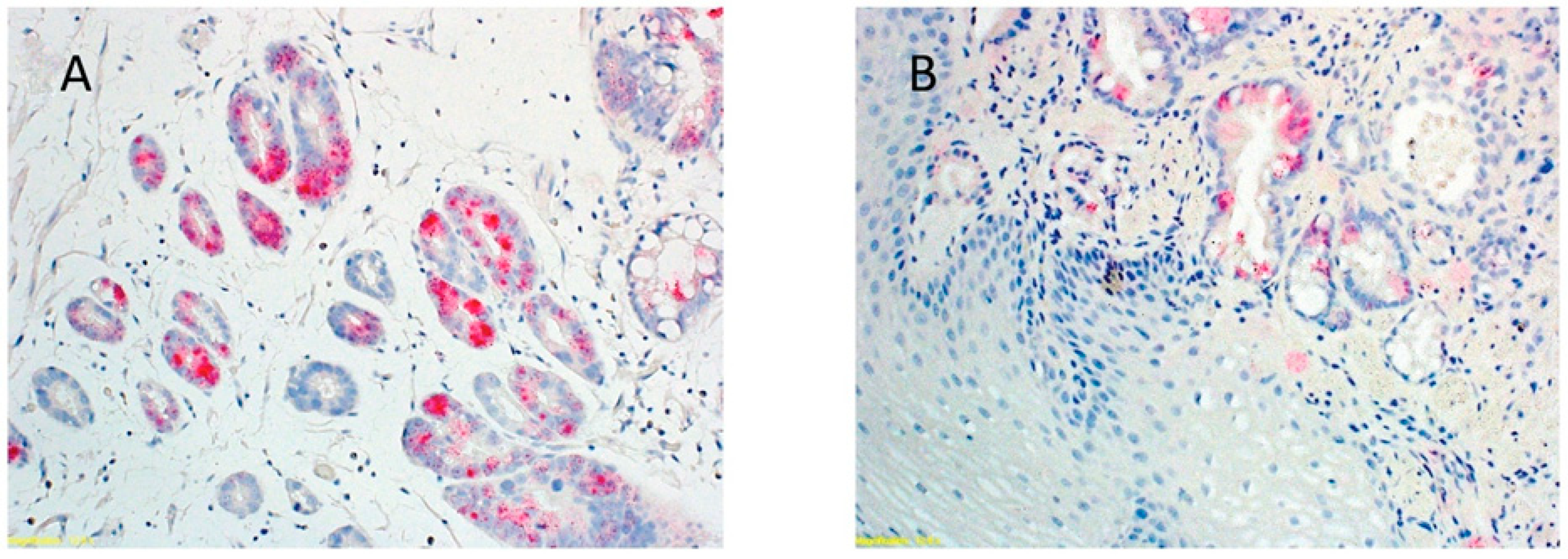
4. Establishing Causality
5. Consistency across Studies and Biological Gradient (Probable Relationship)
6. Strength of Association and Biological Gradient (Probable Relationship)
7. Analogy and Biological Gradient (Probable Relationship)
8. Analogy and Experimentation [Probable Relationship]
9. Comparative Genomic Analysis Reveals Distinct Differences between HPV-Positive and HPV Negative OAC (Probable Relationship)
10. Experimentation (Evidence for Possible Relationship)
11. HPV and Survival in OAC (Probable Relationship)
12. HPV and Esophageal Cancer Cell Lines
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018, a worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- White, M.K.; Pagano, J.S.; Khalili, K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin. Microbiol. Rev. 2014, 27, 463–481. [Google Scholar] [CrossRef]
- Biological Agents. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–441. [Google Scholar]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012, a synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466. [Google Scholar] [CrossRef]
- Humans IWGotEoCRt. Human papillomaviruses. IARC Monogr. Eval. Carcinog. Risks Hum. 1995, 64, 1–378. [Google Scholar]
- Bosch, F.X.; Lorincz, A.; Munoz, N.; Meijer, C.J.; Shah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Castellsague, X.; Diaz, M.; de Sanjose, S.; Muñoz, N.; Herrero, R.; Franceschi, S.; Peeling, R.W.; Ashley, R.; Smith, J.S.; Snijders, P.J.F.; et al. Worldwide human papillomavirus etiology of cervical adenocarcinoma and its cofactors: Implications for screening and prevention. J. Natl. Cancer Inst. 2006, 98, 303–315. [Google Scholar]
- Stolnicu, S.; Barsan, I.; Hoang, L.; Patel, P.; Terinte, C.; Pesci, A.; Aviel-Ronen, S.; Kiyokawa, T.; Alvarado-Cabrero, I.; Pike, M.C.; et al. International Endocervical Adenocarcinoma Criteria and Classification (IECC): A new pathogenetic classification for invasive adenocarcinomas of the endocervix. Am. J. Surg. Pathol. 2018, 42, 214–226. [Google Scholar] [CrossRef]
- Rajendra, S.; Robertson, I.K. Similar immunogenetics of Barrett’s oesophagus and cervical neoplasia: Is HPV the common denominator? J. Clin. Pathol. 2010, 63, 1–3. [Google Scholar] [CrossRef]
- Odze, R.; Antonioli, D.; Shocket, D.; Noble-Topham, S.; Goldman, H.; Upton, M. Esophageal squamous papillomas. A clinicopathologic study of 38 lesions and analysis for human papillomavirus by the polymerase chain reaction. Am. J. Surg. Pathol. 1993, 17, 803–812. [Google Scholar] [CrossRef]
- Takeshita, K.; Murata, S.I.; Mitsufuji, S.; Wakabayashi, N.; Kataoka, K.; Tsuchihashi, Y.; Okanoue, T. Clinicopathological characteristics of esophageal squamous papillomas in Japanese patients—With comparison of findings from Western countries. Acta Histochem. Cytochem. 2006, 39, 23–30. [Google Scholar] [CrossRef]
- Chang, C.; Worrell, S.G. Viruses and esophageal cancer. Dis. Esophagus 2020, 33, doaa036. [Google Scholar] [CrossRef]
- Campo, M.S. Papillomas and cancer in cattle. Cancer Surv. 1987, 6, 39–54. [Google Scholar]
- Jarrett, W.F.H.; McNeil, P.E.; Grimshaw, W.T.R.; Selman, I.E.; McIntyre, W.I.M. High incidence area of cattle cancer with a possible interaction between an environmental carcinogen and a papilloma virus. Nature 1978, 274, 215–217. [Google Scholar] [CrossRef]
- Black, P.H.; Hartley, J.W.; Rowe, W.P.; Huebner, R.J. Transformation of bovine tissue culture cells by bovine papilloma virus. Nature 1963, 199, 1016–1018. [Google Scholar] [CrossRef]
- Lagergren, J.; Bergstrom, R.; Lindgren, A.; Nyren, O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N. Engl. J. Med. 1999, 340, 825–831. [Google Scholar] [CrossRef]
- Bhat, S.; Coleman, H.G.; Yousef, F.; Johnston, B.T.; McManus, D.T.; Gavin, A.T.; Murray, L.J. Risk of malignant progression in Barrett’s esophagus patients: Results from a large population-based study. J. Natl. Cancer Inst. 2011, 103, 1049–1057. [Google Scholar] [CrossRef]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sorensen, H.T.; Funch-Jensen, P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef]
- Shaheen, N.J.; Crosby, M.A.; Bozymski, E.M.; Sandler, R.S. Is there publication bias in the reporting of cancer risk in Barrett’s esophagus? Gastroenterology 2000, 119, 333–338. [Google Scholar] [CrossRef]
- Yousef, F.; Cardwell, C.; Cantwell, M.M.; Galway, K.; Johnston, B.T.; Murray, L. The incidence of esophageal cancer and high-grade dysplasia in Barrett’s esophagus: A systematic review and meta-analysis. Am. J. Epidemiol. 2008, 168, 237–249. [Google Scholar] [CrossRef]
- Pohl, H.; Welch, H.G. The Role of Overdiagnosis and Reclassification in the Marked Increase of Esophageal Adenocarcinoma Incidence. J. Natl. Cancer Inst. 2005, 97, 142–146. [Google Scholar] [CrossRef]
- Pohl, H.; Sirovich, B.; Welch, H.G. Esophageal adenocarcinoma incidence: Are we reaching the peak? Cancer Epidemiol. Biomarkers. Prev. 2010, 19, 1468–1470. [Google Scholar] [CrossRef]
- Lagergren, J.; Mattsson, F. No further increase in the incidence of esophageal adenocarcinoma in Sweden. Int. J. Cancer 2011, 129, 513–516. [Google Scholar] [CrossRef]
- Rajendra, S.; Wang, B.; Snow, E.T.; Sharma, P.; Pavey, D.; Merrett, N.; Ball, M.J.; Brain, T.; Fernando, R.; Robertson, I.K. Transcriptionally active human papillomavirus is strongly associated with Barrett’s dysplasia and esophageal adenocarcinoma. Am. J. Gastroenterol. 2013, 108, 1082–1093. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- Nasman, A.; Attner, P.; Hammarstedt, L.; Du, J.; Eriksson, M.; Giraud, G.; Ahrlund-Richter, S.; Marklund, L.; Romanitan, M.; Lindquist, D.; et al. Incidence of human papillomavirus (HPV) positive tonsillar carcinoma in Stockholm, Sweden: An epidemic of viral-induced carcinoma? Int. J. Cancer 2009, 125, 362–366. [Google Scholar] [CrossRef]
- Hocking, J.S.; Stein, A.; Conway, E.L.; Regan, D.; Grulich, A.; Law, M.; Brotherton, J.M. Head and neck cancer in Australia between 1982 and 2005 show increasing incidence of potentially HPV-associated oropharyngeal cancers. Br. J. Cancer 2011, 104, 886–891. [Google Scholar] [CrossRef]
- Pagano, J.S.; Blaser, M.; Buendia, M.A.; Damania, B.; Khalili, K.; Raab-Traub, N.; Roizman, B. Infectious agents and cancer: Criteria for a causal relation. Semin. Cancer Biol. 2004, 14, 453–471. [Google Scholar] [CrossRef]
- Koch, R. Zur Untersuchung von Pathogenen Organismen. Mitthdungen Uus Dem Kais. Gesundbeitsamte 1881, 1, 1–48. [Google Scholar]
- Hill, A.B. The Environment and Disease: Association or Causation? Proc. R. Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef]
- Lipkin, W.I. The changing face of pathogen discovery and surveillance. Nat. Rev. Microbiol. 2013, 11, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, C.; Yang, Y.; Zhou, F.; Li, M.; Jin, Q.; Gao, L. Systematic review with meta-analysis: The association between human papillomavirus infection and oesophageal cancer. Aliment. Pharm. 2014, 39, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, A.T.; Graham, S.; McShane, C.M.; Doyle, J.; Tommasino, M.; Johnston, B.; Jamison, J.; James, J.A.; McManus, D.; Anderson, L.A. The prevalence of viral agents in esophageal adenocarcinoma and Barrett’s esophagus: A systematic review. Eur. J. Gastroenterol. Hepatol. 2017, 29, 817–825. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Hollier, J.M.; Gravitt, P.; Alsarraj, A.; Younes, M. Human papillomavirus and the risk of Barrett’s esophagus. Dis. Esophagus 2013, 26, 517–521. [Google Scholar] [CrossRef]
- Rai, N.; Jenkins, G.J.; McAdam, E.; Hibbitts, S.J.; Fiander, A.N.; Powell, N.G. Human papillomavirus infection in Barrett’s oesophagus in the UK: An infrequent event. J. Clin. Virol. 2008, 43, 250–252. [Google Scholar] [CrossRef]
- Iyer, A.; Rajendran, V.; Adamson, C.S.; Peng, Z.; Cooper, K.; Evans, M.F. Human papillomavirus is detectable in Barrett’s esophagus and esophageal carcinoma but is unlikely to be of any etiologic significance. J. Clin. Virol. 2011, 50, 205–208. [Google Scholar] [CrossRef]
- Antonsson, A.; Knight, L.; Whiteman, D.C. Human papillomavirus not detected in esophageal adenocarcinoma tumor specimens. Cancer Epidemiol. 2016, 41, 96–98. [Google Scholar] [CrossRef]
- Wang, B.; Rajendra, S.; Pavey, D.; Sharma, P.; Merrett, N.; Wu, X.; Snow, E.T.; Kumbhari, V.; Ball, M.J.; Robertson, I.K. Viral load and integration status of high-risk human papillomaviruses in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Am. J. Gastroenterol. 2013, 108, 1814–1816. [Google Scholar] [CrossRef]
- Hussain, S.; Rani, J.; Tulsyan, S.; Sisodiya, S.; Chikara, A.; Nazir, S.U.; Srivastava, A.; Khan, A.; Dash, N.R.; Anoop Saraya, A.; et al. Influence of HPV infection in esophageal cancer: A systematic review and meta-analysis. Gene Rep. 2022, 28, 101640. [Google Scholar] [CrossRef]
- Rajendra, S.; Robertson, I.K. Barrett’s Oesophagus; Acid Hum. Papilloma Virus? J. Clin. Virol. 2009, 44, 176. [Google Scholar] [CrossRef]
- Baldwin, A.; Münger, K. Molecular events associated with human papillomavirus-induced human cancers. In Viral Oncology: Basic Science and Clinical Applications; Wiley-Blackwell: Hoboken, NJ, USA, 2010; pp. 23–35. [Google Scholar]
- Rajendra, S.; Sharma, P. Transforming human papillomavirus infection and the esophageal transformation zone: Prime time for total excision/ablative therapy? Dis. Esophagus. 2019, 32, doz008. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F. Chapter 2, The burden of HPV-related cancers. Vaccine 2006, 24, S11–S25. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.; Lowy, D. Papillomaviruses. Fields Virol. 2007, 2, 2299–2354. [Google Scholar]
- Ng, W.K.; Cheung, L.K.; Li, A.S.; Cheung, F.M.; Chow, J.C. Transitional cell metaplasia of the uterine cervix is related to human papillomavirus: Molecular analysis in seven patients with cytohistologic correlation. Cancer 2002, 96, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.M.; Graham, T.A.; Simpson, A.; Humphries, A.; Burch, N.; Rodriguez-Justo, M.; Novelli, M.; Harrison, R.; Wright, N.A.; McDonald, S.A.; et al. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut 2012, 61, 1380–1389. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef]
- Josefsson, A.M.; Magnusson, P.K.; Ylitalo, N.; Sørensen, P.; Qwarforth-Tubbin, P.; Andersen, P.K.; Melbye, M.; Adami, H.O.; Gyllensten, U.B. Viral load of human papilloma virus 16 as a determinant for development of cervical carcinoma in situ: A nested case-control study. Lancet 2000, 355, 2189–2193. [Google Scholar] [CrossRef]
- Si, H.X.; Tsao, S.W.; Poon, C.S.; Wang, L.D.; Wong, Y.C.; Cheung, A.L. Viral load of HPV in esophageal squamous cell carcinoma. Int. J. Cancer J. Int. Cancer 2003, 103, 496–500. [Google Scholar] [CrossRef]
- Deng, Z.; Hasegawa, M.; Kiyuna, A.; Matayoshi, S.; Uehara, T.; Agena, S.; Yamashita, Y.; Ogawa, K.; Maeda, H.; Suzuki, M. Viral load, physical status, and E6/E7 mRNA expression of human papillomavirus in head and neck squamous cell carcinoma. Head Neck 2013, 35, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Boshart, M.; Gissmann, L.; Ikenberg, H.; Kleinheinz, A.; Scheurlen, W.; zur Hausen, H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984, 3, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Saunier, M.; Monnier-Benoit, S.; Mauny, F.; Ana Dalstein, V.; Briolat, J.; Riethmuller, D.; Kantelip, B.; Schwarz, E.; Mougin, C.; Prétet, J.L. Analysis of human papillomavirus type 16 (HPV16) DNA load and physical state for identification of HPV16-infected women with high-grade lesions or cervical carcinoma. J. Clin. Microbiol. 2008, 46, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762. [Google Scholar] [CrossRef]
- Smith, J.S.; Lindsay, L.; Hoots, B.; Keys, J.; Franceschi, S.; Winer, R.; Clifford, G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. Cancer 2007, 121, 621–632. [Google Scholar] [CrossRef]
- Chow, L.T.; Broker, T.R.; Steinberg, B.M. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 2010, 118, 422–449. [Google Scholar] [CrossRef]
- Ho, G.Y.F.; Bierman, R.; Beardsley, L.; Chang, C.J.; Burk, R.D. Natural History of Cervicovaginal Papillomavirus Infection in Young Women. N. Engl. J. Med. 1998, 338, 423–428. [Google Scholar] [CrossRef]
- Schiffman, M.; Castle, P.E. Human papillomavirus: Epidemiology and public health. Arch. Pathol. Lab. Med. 2003, 127, 930–934. [Google Scholar] [CrossRef]
- Bae, J.H.; Kim, C.J.; Park, T.C.; Namkoong, S.E.; Park, J.S. Persistence of human papillomavirus as a predictor for treatment failure after loop electrosurgical excision procedure. Int. J. Gynecol. Cancer 2007, 17, 1271–1277. [Google Scholar] [CrossRef]
- Rajendra, S.; Wang, B.; Pavey, D.; Sharma, P.; Yang, T.; Lee, C.S.; Gupta, N.; Ball, M.J.; Gill, R.S.; Wu, X. Persistence of Human Papillomavirus, Overexpression of p53, and Outcomes of Patients After Endoscopic Ablation of Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2015, 13, 1364–1368.e5. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Kaye, P.V.; Haider, S.A.; James, P.D.; Soomro, I.; Catton, J.; Parsons, S.L.; Ragunath, K.; Ilyas, M. Novel staining pattern of p53 in Barrett’s dysplasia--the absent pattern. Histopathology 2010, 57, 933–935. [Google Scholar] [CrossRef]
- Scheffner, M.; Takahashi, T.; Huibregtse, J.M.; Minna, J.D.; Howley, P.M. Interaction of the human papillomavirus type 16 E6 oncoprotein with wild-type and mutant human p53 proteins. J. Virol. 1992, 66, 5100–5105. [Google Scholar] [CrossRef]
- Brennan, J.A.; Boyle, J.O.; Koch, W.M.; Goodman, S.N.; Hruban, R.H.; Eby, Y.J.; Couch, M.J.; Forastiere, A.A.; Sidransky, D. Association between cigarette smoking and mutation of the p53 gene in squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 1995, 332, 712–717. [Google Scholar] [CrossRef]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef]
- Lechner, M.; Frampton, G.M.; Fenton, T.; Feber, A.; Palmer, G.; Jay, A.; Pillay, N.; Forster, M.; Cronin, M.T.; Lipson, D.; et al. Targeted next-generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV+ and HPV− tumors. Genome Med. 2013, 5, 49. [Google Scholar] [CrossRef]
- Agrawal, N.; Jiao, Y.; Bettegowda, C.; Hutfless, S.M.; Wang, Y.; David, S.; Cheng, Y.; Twaddell, W.S.; Latt, N.L.; Shin, E.J.; et al. Comparative Genomic Analysis of Esophageal Adenocarcinoma and Squamous Cell Carcinoma. Cancer Discov. 2012, 2, 899–905. [Google Scholar] [CrossRef]
- Smeets, S.J.; Braakhuis, B.J.; Abbas, S.; Snijders, P.J.; Ylstra, B.; van de Wiel, M.A.; Meijer, G.A.; Leemans, C.R.; Brakenhoff, R.H. Genome-wide DNA copy number alterations in head and neck squamous cell carcinomas with or without oncogene-expressing human papillomavirus. Oncogene 2006, 25, 2558–2564. [Google Scholar] [CrossRef]
- Richards, K.L.; Zhang, B.; Baggerly, K.A.; Colella, S.; Lang, J.C.; Schuller, D.E.; Krahe, R. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLoS ONE 2009, 4, e4941. [Google Scholar] [CrossRef]
- Rajendra, S.; Wang, B.; Merrett, N.; Sharma, P.; Humphris, J.; Lee, H.C.; Wu, J. Genomic analysis of HPV-positive versus HPV-negative oesophageal adenocarcinoma identifies a differential mutational landscape. J. Med. Genet. 2016, 53, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef]
- Rajendra, S.; Yang, T.; Xuan, W.; Sharma, P.; Pavey, D.; Lee, C.S.; Le, S.; Collins, J.; Wang, B. Active human papillomavirus involvement in Barrett’s dysplasia and oesophageal adenocarcinoma is characterized by wild-type p53 and aberrations of the retinoblastoma protein pathway. Int. J. Cancer 2017, 141, 2037–2049. [Google Scholar] [CrossRef]
- Rajendra, S.; Xuan, W.; Hufnagel, K.; Sharma, P.; Pavey, D.; Alhajjiri, N.; Rattan, A.; Wang, B. Antibodies against human papillomavirus proteins in Barrett’s dysplasia and intramucosal esophageal adenocarcinoma. Ann. N. Y. Acad. Sci. 2020, 1470, 44–56. [Google Scholar] [CrossRef]
- Smith, E.M.; Ritchie, J.M.; Pawlita, M.; Rubenstein, L.M.; Haugen, T.H.; Turek, L.P.; Hamsikova, E. Human papillomavirus seropositivity and risks of head and neck cancer. Int. J. Cancer 2007, 120, 825–832. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Johansson, M.; Waterboer, T.; Kaaks, R.; Chang-Claude, J.; Drogen, D.; Tjønneland, A.; Overvad, K.; Quirós, J.R.; González, C.A.; et al. Evaluation of human papillomavirus antibodies and risk of subsequent head and neck cancer. J. Clin. Oncol. 2013, 31, 2708–2715. [Google Scholar] [CrossRef]
- Achour, M.; Zeghal, D.; Kochbati, L.; Kahla, S.; Zouari, F.; Maalej, M.; Oueslati, R. Antibody response for L1, E6 and E7 HPV 16 and HPV 18 antigens in Tunisian women with cervical cancer and controls. J. Immunoass. Immunochem. 2008, 29, 266–280. [Google Scholar] [CrossRef]
- Meschede, W.; Zumbach, K.; Braspenning, J.; Scheffner, M.; Benitez-Bribiesca, L.; Luande, J.; Gissmann, L.; Pawlita, M. Antibodies against early proteins of human papillomaviruses as diagnostic markers for invasive cervical cancer. J. Clin. Microbiol. 1998, 36, 475–480. [Google Scholar] [CrossRef]
- Reuschenbach, M.; Waterboer, T.; Wallin, K.L.; Einenkel, J.; Dillner, J.; Hamsikova, E.; Eschenbach, D.; Zimmer, H.; Heilig, B.; Kopitz, J.; et al. Characterization of humoral immune responses against p16, p53, HPV16 E6 and HPV16 E7 in patients with HPV-associated cancers. Int. J. Cancer J. Int. Cancer 2008, 123, 2626–2631. [Google Scholar] [CrossRef]
- Frazer, I.H. Interaction of human papillomaviruses with the host immune system: A well evolved relationship. Virology 2009, 384, 410–414. [Google Scholar] [CrossRef]
- Wang, S.S.; Schiffman, M.; Herrero, R.; Carreon, J.; Hildesheim, A.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. Determinants of human papillomavirus 16 serological conversion and persistence in a population-based cohort of 10,000 women in Costa Rica. Br. J. Cancer 2004, 91, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, D.; Wichmann, G.; Baboci, L.; Michel, A.; Höfler, D.; Wiesenfarth, M.; Schroeder, L.; Boscolo-Rizzo, P.; Herold-Mende, C.; Dyckhoff, G.; et al. Sensitivity and specificity of antibodies against HPV16 E6 and other early proteins for the detection of HPV16-driven oropharyngeal squamous cell carcinoma. Int. J. Cancer 2017, 140, 2748–2757. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Wang, Z.; Bergström, R.; Dillner, J.; Nyrén, O. Human papillomavirus infection and esophageal cancer: A nationwide seroepidemiologic case-control study in Sweden. J. Natl. Cancer Inst. 1999, 91, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Qiao, G.; Hubbert, N.L.; Li, L.; Sun, C.; Wang, Y.; Yan, M.; Xu, D.; Li, Y.; Lowy, D.R.; et al. Serologic association between human papillomavirus type 16 infection and esophageal cancer in Shaanxi Province, China. J. Natl. Cancer Inst. 1996, 88, 1467–1471. [Google Scholar] [CrossRef]
- Bjørge, T.; Hakulinen, T.; Engeland, A.; Jellum, E.; Koskela, P.; Lehtinen, M.; Luostarinen, T.; Paavonen, J.; Sapp, M.; Schiller, J.; et al. A prospective, seroepidemiological study of the role of human papillomavirus in esophageal cancer in Norway. Cancer Res. 1997, 57, 3989–3992. [Google Scholar]
- Dillner, J.; Knekt, P.; Schiller, J.T.; Hakulinen, T. Prospective seroepidemiological evidence that human papillomavirus type 16 infection is a risk factor for oesophageal squamous cell carcinoma. BMJ 1995, 311, 1346. [Google Scholar] [CrossRef]
- Rajendra, S.; Xuan, W.; Merrett, N.; Sharma, P.; Sharma, P.; Pavey, D.; Yang, T.; Santos, L.D.; Sharaiha, O.; Pande, G.; et al. Survival Rates for Patients with Barrett High-grade Dysplasia and Esophageal Adenocarcinoma with or Without Human Papillomavirus Infection. JAMA Netw. Open 2018, 1, e181054. [Google Scholar] [CrossRef]
- Fakhry, C.; Westra, W.H.; Li, S.; Li, S.; Cmelak, A.; Ridge, J.A.; Pinto, H.; Forastiere, A.; Gillison, M.L. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J. Natl. Cancer Inst. 2008, 100, 261–269. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Cao, F.; Zhang, W.; Zhang, F.; Han, H.; Xu, J.; Cheng, Y. Prognostic significance of high-risk human papillomavirus and p16(INK4A) in patients with esophageal squamous cell carcinoma. Int. J. Clin. Exp. Med. 2014, 7, 3430–3438. [Google Scholar]
- Kumar, R.; Ghosh, S.K.; Verma, A.K.; Talukdar, A.; Deka, M.K.; Wagh, M.; Bahar, H.M.; Tapkire, R.; Chakraborty, K.P.; Kannan, R.R.; et al. p16 Expression as a Surrogate Marker for HPV Infection in Esophageal Squamous Cell Carcinoma can Predict Response to Neo-Adjuvant Chemotherapy. Asian Pac. J. Cancer Prev. 2015, 16, 7161–7165. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Wang, Y.C.; Lee, C.T.; Chang, C.Y.; Lo, J.L.; Kuo, Y.H.; Hsu, Y.C.; Mo, L.R. The impact of human papillomavirus infection on the survival and treatment response of patients with esophageal cancers. J. Dig. Dis. 2015, 16, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Furihata, M.; Ohtsuki, Y.; Ogoshi, S.; Takahashi, A.; Tamiya, T.; Ogata, T. Prognostic significance of human papillomavirus genomes (type-16,-18) and aberrant expression of p53 protein in human esophageal cancer. Int. J. Cancer 1993, 54, 226–230. [Google Scholar] [CrossRef]
- Da Costa, A.M.; Fregnani, J.H.T.G.; Pastrez, P.R.A.; Mariano, V.S.; Silva, E.M.; Neto, C.S.; Guimarães, D.P.; Villa, L.L.; Sichero, L.; Syrjanen, K.J.; et al. HPV infection and p53 and p16 expression in esophageal cancer: Are they prognostic factors? Infect. Agents Cancer 2017, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Dreilich, M.; Bergqvist, M.; Moberg, M.; Brattström, D.; Gustavsson, I.; Bergström, S.; Wanders, A.; Hesselius, P.; Wagenius, G.; Gyllensten, U. High-Risk Hum. Papilloma Virus (HPV) Surviv. Patients Esophageal Carcinoma: A Pilot Study. BMC Cancer 2006, 6, 94. [Google Scholar] [CrossRef]
- Antonsson, A.; Green, A.C.; Mallitt, K.A.; O’Rourke, P.K.; Pandeya, N.; Pawlita, M.; Waterboer, T.; Neale, R.E. Prevalence and stability of antibodies to 37 human papillomavirus types—A population-based longitudinal study. Virology 2010, 407, 26–32. [Google Scholar] [CrossRef]
- Hippeläinen, M.; Eskelinen, M.; Lipponen, P.K.; Chang, F.; Syrjänen, K. Mitotic activity index, volume corrected mitotic index and human papilloma-virus suggestive morphology are not prognostic factors in carcinoma of the oesophagus. Anticancer Res. 1993, 13, 677–681. [Google Scholar]
- Rajendra, S.; Sharma, P.; Gautam, S.D.; Saxena, M.; Kapur, A.; Sharma, P.; Merrett, N.; Yang, T.; Santos, L.D.; Pavey, D.; et al. Association of Biomarkers for Human Papillomavirus with Survival Among Adults with Barrett High-grade Dysplasia and Esophageal Adenocarcinoma. JAMA Netw. Open 2020, 3, e1921189. [Google Scholar] [CrossRef]
- Arber, N.; Gammon, M.D.; Hibshoosh, H.; Britton, J.A.; Zhang, Y.; Schonberg, J.B.; Roterdam, H.; Fabian, I.; Holt, P.R.; Weinstein, I.B.; et al. Overexpression of cyclin D1 occurs in both squamous carcinomas and adenocarcinomas of the esophagus and in adenocarcinomas of the stomach. Hum. Pathol. 1999, 30, 1087–1092. [Google Scholar] [CrossRef]
- Langer, R.; Von Rahden, B.H.; Nahrig, J.; Von Weyhern, C.; Reiter, R.; Feith, M.; Stein, H.J.; Siewert, J.R.; Höfler, H.; Sarbia, M. Prognostic significance of expression patterns of c-erbB-2, p53, p16INK4A, p27KIP1, cyclin D1 and epidermal growth factor receptor in oesophageal adenocarcinoma: A tissue microarray study. J. Clin. Pathol. 2006, 59, 631–634. [Google Scholar] [CrossRef]
- Zhang, K.; Li, J.-T.; Li, S.-Y.; Zhu, L.-H.; Zhou, L.; Zeng, Y. Integration of human papillomavirus 18 DNA in esophageal carcinoma 109 cells. World J. Gastroenterol. 2011, 17, 4242–4246. [Google Scholar] [CrossRef] [PubMed]
- Boon, S.S.; Chen, Z.; Li, J.; Lee, K.Y.C.; Cai, L.; Zhong, R.; Chan, P.K.S. Human papillomavirus type 18 oncoproteins exert their oncogenicity in esophageal and tongue squamous cell carcinoma cell lines distinctly. BMC Cancer 2019, 19, 1211. [Google Scholar] [CrossRef]
- IARC Monographs Priorities Group. Advisory Group recommendations on priorities for the IARC Monographs. Lancet Oncol. 2019, 20, 763–764. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.K. Epidemiology and Pathobiology of Esophageal Cancer; Tanabe, K.K., Savarese, D.M.F., Eds.; UpToDate: Waltham, MA, USA, 2022; Available online: https://www.uptodate.com/contents/epidemiology-and-pathobiology-of-esophageal-cancer (accessed on 23 January 2022).
- Spechler, S.J.; Souza, R.F. Barrett’s Esophagus. N. Engl. J. Med. 2014, 371, 836–845. [Google Scholar] [CrossRef]
- Li, S.; Luk, H.Y.; Xia, C.; Chen, Z.; Chan, P.K.S.; Boon, S.S. Oesophageal carcinoma: The prevalence of DNA tumour viruses and therapy. Tumour Virus Res. 2021, 13, 200231. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Ragunath, K.; Whitton, A.; Marsh, E.; Kaye, P.; Knight, G. Study to investigate the prevalence of human papillomavirus in Barrett’s oesophagus using a novel screening methodology. BMJ Open Gastroenterol. 2022, 9, e000840. [Google Scholar] [CrossRef]


{kind=link}
| Geographic Region | No. of Samples Tested | OAC | BO | No. of HPV-Positive Samples | HPV Prevalence (%) | No. of Reports |
|---|---|---|---|---|---|---|
| USA | 231 | 16 | 23 | 39 | 16.8 | 8 |
| UK | 141 | 68 | 73 | 9 | 6.38 | 5 |
| Germany | 211 | 8 | 0 | 8 | 3.79 | 4 |
| Australia | 340 | 228 | 112 | 22 | 6.4 | 2 |
| Italy | 23 | 23 | 0 | 3 | 13 | 2 |
| Turkey | 29 | 29 | 0 | 6 | 20.7 | 2 |
| Mexico | 45 | 17 | 28 | 42 | 93.3 | 1 |
| Netherlands | 48 | 48 | 0 | 11 | 22.9 | 1 |
| Iran | 4 | 0 | 4 | 1 | 25 | 1 |
| Sweden | 27 | 27 | 0 | 5 | 18.5 | 1 |
| South Africa | 1 | 1 | 0 | 0 | 0 | 1 |
| China | 57 | 0 | 0 | 0 | 0 | 1 |
| Korea | 3 | 3 | 0 | 0 | 0 | 1 |
| France | 40 | 40 | 0 | 0 | 0 | 1 |
| India | 5 | 5 | 0 | 0 | 0 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajendra, S.; Sharma, P. Causal Link of Human Papillomavirus in Barrett Esophagus and Adenocarcinoma: Are We There Yet? Cancers 2023, 15, 873. https://doi.org/10.3390/cancers15030873
Rajendra S, Sharma P. Causal Link of Human Papillomavirus in Barrett Esophagus and Adenocarcinoma: Are We There Yet? Cancers. 2023; 15(3):873. https://doi.org/10.3390/cancers15030873
Chicago/Turabian StyleRajendra, Shanmugarajah, and Prateek Sharma. 2023. "Causal Link of Human Papillomavirus in Barrett Esophagus and Adenocarcinoma: Are We There Yet?" Cancers 15, no. 3: 873. https://doi.org/10.3390/cancers15030873
APA StyleRajendra, S., & Sharma, P. (2023). Causal Link of Human Papillomavirus in Barrett Esophagus and Adenocarcinoma: Are We There Yet? Cancers, 15(3), 873. https://doi.org/10.3390/cancers15030873